

Abstract
Of the features that characterize glioblastoma, arguably none is more clinically significant than the propensity of malignant glioma cells to aggressively invade into the surrounding normal brain tissue. These invasive cells render complete resection impossible, confer significant resistance to chemo- and radiation-therapy, and virtually assure tumor recurrence. Expression of TROY (TNFRSF19), a member of the tumor necrosis factor receptor superfamily, inversely correlates with patient survival and stimulates glioblastoma cell migration and invasion in vitro. In this study, we report that TROY is overexpressed in glioblastoma tumor specimens and TROY mRNA expression is increased in the invasive cell population in vivo. In addition, inappropriate expression of TROY in mouse astrocytes in vivo using glial-specific gene transfer in transgenic mice induces astrocyte migration within the brain validating the importance of the TROY signaling cascade in glioblastoma cell migration and invasion. Moreover, knockdown of TROY expression in a primary glioblastoma xenograft significantly prolonged survival in vivo. We also report that TROY expression significantly increases resistance of glioblastoma cells to both IR- and TMZ- induced apoptosis in vitro via activation of Akt and NF-κB. Inhibition of either Akt or NF-κB activity suppressed the survival benefits of TROY signaling in response to TMZ treatment. These findings position aberrant expression and/or signaling by TROY as a contributor to the dispersion of glioblastoma cells and therapeutic resistance.
Keywords: TROY, glioblastoma, invasion, resistance
Introduction
Glioblastoma multiforme (GBM) is the most common primary central nervous system tumor accounting for approximately 40% of all primary malignant brain tumors. The mechanism driving the development and recurrence of GBM is still largely unknown which greatly limits the successful treatment of this disease. Indeed, available treatment options for glioblastoma patients remain limited and largely ineffective. Currently, standard treatment includes surgical resection followed by radiation together with concurrent and adjuvant temozolomide (TMZ) chemotherapy which extends the current median survival to 14.6 months (1). Unfortunately, GBM exhibits a high resistance to these standard therapies and recurrence is virtually assured. It is therefore essential to develop new therapeutic strategies for the management of GBM. The development of new combinational therapies, together with an increase in the selectivity of the treatments based on a detailed molecular characterization of these tumors (2) has significant potential to enhance the survival of GBM patients.
The tumor necrosis factor receptor superfamily member TROY (TNFRSF19) is a type I cell surface receptor protein containing the highly conserved TNFR cysteine–rich motifs in the extracellular domain and a tumor necrosis factor-receptor-associated factor (TRAF) – binding sequence in the cytoplasmic domain (3). Although expression of TROY has been described in progenitor cells of the hippocampus, thalamus, and cerebral cortex (3–8), its role in neuronal and glial cell function remains largely undefined. We have demonstrated that increased expression of TROY stimulated glioma cell migration in vitro and increased cell invasion in an organotypic brain slice model (9). Conversely, siRNA mediated knockdown of TROY expression significantly inhibited glioma cell migration and invasion. Furthermore, gene expression profiling of TROY in brain tumor samples indicated that TROY mRNA expression directly correlated with increasing glial tumor grade and was significantly increased in GBM tumor samples. Notably, we demonstrated that TROY expression inversely correlates with patient survival suggesting that TROY expression may play a role in GBM progression and is a good indicator of survival outcome.
The mechanistic basis for TROY mediated stimulation of glioma migration and invasion remains to be defined. We recently demonstrated that increased expression of TROY activates Rac1 signaling in a Pyk2-dependent mechanism (9) linking TROY signaling to cytoskeletal reorganization required for cell motility. Rac1 activation has previously been linked to cell invasion in cancer (10–12) and the activation of Rac1 by the TNFRSF member Fn14 stimulates glioma cell migration and invasion (13). While activation of Rac1 suggests a mechanism for TROY mediated glioma invasion, the role of TROY in survival signaling has not been determined. Previous studies have demonstrated that invasive cells exhibit increased therapeutic resistance as the process of invasion strongly upregulates survival pathways and downregulates pro-apoptotic pathways in the invading cells (14–16). Thus, TROY signaling may coordinately activate signaling pathways important for glioma cell invasion and cell survival that increase resistance and contribute to tumor recurrence.
In this study, we investigated the role of TROY in therapeutic resistance and survival signaling. We show that TROY expression is increased in GBM tumor samples and enhanced in the invasive cell population. We provide evidence that TROY expression increases resistance to radiation and TMZ which is associated with increased survival signaling dependent upon activation of Akt and NF-κB. In addition, we demonstrate that knockdown of TROY expression increases survival in a glioma intracranial xenograft model. These results further support a role for TROY in GBM pathobiology and suggests that targeting TROY and its signaling pathway represents a novel approach to increase tumor vulnerability to cytotoxic therapies and improve the therapeutic response of glioblastoma.
