

Abstract
Cold allodynia, pain in response to cooling, occurs during or within hours of oxaliplatin infusion and is thought to arise from a direct effect of oxaliplatin on peripheral sensory neurons. To characterize the pathophysiological mechanisms underlying acute oxaliplatin-induced cold allodynia, we established a new intraplantar oxaliplatin mouse model that rapidly developed long-lasting cold allodynia mediated entirely through tetrodotoxin-sensitive Nav pathways. Using selective inhibitors and knockout animals, we found that Nav1.6 was the key isoform involved, while thermosensitive transient receptor potential channels were not involved. Consistent with a crucial role for delayed-rectifier potassium channels in excitability in response to cold, intraplantar administration of the K+-channel blocker 4-aminopyridine mimicked oxaliplatin-induced cold allodynia and was also inhibited by Navl.6 blockers. Intraplantar injection of the Nav1.6-activator Cn2 elicited spontaneous pain, mechanical allodynia and enhanced 4-aminopyridine-induced cold allodynia. These findings provide behavioural evidence for a crucial role of Nav1.6 in multiple peripheral pain pathways including cold allodynia.
Introduction
Oxaliplatin, a third-generation platinum chemotherapeutic agent, is associated with acute dose-limiting neurotoxicity, which manifests as cooling-induced peripheral dysaesthesias and paraesthesias including cold allodynia [6; 12]. Acute oxaliplatin-induced cold allodynia is characterized by a rapid onset, with symptoms occurring during or shortly after infusion, and typically resolves within several days of treatment [5]. Many currently used animal models of oxaliplatin-induced neuropathy poorly reflect these characteristics, and often require multiple injections of oxaliplatin to elicit pain behaviours which develop slowly and are of prolonged duration [29; 39; 54]. Mechanistic studies in these animal models have attributed expressional changes and altered function of ion channels expressed on unmyelinated C-fiber nociceptors to the development of cold allodynia, such as the transient receptor potential (TRP) channels TRPM8, TRPA1 and the two-pore domain potassium (K+) channels TREK1 and TRAAK [16; 21; 34; 58]. However, these findings are inconsistent with the clinical time course of acute oxaliplatin-induced cold allodynia and the predominant effects of oxaliplatin on myelinated A-fibers [2; 6; 26; 45; 46]. Thus, the pathophysiological mechanisms underlying acute oxaliplatin-induced cold allodynia remain unclear. While oxaliplatin-induced allodynia has been described as an axonal channelopathy resulting from modulation of neuronal Nav channels [35], the contributions of the nine described isoforms (Nav1.1 – Nav1.9) have not been systematically assessed.
Dorsal root ganglion (DRG) neurons express several Nav isoforms, including the tetrodotoxin (TTX) resistant isoforms Nav1.8 and Nav1.9, as well as the TTX-sensitive isoforms Nav1.1, Nav1.2, Nav1.3, Nav1.6 and Nav1.7 [40]. The TTX-resistant Nav isoform Nav1.8 in particular has been found to be crucial for pain evoked by noxious cold [59], while Navl.9 has been suggested to contribute to the pathogenesis of neuropathic pain [28]. In addition, Nav1.7 is known to be crucial in pain pathways, as loss-of-function mutations in humans cause congenital insensitivity to pain [14], while gain-of-function mutations are associated with painful conditions such as erythromelalgia and paroxysmal extreme pain disorder [19]. In contrast, the functional roles of Nav1.1 and Nav1.6 in peripheral sensory neurons are less clear, and no evidence for involvement of these Nav isoforms in pain phenotypes has been reported to date, as both homozygous Scn1a−/− and Scn8a−/− mice develop motor deficits and die around postnatal day 15 to 20, preventing assessment of behavioural effects in mature animals [9; 55].
We established an animal model of oxaliplatin that more closely mimics acute chemotherapy-induced peripheral neuropathy. We found that intraplantar oxaliplatin rapidly induced a long-lasting cold allodynia that was mediated entirely through TTX-sensitive Nav isoform-dependent pathways. Surprisingly, Nav1.6 was implicated as the key Nav isoform involved, whereas thermosensitive TRP channels were not found to be involved. Consistent with reports of a crucial role for delayed-rectifier potassium channels in excitability in response to cold [52], intraplantar administration of the K+ channel blocker 4-aminopyridine (4-AP) mimicked oxaliplatin-induced cold allodynia and was inhibited by Navl.6 blockers or potentiated by Nav1.6 activators, supporting a crucial role for Navl.6 in chemically-mediated cold pain pathways.
Methods
Chemicals
Oxaliplatin and Dichloro(1,2-diaminocyclohexane)platinum(II) (Pt(DACH)Cl2) were obtained from Sigma Aldrich (Castle Hill, New South Wales, Australia) and dissolved in 5% glucose/H2O to a stock solution of 1 mg/mL to avoid spontaneous hydrolysis arising from the presence of Cl− in physiological solutions. μ-Conotoxins GIIIA and TIIIA were a kind gift from Professor Paul F. Alewood, The University of Queensland, Australia. Cn2 was isolated from the venom of the scorpion Centruroides noxius as previously described [43; 56]. M8-B (N-(2-aminoethyl)-N-(4-(benzyloxy)-3-methoxybenzyl)thiophene-2-carboxamide hydrochloride), a selective and potent antagonist of TRPM8), was synthesized and kindly provided by Amgen, Inc. [4]. The TRPM8 antagonist AMTB (N-(3-Aminopropy1)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride) and tetrodotoxin were from Tocris Bioscience (Bristol, United Kingdom). ProTxII was from Peptides International (Louisville, KY, USA). Peptides were routinely diluted in 0.1–0.3% albumin in phosphate-buffered saline to avoid adsorption to plastic surfaces. All other drugs and pharmacological modulators were diluted in phosphate-buffered saline. All other reagents were from Sigma Aldrich unless otherwise stated.
Animals
Ethical approval for in vivo experiments in animals was obtained from the local institutional animal ethics committee. Experiments involving animals were conducted in accordance with the Animal Care and Protection Act Qld (2002), the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 7th edition (2004) and the International Association for the Study of Pain Guidelines for the Use of Animals in Research.
For behavioural assessment of oxaliplatin-induced neuropathy, we used adult male C57BL/6J mice, age 5–12 weeks. Age-matched controls were used for studies involving knockout animals, and all mouse strains were back-crossed for a minimum of 5 (5–9) generations on C57BL/6 background. Knockout animals were kindly provided by the following researchers: TRPA1−/− mice (D. Corey, Harvard Medical School, Boston, MA, USA), TRPM8−/− mice (A. Patapoutian, The Scripps Research Institute, La Jolla, CA, USA), global Navl.8−/−, Navl.9−/−, and Navl.3−/− mice (J. Wood, University College London, London, UK).
