

Abstract
The Ca2+ sensor stromal interacting molecule 1 (STIM1) and the Ca2+ channel Orai1 mediate the ubiquitous store-operated Ca2+ entry (SOCE) pathway activated by depletion of internal Ca2+ stores and mediated through the highly Ca2+ selective, Ca2+ release-activated Ca2+ (CRAC) current. Furthermore, STIM1 and Orai1, along with Orai3, encode store-independent Ca2+ currents regulated by either arachidonate or its metabolite, leukotrieneC4. Orai channels are emerging as important contributors to numerous cell functions, including proliferation, migration, differentiation, and apoptosis. Recent studies suggest critical involvement of STIM/Orai proteins in controlling the development of several cancers, including malignancies of breast, prostate and cervix. Here, we quantitatively compared the magnitude of SOCE and the expression levels of STIM1 and Orai1 in non-malignant human primary astrocytes (HPA), and in primary human cell lines established from surgical samples of the brain tumor glioblastoma multiforme (GBM). Using Ca2+ imaging, patch clamp electrophysiology, pharmacological reagents, and gene silencing, we established that in GBM cells SOCE and CRAC are mediated by STIM1 and Orai1. We further found that GBM cells show upregulation of SOCE and increased Orai1 levels compared to HPA. The functional significance of SOCE was evaluated by studying the effects of STIM1 and Orai1 knockdown on cell proliferation and invasion. Utilizing Matrigel assays we demonstrated that in GBM, but not in HPA, downregulation of STIM1 and Orai1 caused a dramatic decrease in cell invasion. In contrast, the effects of STIM1 and Orai1 knockdown on GBM cell proliferation were marginal. Overall, these results demonstrate that STIM1 and Orai1 encode SOCE and CRAC currents and control invasion of GBM cells. Our work further supports the potential use of channels contributed by Orai isoforms as therapeutic targets in cancer.
Introduction
Gliomas, and particularly their most aggressive form glioblastoma multiforme (GBM), represent the vast majority of primary brain tumors [35,4]. Among the primary brain malignancies, GBM is characterized by extremely poor prognosis and high resistance to all forms of treatments, including combination of surgical resection, radiotherapy and chemotherapy. The median survival rates for GBM patients seldom exceed one year and have not shown substantial improvement over the past few decades[35,3]. The aggressiveness of GBM is determined by numerous factors, including high genetic diversity, enhanced proliferative and invasive potential, and their ability to develop resistance to presently available drugs [4,35,14]. The current absence of effective therapies creates a pressing need for increased understanding of GBM biology and characterizations of novel signaling pathways that control GBM proliferation and invasion.
Ca2+ signaling controls a wide variety of cell functions and Ca2+ channels have emerged recently as important players in cancer progression [22,32]. Store-operated Ca2+ entry (SOCE) is the Ca2+ entry route into non-excitable cells that is activated upon depletion of internal Ca2+ stores [33,34,28,26]. Under physiological conditions, agonists binding to various growth factor receptors, including G-protein coupled receptors and receptor tyrosine kinases, trigger activation of phospholipases C (PLC) enzymes and leads to the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol and inositol 1,4,5-trisphosphate (IP3) [5,40]. IP3 causes Ca2+ release from the endoplasmic reticulum (ER) and subsequent store depletion. Store depletion is then sensed by stromal interacting molecule 1 (STIM1), which aggregates and moves into discrete areas of junctional ER-plasma membrane to interact directly with Orai1 protein [16,29]. Orai1 is the pore forming unit of the SOCE channel that mediates a highly Ca2+-selective Ca2+ release-activated Ca2+ (CRAC) current [17,25,9]. In mammalian cells, Orai1, in combination with Orai3 also encode store-independent Ca2+ selective channels that are regulated by STIM1 and gated by either arachidonic acid (AA) or its metabolite, leukotrieneC4 (LTC4) [20,21,15].
The STIM/Orai signaling has been recently implicated in migration and cell cycle progression of a number of human cancers, including malignancies of breast, prostate and cervix [19,23,11,10,41,8,12,13,24]. Furthermore, one recent study suggested that Ca2+ entry through Orai1 channels are important for proliferation and survival of the rat glioblastoma cell line C6 [18]. In the present study, we focused on the role of STIM1/Orai1 in SOCE and established two primary glioblastoma cell lines derived from surgical samples of GBM (GBM1 and GBM8). We compared them to cultured human primary astrocytes in terms of STIM1/Orai1 expression, SOCE magnitude and pharmacology. We showed that GBM1, GBM8, the commercially available and widely used human glioblastoma cell line U251 and human primary astrocytes (HPA) display SOCE, upon passive store depletion with thapsigargin, with identical pharmacological profile, namely inhibition with 5μM Gd3+ and 30μM 2-aminoethoxyphenyl-borane (2-APB). The magnitude of SOCE was 2 fold higher in GBM1, GBM8 and U251 compared with HPA. siRNA knockdown showed that SOCE in GBM1 cells is encoded by STIM1 and Orai1. Whole cell patch clamp electrophysiology showed that upon passive store depletion with 20mM of the chelator BAPTA, GBM1 cells activate a highly Ca2+ selective CRAC current which was dependent on STIM1 and Orai1 expression. Finally, using matrigel invasion assay we showed that STIM1 and Orai1 knockdown caused a significant decrease in GBM1 and GBM8 cell invasion with no effect on HPA. However, the effects of STIM1 and Orai1 knockdown on GBM1 and GBM8 proliferation were rather modest; no significant effect on GBM cell proliferation was detected with STIM1 knockdown while Orai1 knockdown caused a small but statistically significant inhibition (by ~20%) of GBM1 and GBM8 cell proliferation.