Materials and Methods
Antibodies and reagents
The anti-HA epitope antibody was obtained from Cell Signaling Technology. The anti-TROY polyclonal antibody was obtained from Abcam. Antibodies to Akt, phospho-Akt, IκB, phospho-IκB, NF-κB, phospho-NF-κB, and cleaved PARP (Asp214) were from Cell Signaling Technology (Beverly, MA). Antibodies to α-tubulin and β-actin were from Millipore (Billerica, MA). The NF-κB inhibitor BAY-11-7082, the AKT inhibitor LY294002, and temozolomide were obtained from Sigma (St Louis, MO). Human placenta laminin was obtained from Sigma.
Cell culture
The human glioblastoma cell lines T98G, SNB19, U118 (American Type Culture Collection), the 293FT lentiviral packaging cell line (Life Technologies), and DF-1 chicken fibroblasts were passaged in DMEM supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 2 mM glutamine, 100 units/ml penicillin, and 10 mg/ml streptomycin. When indicated, cells were serum starved by replacing the culture medium with DMEM supplemented with 0.1% bovine serum albumin. The primary GBM xenograft line 10 (GBM10) was established from a patient surgical sample and maintained as a flank xenograft in immune deficient mice (17, 18). GBM10 flank tumor xenografts were harvested, mechanically disaggregated, and grown in short term culture for 5–7 days in DMEM media for lentiviral transduction before intracranial implantation.
Clinical samples, laser capture microdissection, and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Snap-frozen human non-neoplastic brain specimens from epileptogenic patients and human glioblastoma tumor samples (WHO Grade IV) obtained from patients who underwent primary therapeutic subtotal or total tumor resection under image guidance were obtained from Dr. Timothy Ryken (Department of Neurosurgery) at the University of Iowa. All specimens were collected under an Institutional Review Board approved protocol and de-identified for patient confidentiality. Histological diagnosis was made by standard light microscopic evaluation of hematoxylin and eosin (H&E)-stained sections. Sample integrity, estimated tumor content, and extent of tissue heterogeneity was determined by a board certified pathologist (JE). Laser capture microdissection (LCM) to isolate the invasive cell population from the tumor edge and the matched tumor core population was performed as described (19). Total RNA was isolated from the LCM cells using the Paradise Reagent System (Arcturus, Mountain view, CA). PCR analysis of TROY (sense, 5′-TGCTTGCCAGGATTTTATAGGAA-3′; antisense, 5′-GACGCGATCTTCACGAGGTT-3′) and histone H3.3 (sense, 5′-CCACTGAACTTCTGATTCGC-3′; antisense, 5′-GCGTGCTAGCTGGATGTCTT-3′) was quantified by RT-PCR using a Light Cycler 480 (Roche) with SYBR Green fluorescence signal detection after each cycle of amplification. The results were quantified and analyzed as described previously (20).
Colony formation assay
A clonogenic assay was used to assess cell survival after radiation treatment of cell lines as described previously (21). Briefly, 5.0 × 105 cells were seeded in 100 mm diameter culture dishes and incubated overnight at 37ºC. Subsequently, cells were irradiated in the culture dish at 0, 2, 4, and 6 Gy radiation dose using a RS 2000 X-ray irradiator (Rad Source Technologies Inc., Suwanee, GA). Immediately after irradiation, cells were trypsinized, counted, and plated in a 6-well culture dish at densities of 100, 250, and 500 cells/well in triplicate. Cells were incubated for 12 days then fixed with a 10% acetic acid + 10% methanol solution. Cell colonies were stained with 0.5% crystal violet solution and counted manually by blinded observers.
Cell proliferation and viability assays
The Alamar Blue assay (Biosource, Camarillo, CA) was used to assess cell proliferation as previously described (22). Briefly, 750 cells were seeded into wells of a 96-well plate in 100 μl of DMEM supplemented with 10% fetal bovine serum and incubated at 37ºC. At 24 hour intervals up to 144 hours, 10 μl (10% of total volume) of Alamar Blue reagent was added to the cells and incubated at 37ºC for 5 hrs. A standard curve using an 8-point serial dilution of cells (beginning with 750 cells/well) was prepared. The plates were read on a fluorescence plate reader (Biotek Synergy HT) excitation 560 nm; emission 600 nm. Averages of the fluorescence values were calculated and the cell numbers were determined from the standard curve. For temozolomide (TMZ) experiment, cells were treated with TMZ (250 μM) for 48 hours prior to addition of Alamar Blue.
GBM tissue microarray and immunohistochemistry
The preparation of the GBM tissue microarray and immunohistochemistry protocols used to examine TROY expression in glioblastoma tumor samples has been described previously (23). A scoring system of 0, negative; 1, weak; 2, moderate; 3, strong was used to grade the staining.