Induction of oxaliplatin-induced neuropathy and behavioural assessment
To characterize nociceptive effects in wild-type C57BL/6J and age-matched TRPA1−/−, TRPM8−/−, Navl.8−/−, Navl.9−/− and Nav1.3−/− mice, a single dose of oxaliplatin, oxalate, Pt(DACH)Cl2, 4-AP, Cn2 or allyl isothiocyanate (AITC) was administered by shallow subcutaneous injection to the left hind paw in a volume of 40 μl (intraplantar injection, i.pl.) under light isoflurane anaesthesia. Quantification of spontaneous pain, cold and heat allodynia as well as mechanical allodynia was performed by a blinded observer unaware of the genotype and/or treatments received. Spontaneous nocifensive behaviour was quantified by counting the number of paw lifts, licks, shakes and flinches at room temperature (22–25°C) on a soft padded surface over a period of 5 min. Thermal allodynia was assessed by quantification of nocifensive behaviours over a 5 min period on a temperature-controlled Peltier plate (Hot/Cold Plate, Ugo Basile, Comerio, Italy). Mechanical allodynia was assessed by determining the paw withdrawal threshold to mechanical stimulation using an electronic von Frey apparatus (MouseMet Electronic von Frey, TopCat Metrology, Little Downham, United Kingdom). Briefly, mice were habituated in individual mouse runs for at least 10 min, and the paw withdrawal threshold was determined from the ipsilateral and controlateral paws in three separate trials, at least 5 min apart. The pressure applied through a soft-tipped probe was increased slowly over a pre-determined force rise rate (1 g/s). The force that elicited paw withdrawal was determined using the MouseMet Software and designated as the paw withdrawal threshold.
Where intraplantar injection elicited nocifensive behaviour at room temperature (Cn2, BAPTA, oxalate, 4-AP, AITC), thermal and mechanical allodynia was assessed after cessation of spontaneous pain (15 min to 1 h). To assess the effects of pharmacological modulators on the development of oxaliplatin-induced cold allodynia, compounds were administered by intraplantar injection of appropriately concentrated solutions (HC030031, 100 μM; AMTB, 10 & mu;M; M8-B, 1 μM; TTX, 3 μM; A803467, 10 μM; ProTxII, 3 nM; GIIIA, 10 μM; TIIIA, 10 μM) 5–15 min prior to behavioural quantification. To assess the effect of pharmacological modulators on the nocifensive responses elicited by 4-AP, compounds were co-administered by intraplantar injection as appropriately concentrated solutions (Cn2, l nM; GIIIA, 10 μM; TTX, 3 μM; AMTB, 10 μM; HC030031, 100 μM) in a final volume of 40 μl. No systemic effects, including ataxia, altered gait or motor paralysis were apparent in any mice or after intraplantar injection of any pharmacological modulators. In addition, no sustained hind paw favouring, inflammation, swelling or ulceration of the oxaliplatin-injected paw was visible. Injection of equal volumes of 5% glucose/H2O and phosphate-buffered saline with or without albumin did not elicit any nocifensive behaviour.
FLIPR Membrane Potential Assays
To verify the in vitro potency of compounds with activity Navl.6 channels, inhibition of veratridine-induced membrane potential responses were assessed using the FLIPRTETRA (Molecular Devices, Sunnyvale, CA) plate reader. Nav1.6-expressing CHO cells (EZcells, Chantest, Cleveland, OH) were loaded with Red Membrane Potential dye (Molecular Devices), and responses to stimulation with veratridine (50 μM) were assessed after 5 min pre-treatment with antagonists as previously described [50].
Data and statistical analysis
Fluorescence values from membrane potential imaging experiments were converted to response over baseline values using Screen Works 3.2.0.14 as previously described [50]. For concentration-response curves, maximum values from the response after addition of agonist were plotted against agonist concentration and a 4-parameter logistic Hill equation was fitted to the data using GraphPad Prism Version 5.03 (San Diego, CA). Statistical significance was defined as p < 0.05 and was determined using paired or unpaired Student’s t-tests and one-way ANOVA analysis with Dunnett’s post test as indicated. Statistical analysis was performed using GraphPad Prism Version 5.03.
Results
A mouse model of chemotherapy-induced cold allodynia based on intraplantar administration of oxaliplatin
In humans, cold allodynia generally occurs during or within hours of oxaliplatin infusion and is characterized by pain in response to normally innocuous cooling, presumably resulting from a direct effect of oxaliplatin on peripheral sensory neurons. To isolate the actions of oxaliplatin on peripheral sensory neurons, we established a novel mouse model of oxaliplatin-induced cold allodynia based on the administration of oxaliplatin by shallow subcutaneous (intraplantar, i.pl.) injection into the hind paw of C57/BL6J mice. Intraplantar injection of oxaliplatin (4–40 μg) elicited rapid, dose-dependent development of cold allodynia, evidenced by flinching, lifting, licking and shaking of the affected hind paw upon exposure to a cooled surface (10°C) (Fig. 1a).
Figure 1. A novel animal model of chemotherapy-induced neuropathy based on the intraplantar injection of oxaliplatin.

(a) Intraplantar injection of oxaliplatin (4 – 40 μg/paw) rapidly elicits cold allodynia, with increased paw lifting, licking, shaking and flinching evident 1 h after injection upon exposure to a temperature-controlled surface maintained at 10°C. Injection of vehicle (Control; 5% glucose/H2O) did not elicit any nocifensive responses. (b) Oxaliplatin-induced cold allodynia has a rapid onset (left panel, 0–4 h post-injection), with nocifensive responses upon exposure of the injected hind paw to cool temperatures becoming apparent within minutes after injection (arrow). Cold allodynia after a single injection persists for several days (right panel, 4 h - 9 days post-injection) after intraplantar injection (arrow). (c) Nocifensive responses evoked by intraplantar injection of oxaliplatin (40 μg/paw) are temperature-dependent, with significant paw withdrawals elicited upon exposure to temperatures below 15°C (24 h after injection). No withdrawal responses were evident at elevated temperatures up to 42°C. White bar; for all subsequent experiments, cold allodynia was assessed 24 h after injection of 40 μg oxaliplatin/paw by quantifying paw withdrawal responses at 10°C. (d) Intraplantar injection of oxaliplatin (40 μg/paw) elicited mild mechanical allodynia, with a significant decrease in paw withdrawal threshold to mechanical stimulation compared to control (5% glucose/H2O). Left panel, decreased mechanical threshold was apparent at 1 h after injection and persisted 24 h after injection (right panel). Statistical significance was determined using an unpaired Student’s t-test; ***, p < 0.001; **, p < 0.01 compared to vehicle. Data are presented as mean ± SEM (n = 5- 12 animals/group).
This dose (1.6–2.0 mg/kg) is approximately equivalent to human therapeutic doses (2.5–3.5 mg/kg), and considerably lower than systemic doses previously reported to elicit acute cold allodynia in rodents (5 – 10 mg/kg) [58].
Strikingly, cold allodynia induced by a single dose of oxaliplatin became apparent within minutes of injection and persisted for several days, with significant pain behaviour evident for up to 7 days after injection of the highest dose (2.5 mM; 40 μg; Fig. 1b). The terminal elimination phase of platinum-containing metabolites is long, suggesting that the prolonged effect of a single intraplantar injection of oxaliplatin could arise from the pharmacokinetics of these oxaliplatin metabolites. Alternatively, oxaliplatin, or platinum metabolites, may elicit irreversible changes in neuronal proteins which are involved in mediating increased excitability to cool stimuli.
