

Abstract
Inflammatory Bowel Disease (IBD) is a chronic inflammatory condition thought to reflect a failure of the enteral immune system to adequately regulate itself. Inflammatory stress drives up-regulation of heat-shock proteins (HSP) including the pro-inflammatory chaperone, HSP90. This protein sequesters the transcription factor, heat-shock factor 1 (HSF1) in the cytoplasm preventing transcription of a number of anti-inflammatory proteins. We hypothesized that inhibition of HSP90 would exert an anti-inflammatory effect and thereby attenuate intestinal inflammation in murine models of IBD.
Inhibition of HSP90 with 17-AAG reduced inflammation in acute DSS and chronic CD45RBHigh colitis models coinciding with increased IL-10 production in the colon. Regulatory T cells (Tregs) from mice treated with 17-AAG, demonstrated significantly greater suppressive capacity in vitro abolished in HSF1−/− or IL-10−/− cells. Finally, Tregs treated with 17-AAG exhibited increased nuclear localization of HSF1 with resultant up-regulation of HSF1 response genes including HSP70, HSP90 and IL-10.
Keywords: Heat shock, inflammation, inflammatory bowel disease, interleukin-10
Introduction
Constant exposure of the gut mucosa to luminal antigens makes regulation of the inflammatory response crucial to maintaining a state of hyporesponsiveness. Animal studies indicate that CD4+CD25+Foxp3+ regulatory T cells (Tregs) provide balance for the immune response in normal intestinal mucosa1,2. A breakdown of this tolerance plays a pivotal role in the development of inflammatory bowel diseases (IBD)3. Both mice and humans with a defect in the forkhead box p3 (Foxp3) gene develop a wide range of autoimmune and inflammatory pathologies including enteritis, providing evidence that this pathway may be relevant for intestinal homeostasis.
Previous studies using inhibitors of histone deacetylase (HDAC) to enhance Treg function and thereby attenuate murine colitis suggest a role for HDAC6-dependent regulation of heat shock protein (HSP) 904. Inducible heat shock proteins are activated under conditions of cellular stress; these stresses include chemical, heat, hypoxia and exposure to cytokines. These proteins, once expressed, act as chaperones to either allow for normal protein folding or to stabilize the protein and prevent degradation. Transcriptional activation of the heat shock response requires release of heat shock factor 1 (HSF1) from its chaperone HSP90, and trimerization of HSF1. It has previously been demonstrated that HSF-1 and HSPs are involved in the repression of pro-inflammatory cytokines such as IL-1β, TNFα and IFNγ5–7, and activation of the anti-inflammatory gene IL-108,9. In genome wide association studies, HSF1 and HSPs have been associated with IBD10,11, generally as protective factors.
Inhibitors of HSP90 such as 17-allylaminogeldanamycin (17-AAG) have been shown to compete with ATP for binding to the N-terminus of the protein, causing release of HSF1 from the inhibitory complex with HSP90, thus allowing trimerization of HSF1 and translocation to the nucleus12. Nuclear HSF1 binds to heat shock response elements and induces transcription of heat shock response genes13.
HSP90 inhibitors have proven effective in treatment of murine models of sepsis6, arthritis14, uveitis15 and multiple sclerosis16, but their potential in IBD has yet to be established. Here we examined the therapeutic benefit of 17-AAG, a potent inhibitor of HSP90 for the regulation of inflammation in two distinct murine models of colitis. First we investigated whether inhibition of HSP90 affected colitis onset and severity. We next assessed if HSP90 inhibition could reverse established adoptive-transfer colitis. We then investigated the capacity of HSP90 inhibition to enhance Treg function ex vivo, and thereby offer a Treg-dependent mechanism to modulate disease severity. Finally, we assessed the effect of HSP90 inhibition on nuclear translocation of HSF1 in activated Tregs and subsequent changes in gene transcription. Our data further define a role for the heat shock response pathway (HSP90 and HSF1) in the maintenance of mucosal immune homeostasis.
Materials and Methods
Mice
A colony of C57/Bl6J mice were maintained in house kept under specific-pathogen-free conditions. Foxp3-green fluorescent protein (GFP), RAG1−/− (2216), IL-10−/− (2251) and HSF1−/− mice (129/SvEvTac; 10543) were obtained from Jackson laboratories (Bar Harbor, ME); HSF1+/+ littermates were used as controls for HSF1−/− mice. Fecal samples from these mice were consistently negative for Helicobacter, protozoa and helminthes. All animals were handled according to procedures approved by the institutional committee for animal use.
Chemically-induced acute murine colitis model
Mice were treated with dextran sodium sulfate (DSS) ad libitum (3% w/v; MP Biomedicals, Santa Ana, CA; 36–50 kDa) in drinking water for 7 d. Water alone groups were included as a control. DSS groups received daily i.p. injections of 17-AAG (40 mg/kg), or vehicle DMSO. Body weight, stool consistency and occult bleeding was assessed daily to construct a disease activity index. At the time of sacrifice, spleen, mesenteric lymph nodes and segments of colonic tissue were excised for flow cytometric analysis of leukocyte subsets, colon lengths were assessed and colon sections processed for histology, RNA extraction and cytokine analyses to assess disease activity. Histology was evaluated and scored by a trained pathologist blinded to the conditions of the experiments (PJ) according to a previously described scoring system17. Colonic tissues were snap frozen and homogenized in RIPA buffer or paraffin embedded for western blot and immunofluorescence respectively using anti-HSP90 antibody (C45G5; Cell Signaling; Danvers, MA) according to manufacturers instructions.
Adoptive-transfer murine colitis model
Naïve T cells from Foxp3-GFP or IL-10−/− donor mice were isolated by magnetic enrichment of CD4+ cells from splenocytes (CD4 T cell isolation kit, Miltenyi Biotec, Auburn, CA) followed by fluorescence-activated sorting of CD4+CD45RBHighFoxp3Neg naïve T cells. RAG1−/− mice received 5×105 naïve T cells by i.p. injection. Establishment of colitis was determined by weekly weight monitoring. Upon establishment of disease, mice were treated with 17-AAG (40 mg/kg) or vehicle (DMSO), receiving daily injections for 10–12 days.