Material and Methods
Reagents
Thapsigargin and 2-aminoethyl diphenylborinate (2-APB) were purchased from Calbiochem, Fura-2AM was from Molecular Probes, Cs-BAPTA was from Life Technologies/Invitrogen, and GdCl3 was from Thermo Fisher Scientific. Na-methanesulfonate and Cs-methanesulfonate were obtained from Sigma. Gene-specific STIM1 and Orai1 siRNAs were acquired from Thermo Fisher Scientific/Dharmacon and additionally validated in our previous work (see [23,24] for sequences and specificity controls). All gene specific primers were synthesized by Integrated DNA Technologies (see [23,24] for primer sequences). Anti-GFAP monoclonal antibody (glial fibrillary acidic protein; G3893) was from Sigma. Matrigel Boyden chambers were obtained from BD Biosciences. All other chemicals and reagents, unless otherwise specified, were from Thermo Fisher Scientific.
Cell cultures
Human glioblastoma cells U251-MG (U251; gift of Dr. M.G. Kaplitt, Weill-Cornell Medical Center, New York, NY), and primary human astrocytes (ScienCell Research Laboratories, Carlsbad, CA), were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin. Cells were grown at 37°C in a humidified atmosphere of 5% CO2/95% air. Cell culture medium was replaced twice a week, and cells were passaged as necessary using recombinant protease TrypLE Express. All culture reagents were from Life Technologies/Invitrogen.
Primary glioblastoma cells (GBM1 and GBM8) were prepared from surgical samples of pathologically confirmed glioblastoma multiforme as previously described [2]. Tissue was obtained with written consent directly from the surgical suite under a protocol approved by the Albany Medical Center Institutional Review Board. Dissected samples of tumor tissue (~300 mg) were thoroughly washed with ice-cold Ca2+/Mg2+ free phosphate-buffered saline (PBS, pH 7.4) and minced to small pieces. Tissue fragments were digested in the solution of 0.125% Trypsin/0.015% EDTA in PBS containing 250 μg/ml DNAse I, and gently triturated using a fire-polished glass Pasteur pipette. The resulting cell suspension was filtered through a Nylon cell strainer (70 μm, BD Biosciences). Cells were initially grown in T75 cell culture flasks (TPP, Trasadingen, Switzerland) in DMEM plus 20% FBS supplemented with 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 95% air and 5% CO2. After 2–3 passages FBS content was reduced to 10%, and penicillin and streptomycin concentrations were reduced to those described above. The astroglial origin and homogeneity of the resulting cultures were confirmed by immunocytochemical staining for glial fibrillary acidic protein (anti-GFAP monoclonal antibody, G3893, Sigma).
siRNA transfections
GBM1, GBM8, U251 and HPA cells were plated at ~60% confluence on 60 mm dishes (TPP) for mRNA expression analysis and Western blotting, or at ~20% density in poly-D-lysine coated 12-well plates (BD Biosciences) for proliferation assays. Sub-confluent cells were transfected using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s instructions, using STIM1 (siSTIM1), Orai1 (siOrai1), or non-targeting (siControl) siRNA constructs. In the majority of experiments, transfection was done together with pmaxGFP expressing plasmid (Lonza). Co-transfection with pmaxGFP was utilized to assess transfection efficacy and select cells for electrophysiological experiments. After transfection, cells were grown under standard culturing conditions for 72 hrs before mRNA expression assays, or for 96 hrs for Western blot analysis of protein expression and all functional assays.
STIM1 and Orai1 mRNA expression
Expression of genes of interest was determined using quantitative RT-PCR. mRNA samples were isolated from cultured cells using the RNAqueous-4PCR kit (Applied Biosystems/Ambion) according to the manufacturer’s instructions. Concentrations of mRNA were quantified using a NanoDrop 1000 (Thermo Fisher Scientific). cDNA samples were prepared using the iScript cDNA Synthesis kit (BioRad) following the manufacturer’s instructions. The RT reaction ratio was 1 μg of mRNA per each 20 μl of reaction mix. One μl of resulting cDNA was utilized for qPCR. Gene expression was quantified by using the CFX96 Real-Time PCR Detection System (BioRad) and iQ SYBR Green Supermix kit (BioRad) according to the manufacturer’s instructions. Two housekeeping genes, RPL13A and RPS20, were used as internal reference. Validated primers for reference genes were obtained from Qiagen; their sequences were proprietary.
Western blotting
Transfected cells were lysed using RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 0.2 mM EDTA). Protein samples (50–100 μg of protein per lane) were subjected to SDS-PAGE (7.5–10%) and then electrotransferred onto polyvinylidene fluoride membranes. After blocking with 5% nonfat dry milk (NFDM) dissolved in Tris-buffered saline containing 0.1% Tween 20 (TTBS) for 2 hr at room temperature, blots were washed 3 times with TTBS and incubated overnight at 4°C, with specific primary antibodies in TTBS containing 2% NFDM. The primary antibodies used were anti-hOrai1 (1:1000; Alomone Labs, Jerusalem, Israel), anti-hSTIM1 (1:250; BD Biosciences), and anti-β-actin-NT domain (1:2000; Sigma). Next day membranes were washed three times with TTBS and incubated for 1hr at room temperature with secondary horseradish peroxidase-conjugated anti-mouse (1:20,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or anti-rabbit IgG (1:10,000; Jackson ImmunoResearch Laboratories) in TTBS containing 2% NFDM. Detection was performed using the enhanced chemiluminescence reagent (ECL; GE Healthcare Life Sciences, Piscataway, NJ, USA) and a bioluminescent image analyzer LAS-4000 (FujiFilm Medical Systems, Stamford, CT, USA).