Nocifensive behaviour evoked by intraplantar injection of oxaliplatin became apparent at temperatures below 15°C (18.2 ± 6.5 flinches/5 min), but no heat allodynia was evident, with animals displaying little or no nocifensive behaviour at elevated temperatures up to 42°C (1.0 ± 0.7 flinches/5 min) (Fig. 1c). In addition, intraplantar injection of oxaliplatin also elicited mechanical allodynia, evidenced by decreased paw withdrawal threshold to mechanical stimulation (Fig. 1d; Control, 4.7± 0.3 g; oxaliplatin 2.5 ± 0.3 g). Therefore, this novel animal model of intraplantar oxaliplatin produces behavioural responses that parallel the human symptomatology of oxaliplatin-induced neuropathy, confirming a direct peripheral effect of oxaliplatin on sensory nerve endings as the basis of cold-evoked paraesthesias and dysaesthesias.
Oxaliplatin metabolites contribute to mechanical, but not cold allodynia
Oxaliplatin is rapidly hydrolyzed in vivo to bioactive derivatives through displacement of the oxalate group by H2O and Cl− to produce oxalate as well as reactive monochloro-, dichloro- and diaquo-diaminocyclohexane platinum metabolites [18; 22]. As these oxaliplatin metabolites have previously been suggested to contribute to oxaliplatin-induced neuropathy, we sought to characterize the contribution of oxalate and the oxaliplatin metabolite Pt(DACH)Cl2 to oxaliplatin-induced cold allodynia in our model. We found that intraplantar injection of equivalent doses of Pt(DACH)Cl2, (2.5 mM; 38 μg) or oxalate (2.5 mM; 14 μg) did not cause cold allodynia (Fig. 2a). However, injection of a higher dose of oxalate (150 mM; 810 μg/paw) caused short-lived (< 1 h) spontaneous nocifensive behaviour, evidenced by lifting, licking and shaking of the paw (Fig. 2b), as well as mechanical allodynia which remained apparent 24 h after a single intraplantar injection of oxalate (Fig. 2c; Control, 4.4 ± 0.5 g; oxalate 0.9± 0.3 g). Consistent with the ability of oxalate to chelate Ca2+ [23], both the spontaneous pain (29.1 ± 5.1 flinches/5 min) and mechanical allodynia (2.6 ± 0.3 g) were mimicked by intraplantar injection of the Ca2+ chelator BAPTA (10 mM; 191 μg) (Fig. 2b and 2c). In contrast, intraplantar injection of the cell membrane-permeable BAPTA-AM [48] (10 μM; 310 ng) had no effect on spontaneous nocifensive behaviour and did not elicit cold allodynia (Fig. 2a-c). The effect of oxalate and BAPTA on sensory nerve endings likely arises from the destabilizing effect of Ca2+ chelation on neuronal membranes which interferes with the surface screening charge. Thus, removal of extracellular Ca2+ results in increased excitability by decreasing the threshold potential and membrane resistance, and increasing Na+ conductance [20]. These effects have been shown in ex vivo preparations to result in spontaneous action potential discharge and an increase in mean firing frequency [24; 42], corroborating the proalgesic effect of extracellular Ca2+ chelation we observed in our animal model.
Figure 2. Oxaliplatin metabolites contribute to mechanical, but not cold allodynia.
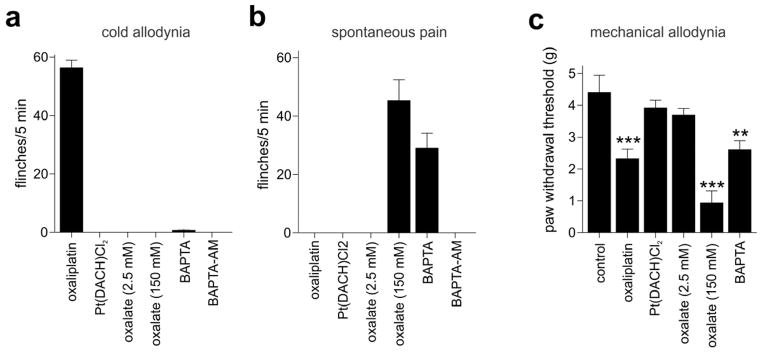
a) Intraplantar injection of equimolar doses of the oxaliplatin metabolites Pt(DACH)Cl2 (2.5 mM; 38 μg/paw) and oxalate (2.5 mM; 14 μg/paw) did not elicit paw withdrawal responses at 10°C, compared to the pronounced cold allodynia elicited by oxaliplatin (2.5 mM; 40 μg/paw). Cold allodynia was also not elicited by intraplantar injection of the Ca2+ chelator BAPTA (10 mM; 191 μg/paw) or the membrane-permeable BAPTA-AM (10 μM; 310 ng/paw). (b) Intraplantar injection of high doses of oxalate (150 mM; 810 μg/paw) and BAPTA (10 mM; 191 μg/paw), but not low doses of oxalate, oxaliplatin, BAPTA-AM or Pt(DACH)Cl2, elicited short-lasting (< 1 h) spontaneous nocifensive behaviour (increased number of paw lifts, licks, shakes and flinches) evident at room temperature. (c) Intraplantar injection of high doses of oxalate (150 mM; 810 μg/paw), BAPTA (10 mM; 191 μg/paw) and oxaliplatin (2.5 mM; 40 μg/paw) caused a significant decrease in the paw withdrawal threshold to mechanical stimulation, while mechanical responses were unchanged after intraplantar injection of equimolar doses of Pt(DACH)Cl2, and oxalate. Statistical significance was determined using a one-way ANOVA with Dunnett’s post test. ***, p < 0.001; **, p < 0.01 compared to Control (vehicle). Data are presented as mean ± SEM (n = 3 – 5) animals/group.