Lymphocyte isolation
Single-cell suspensions were obtained by gently pressing the mesenteric lymph node (MLN) or spleen against a 70 μm cell-strainer. Splenic red blood cells were lysed by 3 min incubation in ammonium chloride lysing reagent (ACK Lysis Buffer, Invitrogen, Carlsbad, CA). Intestinal segments were opened along the mesentery and rinsed of luminal contents with PBS before cutting into 1 cm sections in PBS containing 15 mM HEPES and 1 mM EDTA with vigorous agitation on a vortex mixer. The tissue was then passed through a 70 μm tissue strainer and the process repeated until the wash remained clear. The remaining lamina propria (LP) was digested in 1 mg/ml Collagenase Type VIII (Sigma Aldrich, St Louis, MO, C9722) for 10 min in an orbital shaker at 270 rpm and 37°C. Tissues were vortexed briefly and filtered to remove any remaining undigested material and cells counted prior to flow cytometric evaluation.
Cytokine production assays
Tissue explants (0.5 cm2) were cultured for 24 h in DMEM (without sodium pyruvate, Cellgro Manassas, VA, supplemented with 5 % FBS, 2 mM glutamine, 100 IU penicillin and 100 μg/ml streptomycin; Invitrogen) culture supernatants were then analyzed for the presence of cytokines using the Milliplex Cytokine Multiplex Assay system (Logan, UT).
Flow cytometry
Cells from indicated compartments were incubated with fluorescent rat anti-mouse antibodies including against: mouse CD3 (17A2), CD4 (RM4-5), CD25 (PC61.5), HSP90α (C-20), HSP90β (D-19), IL-10 (JES5-16E3) and Foxp3 (FJK16S) or their respective isotype controls. Foxp3 staining was performed according to manufacturer's instructions (eBiosciences, San Diego, CA). Heat shock of cells was performed by incubating the cells for 30 min at 42°C. Cells were stained, washed and fixed with 2% paraformaldehyde and analyzed using the FACS® Canto system (Becton-Dickinson Immunocytometry Systems, San José, CA). Post-analyses were performed using FlowJo software (Tree Star Inc., Ashland, OR). Intracellular cytokine staining for IL-10 was performed as previously described18.
T cell proliferation assays
CD4+CD25+ Tregs were isolated by negative selection of CD4+ T cells, followed by positive selection of CD25+ cells using the MACS Treg isolation kit (Miltenyi Biotec). Fifty-thousand CellTrace Violet-labeled (Invitrogen) CD4+CD25Neg effector T cells/well were stimulated with anti-CD3 mAb in the presence of irradiated syngeneic APC and varying ratios of purified CD4+CD25+Treg from HSF1+/+, IL-10−/− or HSF1−/− mice; suppression of proliferation was determined by the CellTrace profile of dividing effector cells at 72 h.
In vitro stimulation assays
Freshly isolated CD4+/CD25+ Tregs from WT mice were stimulated with plate-bound anti-CD3 antibody (2 μg/ml; eBioscience) and soluble anti-CD28 (2 μg/ml; eBioscience) with (out) 17-AAG (250 nM) for either 6 or 24 h. Nuclear and cytoplasmic protein extracts were isolated after 6 h using the NE-PER extraction kit (Thermo Scientific) according to manufacturer's instructions. Western blot of HSF1 (4356S, Cell Signaling) using both denaturing and non-denaturing conditions were performed on 4–15% gradient gels (Biorad; Hercules, CA). RNA was isolated from both timepoints using the RNeasy kit (Qiagen, Valencia, CA) prior to cDNA synthesis and realtime RT-PCR (Taqman Probes; Life Technologies, Grand Island, NY). Individual wells were internally controlled by multiplexing with VIC-labeled 18S Taqman probe.
Statistics
Statistical analyses were performed using Student's t-test or repeated measures ANOVA with Graphpad Prism Data Analysis software (GraphPad Software, La Jolla, CA). Data were expressed as mean ± standard error of the mean (SEM). Statistical significance was set at P<0.05.
Results
HSP90 inhibition attenuates chemically induced acute murine colitis
In order to evaluate the role of HSP90 inhibition on chemically induced murine colitis 10-week-old WT mice were treated with HSP90 inhibitor 17-AAG or vehicle for 7 days while receiving DSS ad libitum in drinking water. Treatment with 17-AAG significantly attenuated weight loss (P<0.05) in the DSS colitis model as measured by repeated measures ANOVA (Figure 1a). This effect coincided with a decreased colon shortening, a surrogate marker of colitis, in 17-AAG-treated mice (51 ± 2mm; P<0.05) compared to vehicle (43 ± 2mm; Figure 1b).
Figure 1.

HSP90 inhibition attenuated chemically-induced murine colitis. (A) Weight loss of mice treated with vehicle or 17-AAG during DSS colitis. Mean % weight loss ± SEM from n≥8 mice, *P<0.05, **P<0.01. (B) Colon length (mm) of mice following DSS colitis measured postmortem, *P<0.05. (C) Histological evaluation of indices of inflammation (injury and inflammation) by a trained pathologist (PJ) in a blinded fashion, **P<0.01. (D) Representative micrographs of colonic H&E sections from mice each treatment group. Black scale bar indicates 100μm.
A pathologist that was blinded to the experimental conditions performed histological assessment of colitis, which identified a significant decrease in tissue injury from vehicle-treated (5.8 ± 1.1) compared with 17-AAG (2.6 ± 0.4; P<0.01; Figure 1c). Inflammation indices also decreased from vehicle-treated (7.6 ± 0.8) compared with 17-AAG (4.7 ± 0.3; P<0.01; Figure 1c). This reduction in tissue injury, loss of crypt architecture and epithelial integrity along with the reduction in inflammatory infiltrate are depicted in the representative H&E micrographs (Figure 1d).
Increased anti-inflammatory cytokines and cells following HSP90 inhibition
To better understand the underlying mechanism by which HSP90 inhibition ameliorates inflammation, cytokine expression analysis was performed on colonic explants from DSS colitic mice treated with or without 17-AAG. These studies corroborated the histological evidence of attenuated inflammation demonstrating a significant reduction in a number of inflammatory cytokines. Release of IL-2 decreased from 22.8 ± 1.2 pg/ml from vehicle-treated mice to 15.7 ± 0.8 pg/ml from 17-AAG-treated mice (P<0.001). IL-4 secretion decreased from 26.7 ± 2.0 pg/ml from vehicle-treated mice to 9.9 ± 0.8 pg/ml from 17-AAG-treated mice (P<0.001). IL-17 secretion decreased from 440.4 ± 39.9 pg/ml from vehicle-treated mice to 30.6 ± 4.2 pg/ml from 17-AAG-treated mice (P<0.001). IFNγ secretion decreased from 74.8 ± 8.6 pg/ml from vehicle-treated mice to 38.2 ± 3.9 pg/ml from 17-AAG-treated mice (P<0.01). Expression of IL-10 was significantly increased by 17-AAG administration with 445.6 ± 44.4 pg/ml released from vehicle-treated mice to 1704 ± 239.1 pg/ml in 17-AAG-treated mice (P<0.001), while release of TNFα was unaltered (Figure 2a). Similarly expression of IL-1β and IL-6 were also not significantly altered (not shown)
Figure 2.