Ca2+ measurements
Ca2+ measurements were performed as described previously [23,24]. Briefly, coverslips with cells plated at medium density were mounted in a Teflon chamber and incubated at 37°C for 40–60 min in culture media containing 4 μM Fura-2/AM for all cell lines. Cells were then washed 3 times with solution containing (in mM) 140 NaCl, 1.13 MgCl2, 4.7 KCl, 2 CaCl2, 10 D-glucose, and 10 HEPES (pH 7.4, adjusted with NaOH). After washing, coverslips were incubated in the same medium for at least 10 min before [Ca2+] measurements were initiated. For [Ca2+] assays, fluorescence images of several cells were recorded and analyzed with a digital fluorescence imaging system InCyt Im2 (Intracellular Imaging Inc., Cincinnati, OH). Fura-2 was excited alternatively at 340 and 380 nm and fluorescence was captured at an emission wavelength of 510 nm. The 340/380 ratio images were obtained on a pixel-by-pixel basis. All experiments were conducted at room temperature. All figures depicting Ca2+ imaging traces are an average from several cells from one coverslip and are representative of several recordings (see figures for exact number of cells and independent recordings) performed on independently transfected cell batches.
Whole cell patch clamp electrophysiology
To measure CRAC currents, we carried out patch clamp recordings in the whole-cell configuration using an Axopatch setup (Digidata 1440A and Axopatch 200B, Molecular Devices) as previously described[23,24,38,42]. Data analysis was performed using Clampfit 10.1 software. Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments) using a P-97 micropipette puller (Sutter Instruments). They were further polished with a DMF1000 microforge (World Precision Instruments) to a resistance of 2.5–4.0 MΩ when filled with pipette solution of the following composition (in mM):105 Cs-methanesulfonate, 20 Cs-1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (Cs-BAPTA), 8 MgCl2, and 10 HEPES (pH adjusted to 7.2 with CsOH). Increased concentration of MgCl2 (8 mM) in the pipette solution was utilized to inhibit TRPM7 currents[31]. Before performing the recordings, cells were washed with bath solution containing (in mM): 105 Na-methanesulfonate, 10 CsCl, 1.2 MgSO4, 20 CaCl2, 5 glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH). Only cells with tight seals (>16 GΩ) were selected to break in. Cells were maintained at a 0 mV holding potential and subjected to voltage ramps from −140 to +100 mV, lasting 250 ms and applied every 2 s. To amplify Ca2+ channel currents during experiments, the bath solution was switched to divalent-free (DVF) solution containing (in mM): 135 Na-methanesulfonate, 10 HEDTA, 1 EDTA, and 10 HEPES (pH 7.4, adjusted with NaOH) was used as previously published [38]. All currents were low-pass filtered at 2 kHz and sampled at a rate of 10 kHz. All experiments were performed at room temperature (20–25 °C).
Proliferation assays
Cell proliferation was quantified using a colorimetric MTT proliferation assay. HPA, GBM1, and GBM8 cells were plated in 12-well plates at the density of 40,000, 30,000 and 40,000 cells per well, respectively. Cells were transfected as described above 24 hr later, and were grown additionally for 96 hr after transfection. For MTT assays, cells were briefly washed with basal solution containing (in mM): 135 NaCl, 3.8 KCl, 1.2 MgSO4, 1.3 CaCl2, 1.2 KH2PO4, 10 HEPES, and 10 glucose (pH = 7.4, adjusted with NaOH). 500μL of basal solution containing 0.5 mg/ml thiazolyl blue tetrazolium bromide (MTT) (Sigma) were then added to each well. After 1 hr incubation at 37°C, MTT solution was removed, and cells were solubilized in acidified isopropanol (Sigma) to dissolve newly formed formazan particles. 200-μl aliquots from each well were then transferred into a 96-well plate. Absorbances at 562 nm were measured using ELx800 absorbance microplate reader (BioTek Instruments), and compared to absorbance in wells transfected with control siRNA constructs.
Matrigel invasion assays
Invasion assays were performed using Matrigel coated Invasion Chambers (BD Biosciences) in accordance with the manufacturer’s protocol. For these experiments cells were transfected with either siControl, siOrai1 or siSTIM1. 72 hr post transfection, they were plated in the upper chamber of rehydrated Matrigel inserts, serum free media at density of 3 × 105 cells/chamber for GBM cells and 6 × 105 cells/chamber for HPA. Cells were allowed to invade Matrigel matrix overnight towards the bottom chamber containing complete culture media with 10% FBS. On the next day, the bottom part of the insert was fixed with paraformaldehyde and stained with DAPI (Vectashield) to visualize the nuclei of invaded cells. Ten random pictures of each insert were taken using a Leica DM IRB microscope at 20× magnification. The number of invaded cells was counted manually. All invasion assays were performed in triplicates and in at least three independent transfections.