Oxaliplatin-induced cold allodynia is mediated through Nav1.6-expressing peripheral sensory fibers
Nav channels are critical for the propagation of action potentials in excitable cells, including peripheral sensory nerves. The tetrodotoxin-resistant isoform Nav1.8 in particular is crucial for neuronal excitability at cold temperatures and is essential for noxious cold pain [59]. Thus, we sought to elucidate the contribution of Navl.8 to oxaliplatin-induced cold allodynia. Surprisingly, the development of cold allodynia was unchanged in Nav1.8−/− animals, and was also not affected by A803467, a Nav1.8-selective small molecule inhibitor (Fig. 3a; Nav1.8−/−, 98 ± 15% of control; A803467 (10 μM), 90 ± 17% of control). Similarly, the tetrodotoxin-resistant Navl.9 has previously been suggested to contribute to the pathogenesis of neuropathic pain and cold allodynia [28]. However, in our model the cold allodynia was unchanged in Nav1.9−/− animals (Fig. 3a; 107 ± 8% of control), suggesting that TTX-sensitive Nav isoforms are crucial for the development of oxaliplatin-induced cold allodynia. Indeed, intraplantar injection of low concentrations of TTX (3 μM) inhibited nocifensive responses upon exposure to a surface cooled to 10°C (Fig. 3a; 19 ± 4% of control). We thus assessed the contribution of Nav1.3 and Nav1.7 using knockout animals or subtype-selective inhibitors. Surprisingly, the development of cold allodynia was also unchanged in Nav1.3−/− animals (103 ± 11% of control), or after intraplantar injection of the Navl.7-selective inhibitor ProTxII (3 nM; 115 ± 17% of control) (Fig. 3b), suggesting involvement of Nav isoforms which have not yet been associated with any prominent function in pain pathways. In addition to Nav1.3, Nav1.7, Nav1.8 and Nav1.9, DRG neurons are known to express other tetrodotoxin-sensitive isoforms, including Nav1.1, Nav1.2 and Nav1.6. Since knockout mouse models of these Nav isoforms are lethal, we used a range of conotoxins with activity at Nav isoforms that allowed dissection of the contribution of these isoforms to oxaliplatin-induced pain pathways. μ-Conotoxin TIIIA specifically inhibits Navl.2 and Navl.4 at low concentrations, while at high concentrations Nav1.1, but not Nav1.6, is also inhibited [53; 57] (Fig. 3c; Nav1.6 pIC50 6.0 ± 0.3). Intraplantar injection of both low (100 nM) or high (10 μM) concentrations of TIIIA did not significantly decrease oxaliplatin-induced cold allodynia (Fig. 3D), suggesting a crucial role for Navl.6 in cold pain pathways activated by oxaliplatin. Indeed, intraplantar injection of GIIIA (10 μM), which in addition to Nav1.1 also inhibits Nav1.6 at high concentrations, but has no effect on Nav1.3 and Nav1.7 [53], achieved near complete reversal of oxaliplatin-induced cold allodynia (14 ± 9% of control) (Fig. 3d). In contrast, nocifensive behavior elicited by intraplantar administration of the TRPA1 agonist AITC (5 mM) was not affected by GIIIA (30 μM; Fig 3e). Thus, this demonstrates for the first time a functional contribution of Navl.6 to cold pain pathways at the behavioural level.
Figure 3. Nav isoforms involved in the development of cold allodynia after intraplantar injection of oxaliplatin.
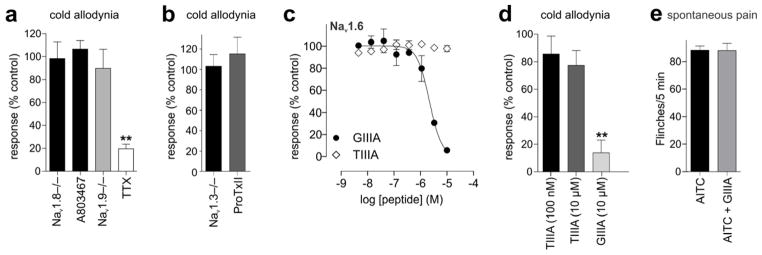
(a) Cold allodynia induced by intraplantar injection of oxaliplatin (24 h after injection of 2.5 mM oxaliplatin; 40 μg/paw) was not significantly different from control in Navl.8−/− animals, or after intraplantar injection of the Navl.8 inhibitor A803467 (10 μM). Paw flinches were also not significantly different from control in Navl.9−/− animals, but were significantly (p < 0.01) inhibited by intraplantar injection of TTX (3 μM). (b) Oxaliplatin-induced cold allodynia was not significantly different from control in Navl.3−/− animals, or after intraplantar injection of the Navl.7 inhibitor ProTxII (3 nM). (c) μ-Conotoxin GIIIA concentration-dependently (pIC50 6.0 ± 0.3) inhibits Navl.6, while TIIIA does not affect Navl.6-mediated responses. Effect of μ-conotoxins on veratridine (50 μM)-induced Navl.6 responses was assessed using a FLIPR membrane-potential assay in HEK cells heterologously expressing Navl.6. (d) Intraplantar injection of TIIIA at concentrations which inhibit Navl.2 (100 nM), or Navl.1 but not Navl.6 (10 μM) did not significantly decrease oxaliplatin-induced cold allodynia. In contrast, GIIIA at a concentration which fully inhibits Navl.6 (10 μM) caused near complete inhibition of cold allodynia. (e) GIIIA (30 μM) had no effect on nocifensive behaviours elicited by intraplantar administration of the TRPA1 agonist AITC (5 mM). Data is presented relative to vehicle-injected wi1d-type animals or age-matched litter controls. Statistical significance was determined using a one-way ANOVA with Dunnett’s post test. **, p < 0.01 compared to Control (vehicle or age-matched litter controls). Data are presented as mean± SEM (n = 4 – 8 animals/group).
Oxaliplatin-induced cold allodynia develops independently of cold-sensitive TRP channels
In peripheral sensory neurons, cold stimuli are transformed to electrical signals through activation of thermosensitive TRP channels, notably TRPM8, TRPA1 and TRPC5. We thus sought to elucidate the contribution of cold-sensitive TRP channels to the development of cold allodynia in our novel model of acute oxaliplatin-induced neuropathy. Surprisingly, oxaliplatin-induced cold allodynia was unaffected in TRPM8−/− animals (128 ± 17% of control) or by the TRPM8-selective inhibitors AMTB (10 μM; 107 ± 13% of control) and M8-B (1 μM; 108 ± 13% of control) (Fig. 4). Cold allodynia was also not significantly decreased in TRPA1−/− animals (115 ± 18% of control) or after treatment with the TRPA1 antagonist HC030031 (100 μM; 76 ± 14% of control), and developed normally in TRPC5−/− animals (data not shown) (Fig. 4). Thus, alternative mechanisms to transform a cool stimulus to an electrical signal are likely to contribute to oxaliplatin-induced cold allodynia.
Figure 4. Oxaliplatin-induced cold allodynia develops independently of cold-sensitive TRP channels.

Oxaliplatin-induced cold allodynia was not changed in TRPM8−/− animals or after intraplantar injection of the TRPM8 antagonists AMTB (10 μM) and M8-B (1 μM). Similarly, no significant difference in the number of paw flinches was observed in TRPA1−/− animals or after intraplantar injection of the TRPA1 antagonist HC030031 (100 μM). Statistical significance was determined using a one-way ANOVA with Dunnett’s post test. Data are presented as mean ± SEM (n = 5–10 animals/group).