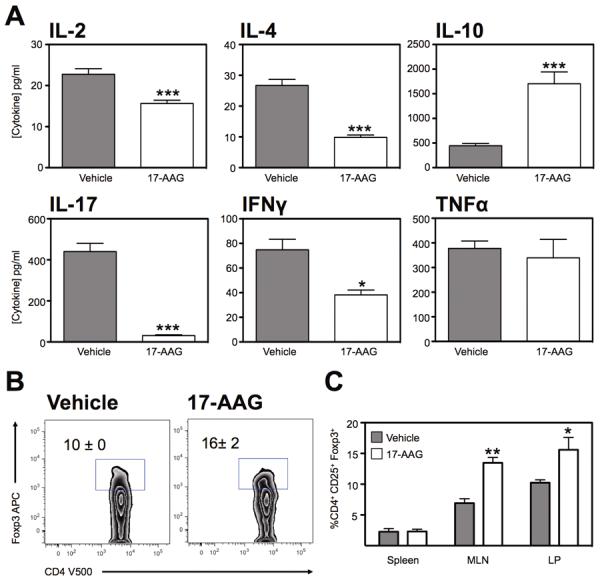
Altered cytokine profile from the inflamed colon following HSP90 inhibition. (A) Multiplex analysis of IL-2, IL-4, IL-10, IL-17, IFNγ and TNFα expression from 24hr culture of colonic explants from DSS colitic mice treated with 17-AAG. Mean ± SEM for n=4 individual mice, *P<0.05, ***P<0.001. (B) Representative zebra plot of CD4 T cells from the lamina propria of DSS colitic mice treated with vehicle or 17-AAG, showing expression of Foxp3. (C) Percentage of CD4+ T cells that are Foxp3+ from indicated organs of colitic mice. Mean ± SEM, n=3, *P<0.05, **P<0.01.
Cell populations were evaluated from both DSS treated groups to assess for the presence of CD4+Foxp3+ cells. Lymphocytes were isolated from the spleen, mesenteric lymph nodes (MLN) and lamina propria (LP) and evaluated by flow cytometry. We noted that there was an increase in the percentage of CD4+Foxp3+ cells in the colonic lamina propria of mice treated with 17-AAG (16% ± 2; P<0.05) compared to vehicle treated mice (10% ± 0; Figure 2b). In addition, we noted a significant increase in the percentage of CD4+Foxp3+ cells in the MLN of 17-AAG (14% ± 1; P<0.01) treated mice compared to vehicle treated mice (7% ± 1; Figure 2c). In summary, 17-AAG had a protective effect on the severity of DSS-mediated colitis partly mediated by decreased inflammatory cytokine production and increased frequency of Tregs.
Expression of HSP90 is increased during murine colitis
We evaluated the expression of HSP90 following induction of murine colitis to determine if it was regulated by inflammation. Western blot analysis demonstrated a significant increase in expression of HSP90 relative to β-actin control in colitic tissue (1.5±0.02; P<0.05; Figure 3a & b) compared with water-treated mice. This was consistent with immunofluorescent assessment of HSP90 expression from colonic tissues of water control and DSS-treated mice. It demonstrates that in the absence of inflammation, HSP90 expression is largely restricted to the base of the crypts whereas during colitis, expression is up-regulated particularly at the mucosal surface (Figure 3c). Our data therefore suggests that there is an increase in HSP90 expression in response to DSS-induced epithelial injury and intestinal barrier disruption.
Figure 3.
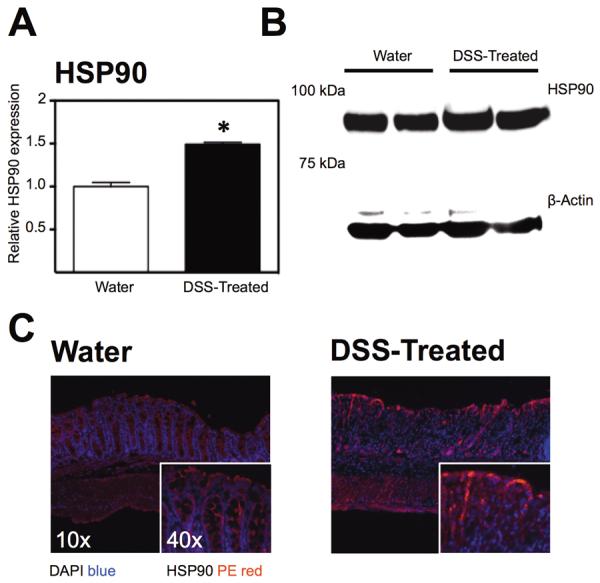
Protein expression and localization of HSP90 in the inflamed mouse colon compared with untreated controls. (A) Densitometric determination of HSP90 expression in colonic tissue from water-treated or DSS-treated mice, *P<0.05. (B) Representative western blot of HSP90 expression from water and DSS-treated mouse colonic homogenates. (C) Representative immunofluorescent micrographs showing staining for HSP90 in PE at 10× and 40× magnification at room temperature using a Zeiss Axio Imager A1 Microscope with Axiovision 4.6 acquisition software.
Expression of HSP90 is enriched in murine regulatory T cells
To determine if HSP90 played a role in Treg function, the expression of HSP90 was analyzed in newly induced Tregs by intracellular flow cytometry. CD4+CD25Neg cells were cultured for 3 days in the presence of TGFβ resulting in a robust induction of Tregs. Expression analysis of cultured Tregs demonstrated an up-regulation of HSP90α following heat shock (MFI 958 ± 73 in CD3/CD28-stimulated Tregs vs MFI 2096 ± 48 in heat-shocked TGFβ-treated Tregs; P<0.001). These changes were not noted in Foxp3Neg cells (Figure 4a). Similarly, expression of HSP90β was also up regulated on Tregs (544 ± 159 vs 1214 ± 64) while not significantly increased in non-Tregs. Thus during heat shock or potentially inflammatory stress, both HSP90 isoforms are preferentially up regulated in Tregs.