Apoptosis assays
Glioma cells (2×105) were seeded into 60 mm culture dishes in culture medium containing 10% FBS. The following day the cells were treated with DMSO or TMZ for 48 h. After treatment, all cells (floating and attached) were collected. The apoptotic cells were detected by fluorescence-activated cell sorting with an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences) according to the manufacturer’s protocol, in which the early- and late-death cells were stained with Annexin V-FITC and PI (propidium iodide). An aliquot of the collected cells was lysed and immunobloted for cleaved PARP using an antibody that recognizes cleaved PARP.
Expression constructs
The 3X HA epitope-tagged WT TROY construct was constructed as previously described (9). The TROY variant designated TROY ΔMPD, containing an in frame deletion of the juxtamembrane residues K194-Q209, was generated by splice overlap extension PCR. For stable transduction of glioma cell lines, epitope tagged TROY fragments were subcloned into the lentiviral transfer vector pCDH GFP (System Biosciences, Mountain View, CA). The empty pCDH lentiviral plasmid expressing only GFP was used as a control. Recombinant lentiviruses encoding epitope tagged TROY fragments were produced by transient transfection of 293T packaging cells with the appropriate TROY expression construct and the pPACKH1 plasmid packaging mix (System Biosciences). Lentivirus constructs encoding a validated shRNA targeting human TROY (Clone ID: V2LHS 30848) or a non-targeting control shRNA were purchased from Thermo Open Biosystems. Recombinant lentiviruses encoding shRNAs were produced by transient transfection of 293T packaging cells with the shRNA expression construct and the Trans-Lentiviral packaging kit (Open Biosystems). For lentiviral transduction, media from the packaging cells was collected 48 hours after transfection, recombinant lentiviruses concentrated by PEG precipitation, and added to target cells with 8 μg/ml polybrene for 6 hours at 37°C. Positively transduced cells were enriched by mass sorting the GFP positive cells on a FACSAria cell sorter (BD Biosciences). Inducible TROY cytoplasmic domain chimeric proteins were generated with the ARGENT Homodimerization kit (24) (Ariad Pharmaceuticals, Cambridge, MA). The wild type TROY cytoplasmic domain or the TROY ΔMPD cytoplasmic domain were generated by PCR and ligated downstream of tandem modified FK506 binding domains (FKBP36V) in the pC4M-Fv2E vector containing a NH2-terminal myristoylation signal. Coding sequences for the chimeric proteins were subcloned into the pCDH lentiviral transfer vector. The small molecule homodimerizer AP20187 was obtained from Ariad and added to serum starved cultures at 50 nM.
RCAS/tv-a system
The coding sequence for TROY was subcloned into the ALV-A expression vector RCAS-Y. RCAS-AP and RCAS-HGF, encoding alkaline phosphatase and hepatocyte growth factor respectively, were described previously (25). Transfected DF-1 chicken fibroblasts were passaged every other day in DMEM containing 10% FBS and 100 units/ml penicillin, and 10 mg/ml streptomycin. Transgenic mice (G-tva) that express the TVA receptor from the glial fibrillary acidic protein (GFAP) promoter have been previously described (26). Viral producing DF-1 cells infected with RCAS-HGF or RCAS-TROY were combined in equal proportion with viral producing DF-1 cells infected with RCAS-AP and co-injected into newborn G-tva mice at the intersection of the coronal and sagittal sutures. Each mouse received a single 5 μl intracranial injection. Ten weeks post-injection, mice were sacrificed, brains resected, and fixed in 10% neutral buffered formalin. For AP staining, the brains were dehydrated in 20% sucrose, 2% glycerol in PBS and cut into 60 μm sections. Sections were stained using the 1-Step NBT/BCIP kit (Thermo Scientific) after treatment at 65°C in PBS pH 9.5 for 1 hour to remove endogenous AP activity.
Intracranial xenograft tumor model
The orthotopic intracranial xenograft model was performed under a protocol approved by the Mayo Institutional Animal Care and Use Committee. The procedure was performed as described in detail (27). Briefly, female athymic nude mice were randomized into groups of 10 that received either GBM10 cells transduced with a shRNA targeting TROY or GBM10 cells transduced with a non-targeting control shRNA. Extent of knockdown of TROY in GBM10 cells was assayed by immunoblot of transduced cells 48 hours after injection. Cells (5×105) were injected into the right basal ganglia of anesthetized mice with a stereotaxic frame. Animals were weighed daily and monitored for the onset of neurological symptoms and euthanized when they reached a moribund condition.
Radial migration assay
Cell migration was assayed with a monolayer migration assay as previously described (13, 28). Cell migration over 24 hours was determined for at least 10 replicate samples on glass slides precoated with laminin (10 μg/ml). Migrations rates were normalized to nonspecific migration on bovine serum albumin.