Statistical analysis
Data are expressed as the mean values ± SE. Statistical analysis was done using paired t-test with Origin 8.1 software (OriginLab, Northampton, MA, USA), except when described otherwise. Differences were considered significant when P values were < 0.05. The P values of < 0.05, <0.01 and <0.001 are indicated as *, **, and ***, respectively.
Results
Store-operated Ca2+ entry (SOCE) in non-malignant human primary astrocytes (HPA) and glioblastoma (GBM) cells
Primary glioblastoma cells (GBM1 and GBM8) were established from surgical samples of pathologically confirmed glioblastoma multiforme patients under an approved protocol as previously described [2]. Figure 1 shows immunostaining of HPA, GBM1 and GBM8 cells with an antibody recognizing the astroglial cell marker, glial fibrillary acidic protein (GFAP; green), thus confirming the astroglial nature of these cells. These images are representative of typical immnostainings from all primary human astroglial cell cultures used in our lab. Please note that despite substantial morphological difference between HPA, GBM1, and GBM8 (Figure 1), in all cases >95% of all cells were GFAP-positive.
Figure 1. Characterization of astroglial origin of HPA and GBM cells.
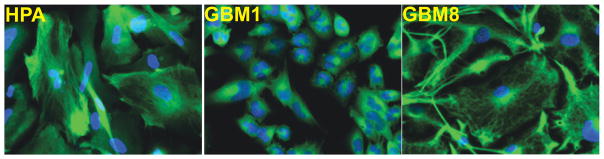
Immunostaining of primary human astrocytes (HPA) and primary glioblastoma cell lines (GBM1 and GBM8) derived from surgical samples of glioblastoma multiforme. Cells were fixed and stained with the antibody recognizing the astroglial cell marker, GFAP (green) and counterstained with DAPI (blue). All images were captured using a Zeiss LSM510META-NLO confocal microscope at the same magnification. In all cultures >95% of cells were GFAP-positive.
To uncover and characterize SOCE in GBM cells, we performed Fura2 Ca2+ imaging assays in cells treated with the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin (2 μM). This standard pharmacological maneuver is used to deplete the internal Ca2+ stores and activate SOCE. We used the classical SOCE protocol to separate the Ca2+ release from intracellular stores from Ca2+ entry through plasmalemmal channels[6,38,27,7]. Intracellular Ca2+ release was assessed upon addition of thapsigargin in nominally Ca2+ free solution. Ca2+ entry was measured upon restoration of 2 mM Ca2+ to the bath solution after depletion of the intracellular stores. In experiments presented in Figure 2, we compared SOCE characteristics in non-malignant human primary astrocytes (HPA) to those in human primary glioblastoma multiforme cells (GBM1 and GBM8) and in the well-established GBM cell line U251. In all three GBM cell lines as well as in HPA, the SOCE displayed the typical pharmacological properties of SOCE reported for many other cell types, namely blockade by low concentration of lanthanides (5 μM Gd3+) and 30 μM 2-APB (Figure 2). These characteristics are consistent with the idea that the SOCE in GBM cells and HPA is encoded by STIM1 and Orai1. Interestingly, the magnitude of SOCE in all three GBM cell lines was significantly larger than in HPA (Figure 2). Statistical analysis on a large number of independent measurements including a total of 100–300 cells per condition showed that the magnitude of SOCE in GBM cells was twice that of HPA (Figure 3A); no statistically significant difference was noted between different cell lines regarding the Ca2+ release phase.
Figure 2. Pharmacological characterization of SOCE in Human primary astrocytes and Glioblastoma cells.

Standard “Ca2+ off/Ca2+ on” protocol was employed using thapsigargin to measure SOCE in human primary astrocytes HPA (A and B), primary glioblastomas GBM1 and GBM8 (C–F) and established glioblastoma cell line U251 (G, H). 5 μM Gd3+ and 30 μM 2-APB were used to block SOCE. Both Gd3+ and 2-APB were able to completely abrogate SOCE in all cell types suggesting that all these cells mediate SOCE via a molecularly similar pathway. Representative traces representing average of several cells assayed simultaneously from the same coverslip (n) are shown in the figure. Each trace is representative of at least three independent coverslips.
Figure 3. Statistical analysis of SOCE and mRNA expression of STIM1 and Orai1.

A, The amplitude of SOCE in all four cell lines (HPA, GBM1, GBM8 and U251) was calculated from all the experiments performed and was statistically analyzed. The number of independent experiments performed and total number of cells analyzed for each cell line are shown in the bar graph as n=x, y where x=number of independent experiments and y=total number of cells analyzed. B, qPCR on reverse-transcribed STIM1 and Orai1 mRNA isolated from all four cell lines and normalized to expression in HPA.
In order to understand if increased SOCE in malignant GBM cells is related to increased expression of STIM1 and Orai1, we performed quantitative PCR assays using the reverse-transcribed mRNA samples, which were prepared from HPA, GBM1, GBM8 and U251 cells. As seen in Figure 3B, GBM1 and GBM8 cells had increased expression of Orai1 mRNA compared to HPA. Furthermore, GBM8 cells showed increased expression of STIM1. We found no statistically significant changes in expression of either Orai1 or STIM1 in U251 cells.