Figure 4.
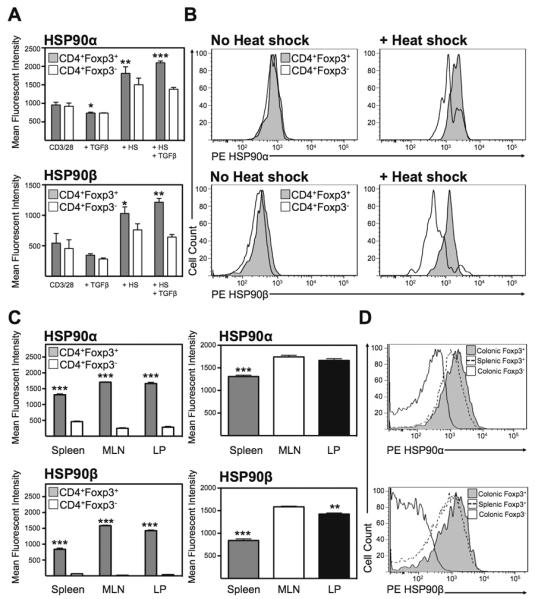
Heat shock-induced HSP90α and HSPβ expression on mouse Tregs. (A) Mean fluorescent intensity of HSP90α and HSP90β expression in permeabilized newly converted CD4+CD25+Foxp3+ Tregs compared with non-Tregs with(out) heat shock. Mean ± SEM, n=3, *P<0.05, **P<0.01, ***P<0.001. (B) Representative histograms of expression of HSP90α and HSP90β on permeabilized CD4+Foxp3+ (grey) or CD4+Foxp3Neg cells (white). (C) Mean fluorescent intensity of HSP90α and HSP90β expression in permeabilized newly converted CD4+CD25+Foxp3+ Tregs compared with non-Tregs with(out) heat shock. Mean ± SEM, n=3, **P<0.01, ***P<0.001. (D) Representative histograms of expression of HSP90α and HSP90β on permeabilized CD4+Foxp3+ from the colonic LP (grey), the spleen (dashed line) or CD4+Foxp3Neg cells (white).
Expression analysis of intracellular HSP90α and HSP90β by flow cytometry from WT mice demonstrated that both HSP90α and β isoforms were significantly upregulated on CD4+Foxp3+ Tregs compared to non-Tregs freshly isolated from the spleen, MLN and colonic LP (Figure 4c). Furthermore, expression of both isoforms was greatest in the MLN followed by the LP and was significantly lower in the spleen (Figure 4d). Thus T cell expression of HSP90 occurs preferentially on Tregs and appears to coincide with Treg activation.
HSP90 inhibition alleviates colitis in the CD45RBHigh adoptive-transfer model
As DSS colitis is not predominantly T cell mediated, we employed the adoptive transfer model of colitis to address the role of HSP90 in a more T cell directed manner. To do this, naïve CD4+/CD45RBHigh cells were adoptively transferred into lymphopenic sex-matched recipients and development of colitis monitored indirectly by continuous assessment of body weight. 17-AAG-treated mice were protected from further significant weight loss and actually gained weight if treated with 17-AAG (101 ± 1%; P<0.001; Figure 5a) compared with vehicle-treated mice (89 ± 1%). Postmortem analysis revealed that 17-AAG treated mice (71 ± 4 mm; P<0.01) displayed significantly less colonic shortening compared with the vehicle control mice (60 ± 2 mm; Figure 5b). The protective effect of 17-AAG also resulted in a visible decrease in inflammation compared with vehicle controls. Active inflammatory indices decreased from vehicle-treated (7.8 ± 1; Figure 5c) compared to 17-AAG–treated (2.6 ± 1; P<0.01). Similarly, chronic inflammatory indices decreased from vehicle-treated (8.6 ± 1) compared to 17-AAG–treated (4.6 ± 1; P<0.01). Furthermore, total inflammatory indices significantly decreased from vehicle (16.4 ± 1.5) compared with 17-AAG-treated mice (7.2 ± 1.4; P<0.001). Tissue from 17-AAG–treated mice also displayed significantly less disruption of crypt architecture as shown by representative H&E micrographs (Figure 5d). This beneficial effect was lost when CD45RBHigh adoptive transfer colitis experiments were conducted using IL-10−/− cells consistent with a pivotal role for increased IL-10 production in the therapeutic effect of 17-AAG (Figure S1).
Figure 5.
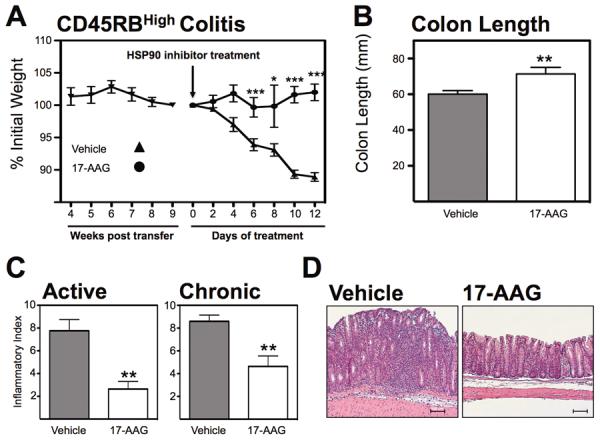
HSP90 inhibition attenuates established adoptive-transfer colitis. (A) Weight-loss curve of RAG1−/− mice adoptively transferred CD45RBHigh cells treated with vehicle or 17-AAG upon establishment of disease, *P<0.05, ***P<0.001 (B) Postmortem measurement of colon length of CD45RBHigh cell recipient with(out) treatment with 17-AAG (40mg/kg/day). Mean ± SEM, **P<0.01. (C) Histological evaluation of indices of active and chronic inflammation in CD45RBHigh model of colitis by a trained pathologist in a blinded fashion. **P<0.01. (D) Representative micrographs of colonic H&E sections from mice each treatment group. Black scale bar indicates 100μm
HSP90 inhibition promotes IL-10 production and Treg induction during chronic colitis
To assess the impact of HSP90 inhibition on cytokine production in this T-cell dependent colitis model, multiplex cytokine analyses were performed on cultured explant tissues as before. Cytokine expression analysis from CD45RBHigh recipient mice demonstrated a similar pattern to those of DSS colitic mice, with a significant decrease in IFNγ from vehicle-treated controls (59 ± 7 pg/ml) compared with 17-AAG-treated mice (29 ± 3 pg/ml; P<0.01; Figure 6a). While TNFα production was higher from vehicle controls (283 ± 58 pg/ml) relative to 17-AAG-treated mice (156 ± 35 pg/ml), this difference was not statistically significant. As in the DSS model, IL-10 production significantly increased from vehicle-treated mice (19 ± 2 pg/ml) to 17-AAG-treated mice (168 ± 27 pg/ml; P<0.001). Therefore 17-AAG induced a similar pattern of cytokine production in CD45RBHigh colitis as with DSS colitis with 17-AAG-treated mice producing significantly more IL-10.