In some sensory and central neurons, cooling elicits enhanced excitability and increased firing frequency as a result of cold-induced closure of background potassium channels [3; 15; 33; 38; 52]. This effect appears to be opposed by continued activity of Kvl channels which act as an excitability break and regulate cold sensitivity in trigeminal neurons in concert with TRPM8 [32; 52]. Since it is known that oxaliplatin inhibits potassium channels [26] in addition to sodium channels [2; 8; 23; 27; 46], we assessed if the oxaliplatin-induced effects could be replicated by inhibition of delayed rectifier potassium channels in sensory nerve endings. Indeed, intraplantar injection of 4-AP (1 mM) elicited cold allodynia (35.2 ± 8.6 flinches/5 min) which was not affected by intraplantar injection of the TRPA1 inhibitor HC030031 (100 μM; 37.4 ± 13.4 flinches/5 min) or the TRPM8 inhibitor AMTB (10 μM; 46.6 ± 10.5 flinches/5 min) (Fig. 5a). Like oxaliplatin-induced cold allodynia, enhanced nocifensive responses to cold elicited by 4-AP were inhibited by the Navl.6 inhibitor GIIIA (8.6 ± 4.4 flinches/5 min), confirming an important role for Navl.6 in cold pain pathways (Fig. 5A).
Figure 5. Inhibition of potassium channels on Nav1.6-expressing pain pathways elicits cold allodynia.
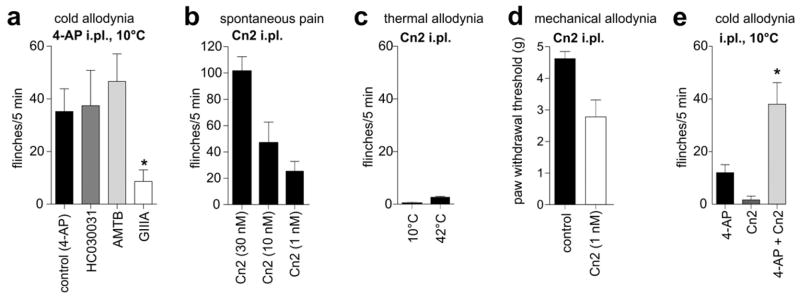
(a) Intraplantar injection of the potassium channel inhibitor 4-AP (1 mM) elicited cold allodynia, evidenced by an increased number of paw lifts, licks, shakes and flinches on exposure to a temperature-controlled plate maintained at 10°C. Cold allodynia induced by 4-AP was not significantly inhibited by concomitant intraplantar injection of the TRPM8 antagonist AMTB (10 μM) or the TRPA1 antagonist HC030031 (100 μM), but was significantly (p < 0.05) inhibited by intraplantar μ-conotoxin GIIIA (10 μM). (b) Intraplantar injection of Cn2 (1 nM – 30 nM) elicited dose-dependent spontaneous pain. Responses were quantified by counting the number of behaviours immediately after injection (c) Activation of peripheral Nav1.6 by Cn2 (1 nM) did not elicit cold (10°C) or heat (42°C) allodynia. (d) Intraplantar Cn2 (1 nM) caused significant (p < 0.01) mechanical allodynia, with the paw withdrawal threshold to mechanical stimulation decreased from 5.4 ± 0.4 g (control) to 2.8 ± 0.6 g (Cn2, 1 nM). (e) Intraplantar injection of the Nav1.6 activator Cn2 (10 nM) alone did not elicit significant cold allodynia (1.7 ± 1.5 flinches/5 min at 10°C), but significantly (p < 0.05) potentiated cold allodynia elicited by intraplantar injection of 4-AP (500 μM; 12.0 ± 3.1 flinches/5 min at 10°C) when co-administered (4-AP + Cn2; 38.0 ± 8.2 flinches/5 min). Statistical significance was determined using a one-way ANOVA with Dunnett’s post test. *, p < 0.05; **, p < 0.01 compared to control. Data are presented as mean ± SEM (n = 4 – 7 animals/group).
Pain behaviors induced by selective Nav1.6 activation
To further characterize the role of Nav1.6 in pain pathways, we also assessed spontaneous pain behaviours, thermal allodynia and mechanical allodynia after intraplantar injection of the Nav1.6-selective activator Cn2. Cn2 is a β-scorpion toxin isolated from the venom of the scorpion Centruroides noxius that specifically enhances activity of Nav1.6 with an EC50 of 39 nM, causing a leftward shift of the voltage-dependence of activation and a transient resurgent current [43]. Intraplantar injection of Cn2 elicited dose-dependent spontaneous pain characterized by licking, lifting and vigorous shaking of the injected paw that was transient for lower concentrations (1 nM, < 15 min). At the highest concentration tested (30 nM; Fig. 5b), the frequency and severity of these responses rapidly diminished after injection, although some nocifensive behaviours remained evident for > 4 h after intraplantar administration. Thus, for subsequent experiments, low concentrations of Cn2 were utilized.
Intraplantar Cn2 (1 nM) did not elicit thermal allodynia, with little or no nocifensive behaviour evident at 10°C or 42°C (Fig. 5c) but caused significant (p < 0.01) mechanical allodynia, evidenced by decreased paw withdrawal thresholds to mechanical stimulation (Fig. 5d; Control, 5.4 ± 0.4 g; Cn2 (1 nM), 2.8 ± 0.6 g). To examine the contribution of potassium channels to Nav1.6-dependent cold-pain, we co-administered Cn2 (10 nM) with 4-AP (500 μM) by intraplantar injection. As opposed to Cn2 alone, this combination potentiated the cold allodynia produced by 4-AP (Fig. 5e; 4-AP, 12.0 ± 3.1 flinches/5 min; 4-AP + Cn2, 38.0 ± 8.2 flinches/5 min), providing evidence that activation of Nav1.6 per se produced only spontaneous pain and mechanical allodynia, but when combined with inhibition of delayed rectifier potassium channels could enhance cold allodynia.
Discussion
Acute oxaliplatin-induced neuropathy occurs in almost all patients and manifests as circumoral and distal sensory and/or motor disturbances including paraesthesias and dysaesthesias and muscle fasciculations. These symptoms are triggered by exposure to cold and are associated with a significant reduction in the cold pain threshold [6]. However, the pathophysiological basis of acute oxaliplatin-induced neuropathy, in particular cold allodynia, is poorly understood. This is in part due to a paucity of animal models that accurately reflect the neuropathic symptomatology encountered clinically and, specifically, the rapid onset of cold allodynia. To better understand chemically-induced cold allodynia, we established an animal model of chemotherapy-induced peripheral neuropathy based on the intraplantar injection of oxaliplatin. This model supports a direct excitatory action of oxaliplatin on peripheral sensory nerve endings as the causative mechanism underlying oxaliplatin-induced neuropathy, with cold allodynia becoming evident within minutes and persisting for several days after a single local injection of oxaliplatin. We were able to show that inhibition of potassium channels leads to increased neuronal excitability at low temperatures independent of activation of TRP channels, and that peripheral Navl.6 was crucial for the propagation of these signals and the appearance of cold allodynia.
The metabolism of oxaliplatin is complex and involves rapid hydrolysis (t½α ~ 14 min) to oxalate and various bioactive platinum compounds, which in turn form adducts with DNA, proteins, peptides and amino acids that undergo slow, triphasic elimination through predominantly renal routes, with a long terminal half-life for platinum of up to 11 days [18; 22]. It is difficult to estimate the concentration of oxaliplatin that sensory neurons are exposed to in human patients. While the plasma concentration of free oxaliplatin after intravenous administration is relatively low [18; 22], consistent with an apparent large volume of distribution, oxaliplatin is likely to accumulate in sensory neurons through active transport by copper transporters and the L-carnitine transporter OCTN1 [25; 30].