SOCE in GBM cells is encoded by STIM1 and Orai1
We showed above that all GBM cell lines display the same pharmacological profile suggesting that SOCE in all cell lines is likely encoded by STIM1 and Orai1. To determine the molecular players of SOCE in GBM cells, we chose to undertake protein knockdown of STIM1 and Orai1 using siRNA silencing approach. We selected one cell line, GBM1, to initially test the efficacy of siRNA constructs in primary GBM cells. Our previous studies validated the selectivity and potency of these siRNA in other human cells lines, such as human breast cancer cells and human endothelial cells [1,23,24]. The efficacy of STIM1 and Orai1 downregulation was documented at the mRNA levels (using qPCR at 72 hr post-transfection) as well as protein levels (using Western blotting at 96 hr post-transfection). As seen in Figure 4A, C, STIM1 siRNA caused ~65% downregulation in mRNA and corresponding ~65% decrease in STIM1 protein. Transfection with Orai1 siRNA (Figure 4B, D) downregulated Orai1 mRNA expression by ~90% and protein levels by ~75%.
Figure 4. STIM1 and Orai1 knockdown in GBM1 cells.

A, B, qPCR on GBM1 cells transfected with siRNA against either STIM1 or Orai1 showing significant decrease in STIM1 and Orai1 expression. C, D, Western blot analysis showing that siRNA targeting STIM1 and Orai1 were able to significantly knockdown their respective targets. Data representative of three independent experiments
The same constructs were used then to test their effects on SOCE. STIM1 siRNA knockdown led to a significant 46% decrease in SOCE (Figure 5A, C) while siRNA targeting Orai1 significantly reduced SOCE by 66% (Figure 5B, D).
Figure 5. STIM1 and Orai1 mediate SOCE in GBM1 cells.

Representative traces from GBM1 cells transfected with either siSTIM1 (A) or siOrai1 (B) and depicting abrogation of SOCE upon STIM1 or Orai1 knockdown compared to cells transfected with non-targeting siRNA (siControl). C, D, Statistical analysis on SOCE from 8 independent experiments originating from at least two separate transfections is shown. Number of independent experiments performed and total number of cells analyzed for each condition are also depicted in the bar graph as n=x, y where x=number of independent experiments and y=total number of cells analyzed.
Store depletion-activated CRAC currents in GBM cells also depend on STIM1 and Orai1
To provide the final proof that SOCE pathway in GBM cells requires STIM1 and Orai1, we performed whole-cell patch clamp recordings of CRAC currents in GBM1 cells transfected with either STIM1 or Orai1 siRNA. To isolate CRAC, the recordings were done with a pipette solution containing 20 mM of the Ca2+ chelator BAPTA to deplete the internal Ca2+ stores and a bath solution containing 20 mM Ca2+ to maximize inward Ca2+ currents. As seen in Figure 6A, in whole-cell mode BAPTA dialysis produced small, slowly developing inward current that was fully activated within 3–4 minutes. This current was potently amplified by replacing the Ca2+-containing bath solution with a divalent-free (DVF) solution (Figure 6A). In DVF solutions, Na+ ions become the charge carrier. As shown in the same panel (Figure 6A), during the second and the third DVF application, this amplified Na+ current immediately and rapidly depotentiated within the short time window of DVF application. Such current behavior is highly consistent with the signature CRAC properties[31]. The first DVF switch that was performed right after whole-cell break-in, at the time when CRAC development was negligible, was used to gauge background currents before store depletion (Figure 6A, note the absence of depotentiation). In Figure 6D, we show the current/voltage (I/V) relationship of currents recorded in DVF bath solutions taken where indicated by the asterisk in Figure 6A, after subtraction of background currents. The resulting currents show all typical properties of classical CRAC, namely inward rectification and positive reversal potential (~+60mV)[17]. Most importantly, knockdown of either STIM1 or Orai1 in GBM1 cells led to a dramatic inhibition of CRAC currents measured in either Ca2+-containing or DVF solutions (Figure 6B–E). Figure 6E present statistical comparison of CRAC current amplitudes recorded from 5–6 cells per group.
Figure 6. STIM1 and Orai1 mediate CRAC currents in GBM1 cells.

CRAC currents activated by BAPTA dialysis through the patch pipette are abolished by STIM1 and Orai1 specific knockdown compared to control siRNA (A–C). A, Whole-cell CRAC currents measured in Ca2+-containing (20mM) bath solutions (at −100mV) and Na+ CRAC currents recorded in divalent free (DVF) external solutions were activated by dialysis of 20mM BAPTA through the patch pipette in cells transfected with control non-targeting siRNA (siControl). Knockdown of either STIM1 (B) or Orai1 (C) dramatically inhibited both Ca2+ and Na+ CRAC currents activated by BAPTA as compared to control (sicontrol 1.395 ± 0.236 vs 0.360 ± 0.105 siSTIM1 vs 0.380 ± 0.039 siOrai1). I/V relationships of Na+ CRAC from GBM1 cells transfected with either siControl, siSTIM1 or siOrai1were taken where indicated by the color-coded asterisks and are shown in D. Statistical analyses on Na+ CRAC currents taken at −100mV from several independent experiments (n number is provided in figure) are shown in E.
Functional effects of STIM1 and Orai1 gene silencing on GBM proliferation and invasiveness
After validation of siRNA efficacy and determination that SOCE and CRAC in GBM cells are mediated by STIM1 and Orai1, we sought to determine the functional significance of these two proteins in non-malignant and malignant glial cells. We focused on two important aspects of cell physiology that have a direct impact on malignancy of transformed cells, namely rate of proliferation and invasiveness.