Figure 6.
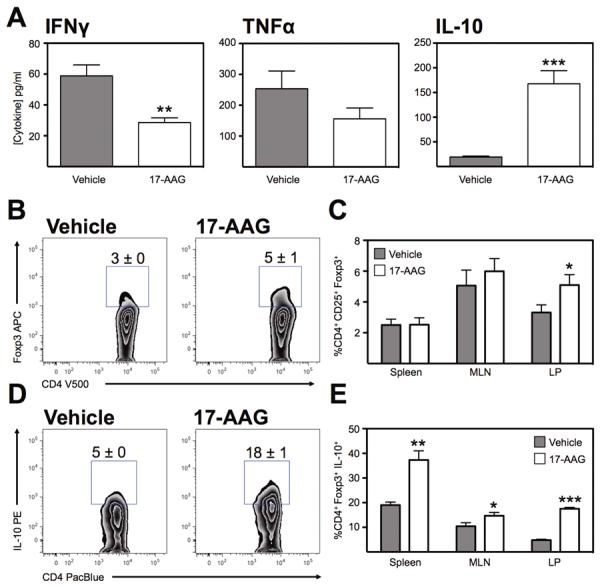
HSP90 inhibition increases IL-10 production and Foxp3 expression in the inflamed colon. (A) Multiplex analysis of IFNγ, TNFα and IL-10 expression from 24hr culture of colonic explants from CD45RBHigh adoptive-transfer colitic mice treated with 17-AAG. Mean ± SEM for n=4 individual mice, **P<0.01, ***P<0.001. (B) Representative zebra plot of CD4+ T cells from the lamina propria of CD45RBHigh colitic mice treated with vehicle or 17-AAG, showing expression of Foxp3. (C) Percentage of CD4+ T cells that are Foxp3+ from indicated organs of colitic mice, *P<0.05. (D) Representative zebra plots of IL-10 expression in CD4+Foxp3+ Tregs isolated from the colonic LP of CD45RBHgh colitis. (E) Percentage of CD4+Foxp3+ IL-10+ Tregs following vehicle or 17-AAG (40mg/kg/day) treatment. Mean ± SEM, n≥4, *P<0.05, **P<0.01, ***P<0.001.
Thereafter, cell populations were evaluated from the above adoptive transfer colitis experiments and treated groups were assessed for the presence of CD4+Foxp3+ cells. Lymphocytes were isolated from the spleen, MLN and colonic LP and evaluated by flow cytometry. Similar to our DSS experiments, we noted that there was an increase in the percentage of LP CD4+Foxp3+ cells from vehicle treated mice (3% ± 0; Figure 6b) to mice treated with 17-AAG (5% ± 1; P<0.01). Moreover, we demonstrated a significant increase in IL-10 production by Tregs isolated from RAG1−/− colitic mice treated with 17-AAG (18%±1; Figure 6d) compared with vehicle controls (5% ± 0; P<0.001). Therefore, 17-AAG attenuated established colitis in this adoptive transfer model and increased the presence of Tregs along with increasing their IL-10 production in the colonic lamina propria.
Suppressive function of Tregs is increased by HSP90 inhibition via HSF1
Above we have noted an increase in Treg numbers after treatment with 17-AAG as well as an increase in IL-10 expression in the two models of colitis. We next addressed the role of HSP90 inhibition in Treg function by analyzing proliferation of fluorescently labeled target cells in the presence of increasing ratios of Tregs. Co-culture of naïve CellTrace-labeled CD25Neg Teffector cells with increasing concentrations of Tregs treated with 17-AAG demonstrated a significant decrease in proliferation in the 17-AAG treated group. For example, at a 1:4 Treg:Teffector ratio, 30% of CellTrace-labeled cells proliferated compared to 37% proliferation in the HSF1+/+ control group (Figure 7). Whereas HSF1−/−Tregs had comparable suppressive capacity as their HSF1+/+ counterparts, lack of HSF1 abrogated the enhancement of suppressive function seen in 17-AAG treated Tregs. Thus 17-AAG enhances Treg function in an HSF1-dependent manner.
Figure 7.
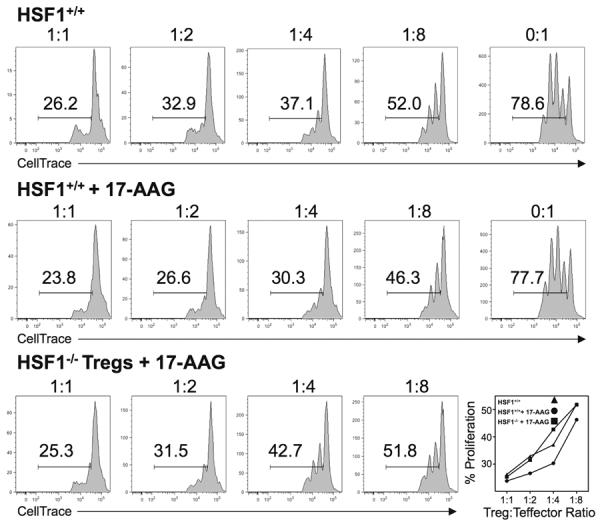
HSP90 inhibition enhances regulatory T cell function in vitro via HSF1. Representative histograms of CellTrace-labeled CD4+ T cells co-cultured with decreasing concentrations of CD4+CD25+ Tregs from HSF1+/+ vehicle-treated mice or HSF1+/+ and HSF1−/− mice treated with 17-AAG. Proliferation of CD3-stimulated CD4+ co-cultured with Tregs from vehicle or 17-AAG-treated mice. Results representative of N=3 individual experiments with similar results.
To further define the mechanism of 17-AAG-mediated enhanced Treg suppression, we repeated the Treg suppression assays using cells from IL-10−/− mice. As has been reported previously, IL-10−/− Tregs have decreased suppressive function, however, more importantly the enhancement seen with 17-AAG was abolished in the absence of IL-10. Therefore the HSF1-dependent 17-AAG-mediated increased suppression appears to be IL-10 dependent as well (Figure 8).