Given the rapid onset and prolonged nature of the sensory disturbances associated with oxaliplatin infusion [5], contribution of various oxaliplatin metabolites to the development of peripheral neuropathy has been suggested. In DRG explants, Pt(DACH)Cl2 was more neurotoxic than oxaliplatin [31], while after repeated intraperitoneal administration, oxalate elicited cold hyperalgesia and increased paw withdrawal responses to application of acetone, but not mechanical allodynia [41]. We thus assessed the effect of equimolar doses of oxalate and Pt(DACH)Cl2, two major oxaliplatin metabolites, on the development of cold and mechanical allodynia after intraplantar injection. However, while neither metabolite elicited cold allodynia after a single local injection, Ca2+ chelation by oxalate elicited spontaneous nocifensive behaviour as well as prolonged mechanical allodynia. This effect was mimicked by intraplantar injection of BAPTA and can be attributed to the effects of extracellular Ca2+ removal on membrane properties, including decreased threshold potential and membrane resistance, as well as increased Na+ conductance [20].
Acute oxaliplatin-induced neuropathy has been postulated to involve the modulation of axonal Nav channels, based on the observation that oxaliplatin infusion elicited changes in Nav-dependent variables in humans [27; 35]. Similarly, in rat DRG neurons, oxaliplatin resulted in increased Na+ currents and a shift of the voltage-response relationship towards more negative potentials, [2] with similar effects observed in cockroach neurons and frog myelinated axons [8; 23].
The lack of contribution of Navl.8 to oxaliplatin-induced cold allodynia, which we demonstrated in both Navl.8 knockout animals and after intraplantar administration of the Navl.8-selective inhibitor A803467, was surprising given the crucial role of this Nav isoform in cold pain. Specifically, Nav1.8 in nociceptive peripheral sensory neurons has previously been demonstrated to be critical for the development of pain evoked by noxious cold [59], and in mice with diphtheria toxin-mediated ablation of Nav1.8-expressing nociceptors, noxious cold responses are virtually abolished [1]. Oxaliplatin induces repetitive firing, broadening of the repolarization phase and after-hyperpolarization in myelinated A-fibers, while non-myelinated C-fibers remain largely unaffected [2; 26; 45; 46]. In contrast, although Navl.8 is widely expressed in peripheral sensory neurons, including a subpopulation of myelinated A-fibers [44], its contribution to cold-evoked pain behavior arises mainly from nociceptive C-fibers [1; 51; 59]. Thus, our finding that Nav1.8 does not contribute to oxaliplatin-induced cold allodynia can be explained by the differential expression of Nav isoforms in peripheral sensory nerve fibers that contribute to the pathophysiology of oxaliplatin-induced cold allodynia. The role of Nav1.8 in pathological cold pain appears to be different from its role in physiological cold pain [59], and in addition differs to other models of chemically-induced cold allodynia, such as ciguatoxin-induced cold pain, where Nav1.8 still contributes significantly to pain behaviours [51].
Consistent with a major role for Navl.6 in the propagation of action potentials in myelinated A-fibers [53; 57], we found no contribution of Navl.3, Nav1.7 and Nav1.9 to oxaliplatin-induced cold allodynia, while pharmacological inhibition of Navl.6 virtually abolished pain behaviour. Supporting a crucial role for Navl.6 in oxaliplatin-induced cold allodynia is the observation that in vitro, oxaliplatin elicits Navl.6-mediated resurgent currents and that oxaliplatin-induced A-fiber effects were abolished in Navl.6 knockout animals [46]. Similarly, we observed significant potentiation of veratridine-induced Nav1.6 responses in our membrane potential assay (data not shown), consistent with the previously reported effect of oxaliplatin on fast inactivation [46].
Thus, we have obtained the first evidence that Navl.6 expressed in peripheral sensory neurons contributes to cold pain behaviours. However, since Nav1.6 is highly expressed at nodes of Ranvier in both peripheral sensory and motor axons, as well as nodes in the central nervous system [10], Nav1.6 would be difficult to target therapeutically. Indeed, mice with loss-of-function mutations in Scn8a, the gene encoding for Navl.6, are characterized by early onset progressive paralysis of the hind limbs, leading to juvenile lethality at approximately postnatal day 20 [9]. Thus, the results presented here support a role for Nav1.6 in pathological pain states. Future experiments in sensory fiber-specific knockout model would be valuable to further dissect the role of Navl.6 in pain pathways.
Thermosensitive TRP channels, in particular TRPM8, TRPA1 and TRPC5, are expressed in peripheral sensory neurons and are activated by cooling [36; 47; 60]. Chronic administration of oxaliplatin in animal models has been shown to elicit changes in TRP channel expression, and both TRPM8 and TRPA1 have been causally implied in the development of chemotherapy-induced cold allodynia [21; 34; 58]. However, the rapid onset of cold allodynia both clinically and in our novel model of acute oxaliplatin-induced cold allodynia suggests that changes in the expression level of TRP channels are unlikely to contribute to the observed symptomatology. Indeed, we found no significant effect of TRPA1, TRPM8 or TRPC5 to oxaliplatin-induced cold allodynia using both genetically modified animals and pharmacological modulators where possible. This finding is consistent with the predominantly A-fiber-mediated origin of oxaliplatin-induced cold allodynia, as cold-sensitive TRP channels are expressed predominantly on peptidergic and isolectin B4-positive C-fibers [17; 47; 60].
Activation of TRPM8 by cooling has been demonstrated in peripheral sensory neurons, trigeminal neurons and corneal neurons [7; 11; 17]. In addition, heterologously expressed TRPM8 is also activated by cooling, and a role for TRPM8 has been demonstrated in environmental cold sensing as well as noxious cold pain in several behavioural studies [7; 13]. We found a significant response to acetone in naïve C57BL/6 mice, consisting of vigorous shaking, licking and aversive behaviours, and for this reason chose to assess cold pain behaviour by exposure to a temperature-controlled plate. Previous studies have also reported sensitivity to acetone in naïve animals, which was decreased in TRPM8 knockout animals [13]. This observation could account for the effect of TRPM8 on oxaliplatin-induced cold allodynia previously reported.
An alternative mechanism of inducing cold sensitivity is based on inhibition of potassium channels. In addition to modulation of Nav, oxaliplatin also inhibits neuronal potassium channels [8; 26]. In peripheral myelinated fibers, the effects of oxaliplatin on compound action potentials were similar to those of 4-AP [26]. 4-AP inhibits delayed-rectifier channels, including Kv1.1 and Kv1.2 which have been shown to be highly expressed in cold-insensitive neurons and contribute to lack of cold-sensitivity in trigeminal neurons [32; 49; 52]. Indeed, intraplantar injection of 4-AP caused behavioural responses similar to oxaliplatin, and elicited cold allodynia that was not modulated by inhibition of TRPM8 or TRPA1, but was decreased by pharmacological inhibition of Nav1.6. These findings are consistent with Kv1.1 and Kv1.2 being expressed predominantly in large DRG neurons which give rise to myelinated A-fibers [37], and corroborate an important role for Kv channels in cold sensing and cold allodynia.