We started with comparing rates of cells proliferation in primary GBM1 and GBM8 cells after STIM1 or Orai1 silencing, using commonly utilized MTT assay. Transfection with siRNA against either STIM1 or Orai1 produced modest effect on GBM1 and GBM8 cell proliferation as compared to the same cells transfected with non-targeting control siRNA, with the effects of siOrai1 being the only statistically significant (~20%; Figure 7). Unlike rapidly proliferating GBM1 and GBM8, which over 4 days increased MTT signal >4 fold, transfected human primary astrocytes proliferated very slowly and produced marginal increase in the MTT signal over the same time frame (tested in three cultures; data not shown). This slow growth and the resulting insufficient sensitivity in MTT assays, did not allow us to determine the effects, if any, of STIM1 and Orai1 gene silencing on HPA proliferation.
Figure 7. STIM1 and Orai1 knockdown marginally affected on GBM1 and GBM8 cell proliferation.
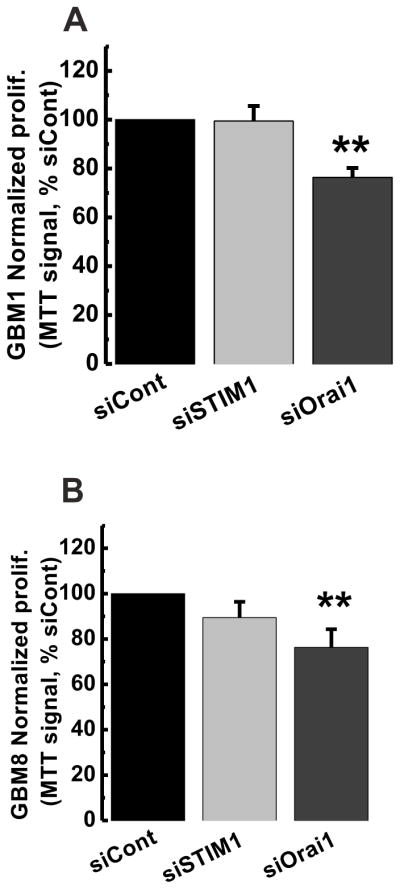
GBM cells were transfected with siRNA against either STIM1 (siSTIM1), Orai1 (siOrai1) or non-targeting siRNA control (siControl). MTT assays were performed as described in the methods section and results are shown for GBM1 (A) and GBM8 (B) cells. STIM1 knockdown has no effect on GBM cell proliferation while Orai1 knockdown caused a small but significant inhibition of proliferation (~20%). Data are representative of four independent experiments.
In a manner similar to our previous studies in breast cancer [24], we next used Matrigel invasion assays with serum as a chemo-attractant to explore the role of STIM1 and Orai1 in GBM invasion. HPA were again used as a non-malignant control. As shown in Figure 8A–C, silencing of either STIM1 or Orai1 potently reduced invasion of GBM1 and GBM8 cells (in the range of 55–65%, P<0.01). Interestingly, STIM1 and Orai1 siRNA had no effects on Matrigel invasion of HPA. This was not due the inability of HPA ability to invade Matrigel matrix. These cells showed some level of invasion, which was significantly less than that of GBM1 and GBM8 cells, but was not affected by either STIM1 or Orai1 gene silencing (Figure 8A, D).
Figure 8. STIM1 and Orai1 regulate invasion of GBM1 and GBM8 cells but not HPA.

Knockdown of either STIM1 or Orai1 significantly decreased invasive capabilities of GBM1 and GBM8 cells in response to serum (A–C), while no effect was observed for HPA (A, D). Cells were seeded in the top chamber in absence of serum and were allowed to invade overnight towards the bottom chamber containing 10% FBS (see methods). On the next day, cells that were able to invade were fixed, stained with DAPI and were manually counted to assess invasion. The statistical analyses of approximately 100 fields originating from 3 independent transfections analyzed in triplicates are also shown (B–D).
Discussion
There are two major findings from this work: 1) identification of STIM1 and Orai1 as essential components of SOCE and CRAC in two primary GBM cell lines established from surgical samples from human patients; and 2) discovery of the critical role for STIM1 and Orai1 in regulation of GBM cell invasiveness, but a modest effect on cell proliferation. These conclusions are supported by gene silencing experiments performed with siRNA constructs thoroughly validated in our previous studies [23,24,1]. They are corroborated by independent measurements of SOCE using Ca2+ imaging, and CRAC current recordings using whole cell patch clamp electrophysiology.
Abrogation by low concentrations of lanthanides (e.g. 5μM Gd3+) and 30–50μM 2-APB are characteristics of STIM1/Orai1-mediated SOCE. SOCE in GBM cells shows typical pharmacological features of STIM1/Orai1-mediated Ca2+ entry identified in numerous non-malignant cell types, including leukocytes, endothelial cells and smooth muscle cells[30,1,27], as well as certain malignant cells, such as breast cancer cells[23]. Specifically, primary GBM cells activate highly Ca2+ selective CRAC currents in response to store depletion, with classical CRAC biophysical properties. Consistent with the idea of STIM1 and Orai1 involvement in SOCE and CRAC, we found STIM1 and Orai1 mRNA and proteins to be expressed in GBM cells. The established and frequently utilized U251 human glioblastoma cells and primary human astrocytes also showed SOCE activity in response to thapsigargin with an identical pharmacological profile to GBM1 and GBM8 cells. Furthermore, siRNA gene silencing of STIM1 and Orai1 in primary GBM1 cells unequivocally demonstrated indispensable role of both proteins in SOCE and CRAC currents.