Figure 8.
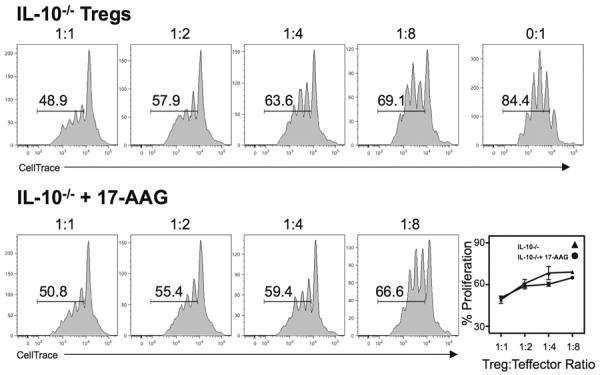
Enhancing suppressive function of 17-AAG is IL-10 dependent. Representative histograms of CellTrace-labeled CD4+ T cells co-cultured with decreasing concentrations of CD4+CD25+ Tregs from IL-10−/− mice treated with(out) 17-AAG. Results representative of N=3 individual experiments with similar results.
Increased translocation of HSF1 in Tregs treated with 17-AAG
With the loss of function of 17-AAG in HSF1−/− mice in the Treg assays and other data suggesting the role of HSP90 in regulation of HSF1 activation and translocation we next assessed the role of inhibition of HSP90 on HSF1 translocation in the Treg cells. To address this we performed western blot analysis of HSF1 expression in cytoplasmic and nuclear Treg fractions under both native and denatured conditions. This demonstrated an increased translocation of the HSF1 trimer to the nucleus of activated Tregs following 6h stimulation with 17-AAG (250nM; Figure 9a) compared with vehicle controls. This increase corresponded with an up-regulation of HSP90aa1, HSPA1A and IL-10 at both 6 h and 24 h time-points whereas HSP90ab1 was only induced at the early time-point and HSPA1B at the late time-point (Figure 9b). Thus HSP90 inhibition with 17-AAG promotes translocation of HSF1 to the nucleus with resultant up-regulation of HSF1 response genes.
Figure 9.
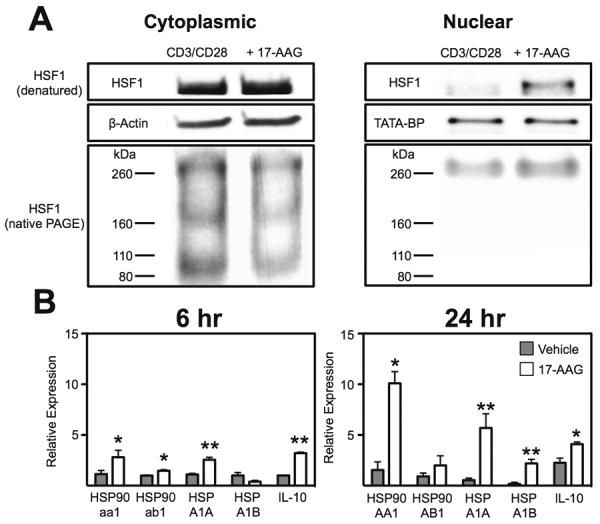
HSP90 inhibition increases HSF1 translocation and transcriptional activation. (A) Representative blot of HSF1 either denatured or non-denatured from Tregs stimulated for 6 h with CD3 and CD28 with(out) 17-AAG (250nM). (B) Realtime RT-PCR analysis of expression of HSP90 isoforms (aa1, ab1), HSP70 isoforms (HSPA1A, HSPA1B) and IL-10 mRNA from isolated activated Tregs treated for 6 or 24h with 17-AAG (250nM). Mean ± SEM, n=3, *P<0.05, **P<0.01.
Discussion
The heat shock response plays a critical role in regulating new protein synthesis in response to cellular stress including during the induction of an inflammatory response. In this study we identify a role of the heat shock response in mucosal homeostasis via Treg control. Considerable progress has been made in recent years in understanding the basic biology of heat shock proteins and in particular HSP90 signaling during inflammation; however, little is known about their role in the induction/maintenance of chronic intestinal inflammatory conditions such as IBD. As such this study aimed to investigate the effect of HSP90 inhibition in both acute and chronic murine models of IBD. Of particular interest was whether pharmacological targeting of HSP90 might be of therapeutic value. In this series of studies we demonstrate that HSP90 inhibition with 17-AAG, which binds to the N-terminal ATPase19, attenuates both new-onset murine colitis using the DSS model and established colitis using the CD45RBHigh adoptive transfer model. Treatment with 17-AAG, enhanced secretion of anti-inflammatory IL-10, increased Treg numbers and improved efficiency of Treg suppression in an HSF1 dependent manner.
Having first identified a successful outcome with HSP90 blockade for the attenuation of colitis, we sought to better understand the expression of HSP90 in response to an inflammatory stimulus. HSP90 expression was increased in response to DSS colitis consistent with previous studies in human IBD demonstrating an up-regulation of HSP90 and HSP70 associated with ulcerative colitis which was decreased by treatment with 5-ASA over 6 months20. While HSP90 is a ubiquitously expressed protein21, we next sought to ascertain whether the up-regulation of HSP90 was detectable in T cells. Flow cytometric analysis demonstrates that HSP90 isoforms are preferentially expressed on Foxp3+ Tregs and Treg induction in vitro drives expression of HSP90 particularly following heat shock. Moreover, expression of HSP90 in Tregs is greater on intestinal Tregs compared with splenic Tregs consistent with it being key for intestinal immunity and upregulated upon Treg activation.
Based on these findings we chose to focus on the therapeutic potential of 17-AAG in a T cell-mediated colitis model using the CD45RBHigh adoptive transfer model22 of IBD. Consistent with our previous findings, we observed an attenuation of inflammation in this model as well. Furthermore 17-AAG once again increased IL-10 production by Tregs along with Treg numbers while attenuating inflammation, pro-inflammatory cytokine secretion and inflammatory cell infiltration into the colon. The increased expression of IL-10 may be sufficient to account for all of these observations as IL-10 has been shown previously to act as a growth factor for Tregs23 and Treg-derived IL-10 is critical for attenuation of colitis in this model24. Similarly we have observed a beneficial effect of increasing Treg frequency in IBD models previously25. Furthermore, in the absence of T cell-derived IL-10, 17-AAG was unable to rescue the colitis in this model consistent with this being a primary therapeutic mechanism of action.