Indeed, activation of Nav1.6 alone was not sufficient to elicit cold allodynia, with the Nav1.6-specific scorpion toxin Cn2 eliciting spontaneous pain and mechanical allodynia but not cold allodynia. However, when combined with inhibition of Kv channels, Cn2 produced profound enhancement of 4-AP-induced cold allodynia. Thus Cn2 not only confirms the pivotal role played by Nav1.6 in chemically-induced cold pain but reveals a role for Nav1.6 in spontaneous pain and mechanical allodynia. In conclusion, the new animal model of oxaliplatin-induced cold allodynia described here reveals an important role for Nav1.6 in pain pathways, with chemically-induced cold allodynia mediated through inhibition of potassium channels on Navl.6-expressing peripheral sensory fibers.
Acknowledgments
This work was supported by an NHMRC Australian Biomedical Postdoctoral Fellowship (569918, IV), NHMRC Fellowship (APP1019761, RJL) and Australian Postgraduate Award (JRD). Funding for this project was obtained through an Australian Research Council LIEF grant for the FLIPRTETRA, a Cancer Council Research Grant (IV), an NHMRC project grant (IV, KZ), an NHMRC Program Grant (RJL) and a National Institutes of Health grant R01NS41233 (AAR).
Footnotes
Conflict of interests
AAR has consulted for TRP programs at several pharmaceutical companies, and his TRP-related research has been supported by Amgen, Inc., Abbott Laboratories, and AbbVie.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321(5889):702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- 2.Adelsberger H, Quasthoff S, Grosskreutz J, Lepier A, Eckel F, Lersch C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur J Pharmacol. 2000;406(1):25–32. doi: 10.1016/s0014-2999(00)00667-1. [DOI] [PubMed] [Google Scholar]
- 3.Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006;25(11):2368–2376. doi: 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida MC, Hew-Butler T, Soriano RN, Rao S, Wang W, Wang J, Tamayo N, Oliveira DL, Nucci TB, Aryal P, Garami A, Bautista D, Gavva NR, Romanovsky AA. Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J Neurosci. 2012;32(6):2086–2099. doi: 10.1523/JNEUROSCI.5606-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev. 2008;34(4):368–377. doi: 10.1016/j.ctrv.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Attal N, Bouhassira D, Gautron M, Vaillant JN, Mitry E, Lepere C, Rougier P, Guirimand F. Thermal hyperalgesia as a marker of oxaliplatin neurotoxicity: a prospective quantified sensory assessment study. Pain. 2009;144(3):245–252. doi: 10.1016/j.pain.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt SE, Julius D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448(7150):204–208. doi: 10.1038/nature05910. [DOI] [PubMed] [Google Scholar]
- 8.Benoit E, Brienza S, Dubois JM. Oxaliplatin, an anticancer agent that affects both Na+ and K+ channels in frog peripheral myelinated axons. Gen Physiol Biophys. 2006;25(3):263–276. [PubMed] [Google Scholar]
- 9.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10(4):461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 10.Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1. 6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97(10):5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr RW, Pianova S, McKemy DD, Brock JA. Action potential initiation in the peripheral terminals of cold-sensitive neurones innervating the guinea-pig cornea. J Physiol. 2009;587(Pt 6):1249–1264. doi: 10.1113/jphysiol.2008.167023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cersosimo RJ. Oxaliplatin-associated neuropathy: a review. Ann Pharmacother. 2005;39(1):128–135. doi: 10.1345/aph.1E319. [DOI] [PubMed] [Google Scholar]
- 13.Colburn RW, Lubin ML, Stone DJ, Jr, Wang Y, Lawrence D, D’Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54(3):379–386. doi: 10.1016/j.neuron.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de la Pena E, Malkia A, Vara H, Caires R, Ballesta JJ, Belmonte C, Viana F. The influence of cold temperature on cellular excitability of hippocampal networks. PLoS One. 2012;7(12):e52475. doi: 10.1371/journal.pone.0052475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, Couette B, Busserolles J, Courteix C, Noel J, Lazdunski M, Eschalier A, Authier N, Bourinet E. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med. 2011;3(5):266–278. doi: 10.1002/emmm.201100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhaka A, Earley TJ, Watson J, Patapoutian A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J Neurosci. 2008;28(3):566–575. doi: 10.1523/JNEUROSCI.3976-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehrsson H, Wallin I, Yachnin J. Pharmacokinetics of oxaliplatin in humans. Med Oncol. 2002;19(4):261–265. doi: 10.1385/MO:19:4:261. [DOI] [PubMed] [Google Scholar]
- 19.Fischer TZ, Waxman SG. Familial pain syndromes from mutations of the NaV1. 7 sodium channel. Ann N Y Acad Sci. 2010;1184:196–207. doi: 10.1111/j.1749-6632.2009.05110.x. [DOI] [PubMed] [Google Scholar]
- 20.Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol. 1957;137(2):218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauchan P, Andoh T, Kato A, Kuraishi Y. Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neurosci Lett. 2009;458(2):93–95. doi: 10.1016/j.neulet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E. Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res. 2000;6(4):1205–1218. [PubMed] [Google Scholar]
- 23.Grolleau F, Gamelin L, Boisdron-Celle M, Lapied B, Pelhate M, Gamelin E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J Neurophysiol. 2001;85(5):2293–2297. doi: 10.1152/jn.2001.85.5.2293. [DOI] [PubMed] [Google Scholar]
- 24.Hensel H, Schafer K. Effects of calcium on warm and cold receptors. Pflugers Arch. 1974;352(1):87–90. doi: 10.1007/BF01061953. [DOI] [PubMed] [Google Scholar]
- 25.Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 2011;338(2):537–547. doi: 10.1124/jpet.111.181297. [DOI] [PubMed] [Google Scholar]
- 26.Kagiava A, Tsingotjidou A, Emmanouilides C, Theophilidis G. The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology. 2008;29(6):1100–1106. doi: 10.1016/j.neuro.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Krishnan AV, Goldstein D, Friedlander M, Kiernan MC. Oxaliplatin and axonal Na+ channel function in vivo. Clin Cancer Res. 2006;12(15):4481–4484. doi: 10.1158/1078-0432.CCR-06-0694. [DOI] [PubMed] [Google Scholar]
- 28.Leo S, D’Hooge R, Meert T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1. 9 knockout mice. Behav Brain Res. 2010;208(1):149–157. doi: 10.1016/j.bbr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 29.Ling B, Authier N, Balayssac D, Eschalier A, Coudore F. Behavioral and pharmacological description of oxaliplatin-induced painful neuropathy in rat. Pain. 2007;128(3):225–234. doi: 10.1016/j.pain.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Liu JJ, Kim Y, Yan F, Ding Q, Ip V, Jong NN, Mercer JF, McKeage MJ. Contributions of rat Ctr1 to the uptake and toxicity of copper and platinum anticancer drugs in dorsal root ganglion neurons. Biochem Pharmacol. 2013;85(2):207–215. doi: 10.1016/j.bcp.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 31.Luo FR, Wyrick SD, Chaney SG. Comparative neurotoxicity of oxaliplatin, ormaplatin, and their biotransformation products utilizing a rat dorsal root ganglia in vitro explant culture model. Cancer Chemother Pharmacol. 1999;44(1):29–38. doi: 10.1007/s002800050941. [DOI] [PubMed] [Google Scholar]