Interestingly, quantitative comparison of SOCE in GBM cells demonstrated approximately two-fold upregulation of SOCE in all malignant cell lines utilized in this study, as compared to non-malignant HPA. These results are consistent with the upregulation of SOCE components in tumor tissues. Indeed, Scrideli et al. recently performed a gene expression profile analysis on primary glioblastomas and identified a number of potential target genes that were significantly overexpressed in GBM, including STIM1 [36]. Previous work by Suyama et al. associated STIM1 expression with metastatic melanomas [39]. Our own qPCR assays in primary GBM cell lines, showed statistically significant upregulation of Orai1 transcripts, and, in one case, STIM1. However, upregulation of SOCE did not necessarily mirror upregulation of STIM1 and Orai1 mRNA. For instance, U251 cells demonstrated statistically significant increase in SOCE with no matching increases in expression of either STIM1 or Orai1 transcripts. These data may point to a more complex regulation of SOCE in malignant cells than merely changes in the mRNA expression level; Orai1 protein levels might be upregulated in U251 cells despite unchanged mRNA levels.
The critical question, of course, is whether STIM1/Orai1 contributes to the cancerous properties of GBM cells. Emerging literature points to major role of STIM1/Orai1 in tumorogenesis, at least in certain tumor types. Thus, Yang et al. discovered critical involvement of STIM1 and Orai1 in invasiveness and metastasis of breast cancers [41]; while Chen et al. obtained qualitatively similar results regarding STIM1 and Orai1 in cervical cancer growth, migration, and angiogenesis[8]. In our own recent work, we found that Orai3, another member of the Orai family that is exclusively expressed in mammals, is a unique component of SOCE pathway in the estrogen receptor α-positive (ERα+) breast cancer cells and plays a critical role in ERα+ tumor progression both in vitro and in vivo [23,24]. Downregulation of Orai3 potently reduced ERα+ breast cancer cell proliferation and invasiveness, as determined by assays similar to those utilized in this study. However, it remains to be determined whether the effects observed on GBM invasion with STIM1 and Orai1 knockdown are due to SOCE or to store-independent pathways[37] (or other alternative pathways) encoded or regulated by STIM and Orai proteins. Further studies are needed to determine the role of Orai2 and Orai3 in native Ca2+ signaling pathways in GBM cells and the potential involvement of Orai2 and Orai3 in GBM development. Our results not only suggest important role of STIM1 and Orai1 in tumor progression, but also indicate potential genetic diversity of STIM1/Orai1-mediated pathways between different tissues. Our present findings highlight the involvement of STIM1 and Orai1 in GBM cell invasion. Knockdown of either gene transcript potently suppressed invasion of primary GBM1 and GBM8 cells in vitro, in Matrigel invasion assays. In contrast, STIM1 or Orai1 silencing had no effect on invasion properties of non-malignant HPA cells.
In contrast to invasion, STIM1 knockdown had no significant effect on GBM cell proliferation, while Orai1 knockdown produced a marginal albeit statistically significant inhibition of cell growth. The limited dependence of GBM cell proliferation on STIM1 and Orai1 expression (~20% decrease in cell proliferation upon Orai1 knockdown) suggests a minimal role for SOCE in this process. Nonetheless, this small effect of Orai1 knockdown might contribute to the overall tumor progression in vivo. A recent study using a rat glioblastoma C6 cell line found that Orai1-mediated SOCE is important for GBM cell proliferation and survival [18]. The same authors reported that Orai1 knockdown in rat C6 cells was more efficient at inhibiting cell proliferation than STIM1 knockdown [18]. In rat C6 cells suppression of proliferation was substantially stronger than that observed by us in human primary GBM cells. Such differences may reflect different contribution of SOCE to proliferation between rat and human cells, or differences between primary tumor cells and immortalized cell lines.
In conclusion, high dependence of GBM cell invasion on expression of STIM1 and Orai1 suggests the potential use these genes as a prospective drug target for new therapies. Future studies are needed to unravel the native channels contributed by STIM/Orai proteins in GBM cells, the precise mechanisms of their activation and the downstream signaling pathways linking these Ca2+ entry routes to migratory responses of GBM cells.
Acknowledgments
This work was supported in part by NIH grants R01 HL097111 to MT, R01 NS061953 to AAM, R01 HL095566 to KM, and AMC translational grant 206-465247 to AAM and YHK.