Our lab has previously demonstrated that 17-AAG enhances Tregs suppressive function in vitro4, however, no mechanism of this enhancement was identified. Accurately identifying which protein chaperoned by HSP90 is altered in response to 17-AAG is difficult, as there are currently over 100 known client proteins for HSP9026. However, HSP90 does form a complex with HSP70 and HSP70 has been shown to attenuate murine arthritis via enhanced IL-10 production from Tregs so it is possible that inhibition of HSP90 increases the availability of HSP70, which then in turn enhances Treg function27. Furthermore 17-AAG up-regulates expression of HSP70 mRNA28 and overexpression of HSP70 has been shown to enhance Treg suppressive function29. However, the ability of 17-AAG to drive HSP70 is actually HSF1-dependent30 and so we sought to determine if the enhanced suppressive function might also be HSF1 dependent.
Using HSF1−/− cells we demonstrated that enhanced suppressive function due to 17-AAG was abolished in the absence of HSF1−/−. Pre-induction of the heat-shock response has been demonstrated previously to protect against acute murine colitis31. While not identified at the time, the anti-inflammatory effect of heat shock is likely mediated by induction of the transcription factor HSF132. HSF1 is known to drive expression of a number of anti-inflammatory genes including IL-1033 and HSP7034 while also suppressing albeit transiently pro-inflammatory gene expression including IL-1β and TNFα35. HSP90 also forms an inhibitory complex with HSF1, preventing trimerization, nuclear translocation and transcriptional activation by HSF132. Inhibition of HSP90 therefore disrupts that complex, increasing HSF1 translocation to the nucleus and enhancing HSF1 transcriptional activation function12. This in turn acts as a negative regulator of pro-inflammatory cytokine gene transcription36 and a positive signal for anti-inflammatory gene transcription. Therefore it is likely that the anti-inflammatory effects of HSP90 inhibitors are at least partly mediated via release of HSF1. The expression of HSP90 is induced in response to heat shock or inflammatory stress31 via STAT1-dependent induction37, which may reflect a mechanism for limiting HSF1 activation. Pharmacological blockade of this endogenous braking mechanism may have therapeutic potential in overactive inflammatory diseases such as IBD and indeed this has been borne out in other models of inflammation. Indeed blockade of HSP90 has proven effective at attenuating inflammation in a number of autoimmune murine models via decreased expression of pro-inflammatory cytokines such as IL-1β15,16,38 consistent with the anti-inflammatory effects seen in our studies.
Having demonstrated that the effect of 17-AAG on Treg suppression was HSF1-dependent we then extended these observations even further to demonstrate that it was also mediated via increased IL-10 output as IL-10−/− cells failed to display this enhanced suppressive function. These in vitro results are further supported by the suppression of adoptively transferred T cell proliferation in CD45RBHigh colitis. Furthermore, this is consistent with studies demonstrating enhanced regulatory T cell function in HDAC6−/− mice4 coinciding with increased HSP90 acetylation correlated with impaired HSP90 function39. Based on our hypothesis that HSP90 inhibition with 17-AAG was driving IL-10 expression in a HSF1 dependent manner, we assessed the localization of HSF1 in activated Tregs treated with 17-AAG in vitro and demonstrated that indeed, blockade of HSP90 caused a shift in HSF1 trimer to the nucleus and a subsequent mRNA upregulation of anti-inflammatory IL-10 and HSP70 isoforms (HSPA1A and HSPA1B) in isolated Tregs. These findings account for the enhanced suppressive function of Tregs treated with 17-AAG as well as the anti-inflammatory effect of 17-AAG in both IBD models tested. Interestingly however, enhanced translocation of HSF1 to the nucleus also led to upregulation of HSP90α and HSP90β mRNA. We believe that this response is part of a negative feedback autoregulatory mechanism whereby HSF1 upregulated HSP90 that in turn sequesters HSF1 in the cytoplasm to prevent overactivation/translocation of the HSF1 anti-inflammatory pathway and thereby reduce risk of opportunistic infection associated with excessive immunosuppression.
The ability of HSP90 inhibition to attenuate murine colitis makes them an attractive candidate for future clinical application in IBD. The ongoing trials of HSP90 inhibitors in cancer chemotherapy in which their safety and pharmacokinetics are firmly established further support the translation of this technology to an IBD context. Finally, as we increase our understanding of the anti-inflammatory mechanisms of HSP90 inhibition in murine models of IBD, these data provide proof of principle for the application of newer more effective HSP90 inhibitors currently in development to use in the treatment of IBD.
Supplementary Material
Figure S1. HSP90 inhibition fails to attenuate established adoptive-transfer colitis in the absence of IL-10. (A) Weight-loss curve of RAG1−/− mice adoptively transferred CD45RBHigh cells from WT or IL-10−/− donor mice and then treated with vehicle or 17-AAG upon establishment of disease (B) Postmortem measurement of colon length of CD45RBHigh cell recipient with(out) treatment with 17-AAG (40mg/kg/day). Mean ± SEM, **P<0.01, ***P<0.001. (C) Histological evaluation of indices of active and chronic inflammation in CD45RBHigh model of colitis by a trained pathologist in a blinded fashion. **P<0.01. (D) Representative micrographs of colonic H&E sections from mice each treatment group. Black scale bar indicates 100μm.
Acknowledgments
Grant Support: K08DK080189 (EDZ) & CCFA-2652 (CBC) and UL1 RR025780 (EDZ & CBC)
Abbreviations used in this paper
- Treg
regulatory T cell
- IBD
inflammatory bowel diseases
- Foxp3
forkhead box p3
- HDAC
histone deacetylase
- HSP
heat shock proteins
- HSF1
heat shock factor 1
- 17-AAG
17-allylaminogeldanamycin
- GFP
green fluorescent protein
- DSS
dextran sodium sulfate
- LP
lamina propria
- MLN
mesenteric lymph node
- WT
wildtype
- MFI
mean fluorescence intensity
Footnotes
Disclosures: No conflicts of interest exist.
Author Contribution: All authors contributed to the generation of data, editing and proofing of this manuscript.