- 32.Madrid R, de la Pena E, Donovan-Rodriguez T, Belmonte C, Viana F. Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J Neurosci. 2009;29(10):3120–3131. doi: 10.1523/JNEUROSCI.4778-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honore E. TREK-1 is a heat-activated background K(+) channel. Embo J. 2000;19(11):2483–2491. doi: 10.1093/emboj/19.11.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, Failli P, Preti D, Marchetti N, Cavazzini A, Mancini F, Pedretti P, Nilius B, Patacchini R, Geppetti P. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain. 2011;152(7):1621–1631. doi: 10.1016/j.pain.2011.02.051. [DOI] [PubMed] [Google Scholar]
- 35.Park SB, Lin CS, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Dose effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS One. 2011;6(4):e18469. doi: 10.1371/journal.pone.0018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108(5):705–715. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- 37.Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98(23):13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reid G, Flonta M. Cold transduction by inhibition of a background potassium conductance in rat primary sensory neurones. Neurosci Lett. 2001;297(3):171–174. doi: 10.1016/s0304-3940(00)01694-3. [DOI] [PubMed] [Google Scholar]
- 39.Renn CL, Carozzi VA, Rhee P, Gallop D, Dorsey SG, Cavaletti G. Multimodal assessment of painful peripheral neuropathy induced by chronic oxaliplatin-based chemotherapy in mice. Mol Pain. 2011;7:29. doi: 10.1186/1744-8069-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol. 2007;579(Pt 1):1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakurai M, Egashira N, Kawashiri T, Yano T, Ikesue H, Oishi R. Oxaliplatin-induced neuropathy in the rat: involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain. 2009;147(1–3):165–174. doi: 10.1016/j.pain.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Schafer K, Braun HA, Hensel H. Static and dynamic activity of cold receptors at various calcium levels. J Neurophysiol. 1982;47(6):1017–1028. doi: 10.1152/jn.1982.47.6.1017. [DOI] [PubMed] [Google Scholar]
- 43.Schiavon E, Sacco T, Cassulini RR, Gurrola G, Tempia F, Possani LD, Wanke E. Resurgent current and voltage sensor trapping enhanced activation by a beta-scorpion toxin solely in Nav1.6 channel. Significance in mice Purkinje neurons. J Biol Chem. 2006;281(29):20326–20337. doi: 10.1074/jbc.M600565200. [DOI] [PubMed] [Google Scholar]
- 44.Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD. Nav1. 8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153(10):2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 45.Sittl R, Carr RW, Fleckenstein J, Grafe P. Enhancement of axonal potassium conductance reduces nerve hyperexcitability in an in vitro model of oxaliplatin-induced acute neuropathy. Neurotoxicology. 2010;31(6):694–700. doi: 10.1016/j.neuro.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P, Carr RW. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1. 6-resurgent and persistent current. Proc Natl Acad Sci U S A. 2012;109(17):6704–6709. doi: 10.1073/pnas.1118058109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112(6):819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 48.Takeshita M, Banno Y, Nakamura M, Otsuka M, Teramachi H, Tsuchiya T, Itoh Y. The pivotal role of intracellular calcium in oxaliplatin-induced inhibition of neurite outgrowth but not cell death in differentiated PC12 cells. Chem Res Toxicol. 2011;24(11):1845–1852. doi: 10.1021/tx200160g. [DOI] [PubMed] [Google Scholar]
- 49.Teichert RW, Raghuraman S, Memon T, Cox JL, Foulkes T, Rivier JE, Olivera BM. Characterization of two neuronal subclasses through constellation pharmacology. Proc Natl Acad Sci U S A. 2012;109(31):12758–12763. doi: 10.1073/pnas.1209759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vetter I, Dekan Z, Knapp O, Adams DJ, Alewood PF, Lewis RJ. Isolation, characterization and total regioselective synthesis of the novel muO-conotoxin MfVIA from Conus magnificus that targets voltage-gated sodium channels. Biochem Pharmacol. 2012;84(4):540–548. doi: 10.1016/j.bcp.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 51.Vetter I, Touska F, Hess A, Hinsbey R, Sattler S, Lampert A, Sergejeva M, Namer B, Sharov A, Collins LS, Eberhardt M, Engel M, Cabot PJ, Wood JN, Vlachová V, Reeh PW, Lewis RJ, Zimmermann K. Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. Embo J. 2012;301(19):3795–3808. doi: 10.1038/emboj.2012.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viana F, de la Pena E, Belmonte C. Specificity of cold thermotransduction is determined by differential ionic channel expression. Nat Neurosci. 2002;5(3):254–260. doi: 10.1038/nn809. [DOI] [PubMed] [Google Scholar]
- 53.Wilson MJ, Yoshikami D, Azam L, Gajewiak J, Olivera BM, Bulaj G, Zhang MM. mu-Conotoxins that differentially block sodium channels NaV1.1 through 1. 8 identify those responsible for action potentials in sciatic nerve. Proc Natl Acad Sci U S A. 2011;108(25):10302–10307. doi: 10.1073/pnas.1107027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao WH, Zheng H, Bennett GJ. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience. 2012;203:194–206. doi: 10.1016/j.neuroscience.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9(9):1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 56.Zamudio F, Saavedra R, Martin BM, Gurrola-Briones G, Herion P, Possani LD. Amino acid sequence and immunological characterization with monoclonal antibodies of two toxins from the venom of the scorpion Centruroides noxius Hoffmann. Eur J Biochem. 1992;204(1):281–292. doi: 10.1111/j.1432-1033.1992.tb16635.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhang MM, Wilson MJ, Gajewiak J, Rivier JE, Bulaj G, Olivera BM, Yoshikami D. Pharmacological Fractionation of Tetrodotoxin-sensitive Sodium Currents in Rat Dorsal Root Ganglion Neurons by mu-Conotoxins. Br J Pharmacol. 2013 doi: 10.1111/bph.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao M, Isami K, Nakamura S, Shirakawa H, Nakagawa T, Kaneko S. Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol Pain. 2012;8:55. doi: 10.1186/1744-8069-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1. 8 is essential for pain at low temperatures. Nature. 2007;447(7146):855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]
- 60.Zimmermann K, Lennerz JK, Hein A, Link AS, Kaczmarek JS, Delling M, Uysal S, Pfeifer JD, Riccio A, Clapham DE. Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc Natl Acad Sci U S A. 2011;108(44):18114–18119. doi: 10.1073/pnas.1115387108. [DOI] [PMC free article] [PubMed] [Google Scholar]