References
- 1.Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circulation research. 2008;103(11):1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. 01.RES.0000338496.95579.56 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdullaev IF, Rudkouskaya A, Mongin AA, Kuo YH. Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulate cell proliferation. PloS one. 2010;5(8):e12304. doi: 10.1371/journal.pone.0012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnholtz-Sloan JS, Sloan AE, Schwartz AG. Relative survival rates and patterns of diagnosis analyzed by time period for individuals with primary malignant brain tumor, 1973–1997. Journal of neurosurgery. 2003;99(3):458–466. doi: 10.3171/jns.2003.99.3.0458. [DOI] [PubMed] [Google Scholar]
- 4.Behin A, Hoang-Xuan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet. 2003;361(9354):323–331. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361 (6410):315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 6.Bird GS, DeHaven WI, Smyth JT, Putney JW., Jr Methods for studying store-operated calcium entry. Methods. 2008;46(3):204–212. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, Barroso M, Singer HA, Jourd’heuil D, Trebak M. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. American journal of physiology Cell physiology. 2010;298 (5):C993–1005. doi: 10.1152/ajpcell.00325.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen YF, Chiu WT, Chen YT, Lin PY, Huang HJ, Chou CY, Chang HC, Tang MJ, Shen MR. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15225–15230. doi: 10.1073/pnas.1103315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Derler I, Schindl R, Fritsch R, Romanin C. Gating and permeation of Orai channels. Frontiers in bioscience: a journal and virtual library. 2012;17:1304–1322. doi: 10.2741/3988. [DOI] [PubMed] [Google Scholar]
- 10.Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. Journal of cellular physiology. 2011;226(2):542–551. doi: 10.1002/jcp.22363. [DOI] [PubMed] [Google Scholar]
- 11.Faouzi M, Kischel P, Hague F, Ahidouch A, Benzerdjeb N, Sevestre H, Penner R, Ouadid-Ahidouch H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochimica et biophysica acta. 2012 doi: 10.1016/j.bbamcr.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Monteith GR, Rao R. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 2010;143(1):84–98. doi: 10.1016/j.cell.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flourakis M, Lehen’kyi V, Beck B, Raphael M, Vandenberghe M, Abeele FV, Roudbaraki M, Lepage G, Mauroy B, Romanin C, Shuba Y, Skryma R, Prevarskaya N. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell death & disease. 2010;1:e75. doi: 10.1038/cddis.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes & development. 2007;21(21):2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle BC, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M. Store-Independent Orai1/3 Channels Activated by Intracrine LeukotrieneC4: Role in Neointimal Hyperplasia. Circulation research. 2013 doi: 10.1161/CIRCRESAHA.111.300220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends in biochemical sciences. 2007;32 (5):235–245. doi: 10.1016/j.tibs.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355 (6358):353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 18.Liu H, Hughes JD, Rollins S, Chen B, Perkins E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Experimental and molecular pathology. 2011;91(3):753–760. doi: 10.1016/j.yexmp.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 19.McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, Monteith GR. ORAI1-mediated calcium influx in lactation and in breast cancer. Molecular cancer therapeutics. 2011;10(3):448–460. doi: 10.1158/1535-7163.MCT-10-0923. [DOI] [PubMed] [Google Scholar]
- 20.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. The Journal of physiology. 2007;579 (Pt 3):703–715. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. The Journal of physiology. 2008;586 (1):185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nature reviews Cancer. 2007;7 (7):519–530. doi: 10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- 23.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. The Journal of biological chemistry. 2010;285 (25):19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27(1):63–75. doi: 10.1096/fj.12-213801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muik M, Schindl R, Fahrner M, Romanin C. Ca(2+) release-activated Ca(2+) (CRAC) current, structure, and function. Cellular and molecular life sciences: CMLS. 2012;69(24):4163–4176. doi: 10.1007/s00018-012-1072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiological reviews. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 27.Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23(8):2425–2437. doi: 10.1096/fj.09-131128. fj.09-131128 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potier M, Trebak M. New developments in the signaling mechanisms of the store-operated calcium entry pathway. Pflugers Archiv: European journal of physiology. 2008;457(2):405–415. doi: 10.1007/s00424-008-0533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443 (7108):230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 30.Prakriya M, Lewis RS. Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. The Journal of physiology. 2001;536 (Pt 1):3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. The Journal of general physiology. 2002;119 (5):487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nature reviews Cancer. 2011;11(8):609–618. doi: 10.1038/nrc3105. [DOI] [PubMed] [Google Scholar]
- 33.Putney JW., Jr A model for receptor-regulated calcium entry. Cell calcium. 1986;7 (1):1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 34.Putney JW., Jr Capacitative calcium entry revisited. Cell calcium. 1990;11 (10):611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 35.Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nature clinical practice Neurology. 2006;2(9):494–503. doi: 10.1038/ncpneuro0289. quiz 491 p following 516. [DOI] [PubMed] [Google Scholar]
- 36.Scrideli CA, Carlotti CG, Jr, Okamoto OK, Andrade VS, Cortez MA, Motta FJ, Lucio-Eterovic AK, Neder L, Rosemberg S, Oba-Shinjo SM, Marie SK, Tone LG. Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. Journal of neuro-oncology. 2008;88(3):281–291. doi: 10.1007/s11060-008-9579-4. [DOI] [PubMed] [Google Scholar]
- 37.Shuttleworth TJ. Arachidonic acid, ARC channels, and Orai proteins. Cell calcium. 2009 doi: 10.1016/j.ceca.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spinelli AM, Gonzalez-Cobos JC, Zhang X, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA, Trebak M. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Archiv: European journal of physiology. 2012;464(5):481–492. doi: 10.1007/s00424-012-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suyama E, Wadhwa R, Kaur K, Miyagishi M, Kaul SC, Kawasaki H, Taira K. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. The Journal of biological chemistry. 2004;279(37):38083–38086. doi: 10.1074/jbc.C400313200. [DOI] [PubMed] [Google Scholar]
- 40.Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW., Jr Phospholipase C-coupled receptors and activation of TRPC channels. Handbook of experimental pharmacology. 2007;(179):593–614. doi: 10.1007/978-3-540-34891-7_35. [DOI] [PubMed] [Google Scholar]
- 41.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer cell. 2009;15(2):124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 42.Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circulation research. 2011;109 (5):534–542. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]