References
- 1.Singh B, et al. Control of intestinal inflammation by regulatory T cells. Immunol Rev. 2001;182:190–200. doi: 10.1034/j.1600-065x.2001.1820115.x. [DOI] [PubMed] [Google Scholar]
- 2.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 3.Duchmann R, et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD) Clin Exp Immunol. 1995;102:448–455. doi: 10.1111/j.1365-2249.1995.tb03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Zoeten EF, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of foxp3+ T-regulatory cells. Mol Cell Biol. 2011;31:2066–2078. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De AK, Kodys KM, Yeh BS, Miller-Graziano C. Exaggerated human monocyte IL-10 concomitant to minimal TNF-alpha induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an antiinflammatory stimulus. J Immunol. 2000;165:3951–3958. doi: 10.4049/jimmunol.165.7.3951. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee A, et al. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med. 2007;176:667–675. doi: 10.1164/rccm.200702-291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Y, Chen C, Stevenson MA, Auron PE, Calderwood SK. Heat shock factor 1 represses transcription of the IL-1beta gene through physical interaction with the nuclear factor of interleukin 6. J Biol Chem. 2002;277:11802–11810. doi: 10.1074/jbc.M109296200. [DOI] [PubMed] [Google Scholar]
- 8.Luo X, et al. Release of heat shock protein 70 and the effects of extracellular heat shock protein 70 on the production of IL-10 in fibroblast-like synoviocytes. Cell Stress Chaperones. 2008;13:365–373. doi: 10.1007/s12192-008-0036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Y, Cahill CM, Asea A, Auron PE, Calderwood SK. Heat shock proteins and regulation of cytokine expression. Infect Dis Obstet Gynecol. 1999;7:26–30. doi: 10.1155/S106474499900006X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nam SY, et al. Heat shock protein gene 70–2 polymorphism is differentially associated with the clinical phenotypes of ulcerative colitis and Crohn's disease. J Gastroenterol Hepatol. 2007;22:1032–1038. doi: 10.1111/j.1440-1746.2007.04927.x. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, et al. Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J Biol Chem. 2007;282:23240–23252. doi: 10.1074/jbc.M704081200. [DOI] [PubMed] [Google Scholar]
- 12.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 13.Bagatell R, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 14.Prakken BJ, et al. Induction of IL-10 and inhibition of experimental arthritis are specific features of microbial heat shock proteins that are absent for other evolutionarily conserved immunodominant proteins. J Immunol. 2001;167:4147–4153. doi: 10.4049/jimmunol.167.8.4147. [DOI] [PubMed] [Google Scholar]
- 15.Poulaki V, et al. Inhibition of Hsp90 attenuates inflammation in endotoxin-induced uveitis. FASEB J. 2007;21:2113–2123. doi: 10.1096/fj.06-7637com. [DOI] [PubMed] [Google Scholar]
- 16.Dello Russo C, et al. The heat-shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin suppresses glial inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J Neurochem. 2006;99:1351–1362. doi: 10.1111/j.1471-4159.2006.04221.x. [DOI] [PubMed] [Google Scholar]
- 17.Smith P, et al. Infection with a helminth parasite prevents experimental colitis via a macrophage-mediated mechanism. J Immunol. 2007;178:4557–4566. doi: 10.4049/jimmunol.178.7.4557. [DOI] [PubMed] [Google Scholar]
- 18.Collins CB, et al. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology. 2011;141:1821–1831. doi: 10.1053/j.gastro.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 20.Tomasello G, et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur J Histochem. 2011;55:e38. doi: 10.4081/ejh.2011.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pratt WB. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med. 1998;217:420–434. doi: 10.3181/00379727-217-44252. [DOI] [PubMed] [Google Scholar]
- 22.Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asseman C, Powrie F. Interleukin 10 is a growth factor for a population of regulatory T cells. Gut. 1998;42:157–158. doi: 10.1136/gut.42.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins CB, et al. Flt3 ligand expands CD103(+) dendritic cells and FoxP3(+) T regulatory cells, and attenuates Crohn's-like murine ileitis. Gut. 2012;61:1154–1162. doi: 10.1136/gutjnl-2011-300820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 27.Wieten L, et al. IL-10 is critically involved in mycobacterial HSP70 induced suppression of proteoglycan-induced arthritis. PLoS One. 2009;4:e4186. doi: 10.1371/journal.pone.0004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pimienta G, Herbert KM, Regan LA. Compound That Inhibits the HOP-Hsp90 Complex Formation and Has Unique Killing Effects in Breast Cancer Cell Lines. Mol Pharm. 2011 doi: 10.1021/mp200346y. [DOI] [PubMed] [Google Scholar]
- 29.de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. 2010;138:583–594. doi: 10.1053/j.gastro.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujikake N, et al. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otani S, et al. Effect of preinduction of heat shock proteins on acetic acid-induced colitis in rats. Dig Dis Sci. 1997;42:833–846. doi: 10.1023/a:1018832618275. [DOI] [PubMed] [Google Scholar]
- 32.Boyault C, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, et al. HSF1 Is a Transcriptional Activator of IL-10 Gene Expression in RAW264.7 Macrophages. Inflammation. 2012 doi: 10.1007/s10753-012-9471-4. [DOI] [PubMed] [Google Scholar]
- 34.Suemasu S, et al. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J Biol Chem. 2009;284:19705–19715. doi: 10.1074/jbc.M109.006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Housby JN, et al. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine. 1999;11:347–358. doi: 10.1006/cyto.1998.0437. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, et al. Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits transcriptional activity and promotes HSP90 binding. J Biol Chem. 2006;281:782–791. doi: 10.1074/jbc.M505822200. [DOI] [PubMed] [Google Scholar]
- 37.Chen XS, et al. Diverse effects of Stat1 on the regulation of hsp90alpha gene under heat shock. J Cell Biochem. 2007;102:1059–1066. doi: 10.1002/jcb.21342. [DOI] [PubMed] [Google Scholar]
- 38.Yun TJ, et al. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol. 2011;186:563–575. doi: 10.4049/jimmunol.1000222. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. HSP90 inhibition fails to attenuate established adoptive-transfer colitis in the absence of IL-10. (A) Weight-loss curve of RAG1−/− mice adoptively transferred CD45RBHigh cells from WT or IL-10−/− donor mice and then treated with vehicle or 17-AAG upon establishment of disease (B) Postmortem measurement of colon length of CD45RBHigh cell recipient with(out) treatment with 17-AAG (40mg/kg/day). Mean ± SEM, **P<0.01, ***P<0.001. (C) Histological evaluation of indices of active and chronic inflammation in CD45RBHigh model of colitis by a trained pathologist in a blinded fashion. **P<0.01. (D) Representative micrographs of colonic H&E sections from mice each treatment group. Black scale bar indicates 100μm.