Immunoblotting
Immunoblotting of cell lysates was performed as described (29). Briefly, cells were lysed on ice in the presence of protease and phosphatase inhibitors, lysates clarified by centrifugation, and protein content determined by BCA assay (Sigma). For immunoblotting, equal amounts of cell lysate (10–20 μg) were resolved by SDS-PAGE, transferred to nitrocellulose, and incubated with the appropriate primary antibody. For clinical GBM biopsy samples, 30 μg of total sample lysate was used for immunoblotting. Detection was performed with HRP-conjugated secondary antibodies and enhanced chemiluminescence (Perkin Elmer Life Sciences, Boston, MA) or by infrared detection using IRDye conjugated secondary antibodies with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Quantitation of band intensity was performed with the Odyssey application software v3.0. Results shown for immunoblots are representative of results from experiments repeated a minimum of three times.
Statistics
Statistical analysis between groups was performed using two sample t-test. A P-value < 0.05 was considered significant. Data are presented as mean (±SE). Survival of mice with intracranial xenografts was determined by Kaplan-Meier analysis using GraphPad Prism 5.0 (GraphPad, Inc).
Results
TROY expression is upregulated in GBM
Previously, we reported that TROY mRNA was elevated in advanced glial tumors with the highest expression observed in GBM (Grade IV) samples (9). In the current study, utilizing laser capture microdissection to select glioma cells from the tumor core or glioma cells from the invasive edge of patient GBM tumor sections, we observed that TROY mRNA expression was increased in the invading glioma cells relative to cells in the matched tumor core in vivo in a majority of samples (Table 1). To corroborate these results, we examined the expression of TROY in GBM tumor samples (Grade IV) and the expression of TROY in GBM tumors in vivo (fig. 1). Immunoblotting of GBM tumor samples indicated that TROY expression was elevated in a majority of the patient tumor samples (12/16) relative to TROY expression in non-neoplastic brain tissue samples. TROY expression in GBM tumors in situ was examined by IHC analysis on a GBM tissue microarray. TROY expression was markedly elevated in tumor biopsy samples relative to non-neoplastic brain further substantiating increased expression of TROY in GBM. Prominent cytoplasmic and membrane staining of TROY (with scores of 2–3 out of 3) was detected in glioblastoma cells, but not in non-neoplastic glial cells, endothelial cells, or neurons.
Table 1.
TROY mRNA expression in invasive cells in vivo
| GBM Specimens | R/C Ratio |
|---|---|
| TB195 | 16.9 |
| VPJT07 | 11.6 |
| BTTB1020 | 10.3 |
| BNI364 | 5.1 |
| VPJT03 | 2.5 |
| BNI371 | 2 |
| BNI758 | 2 |
| BTTB886 | 1.8 |
| TB187 | 1.6 |
| JAP49 | 1.3 |
| BNI9065 | 1.1 |
| BNI660 | 0.9 |
| TB186 | 0.9 |
| VPJT01 | 0.9 |
qRT-PCR analysis of TROY mRNA levels from laser capture microdissected glioma tumor core and rim cells (invasive edge) from cryopreserved biopsy specimens (n=14). Levels of TROY mRNA were normalized to histone H3.3 and expressed as proportions of the mRNA levels of TROY in GBM cells residing in the tumor rim (R) compared in core (C). 9/14 GBM specimens (indicated by bold text) showed a 1.6–16.9-fold increase of TROY mRNA expression in the invading GBM cells at the rim relative to cells in the tumor core (p<0.01).
Figure 1.
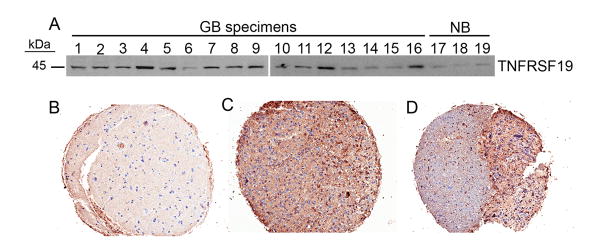
TROY expression is increased in GBM. A, Immunoblot analysis of TROY in GBM tumor samples (1–16) and in non-neoplastic brain tissue samples (NB, 17–19). IHC analysis of TROY expression in non-neoplasic brain (B), GBM biopsy sample from the tumor core (C) and GBM biopsy sample from the tumor edge (D). Samples are representative of 29 matched biopsy samples on a tissue microarray.
TROY expression induces glial specific cell migration in situ
TROY is widely expressed in embryonic tissues, but its postnatal expression is tightly regulated (3–8) suggesting that alterations in TROY expression may be deleterious. To examine whether inappropriate TROY expression stimulated the migration of normal astrocytes in situ, we utilized the G-tva system for glial specific gene transfer in transgenic mice (30). In this system, the avian retroviral receptor, TVA, is expressed under the control of the astrocyte-specific GFAP promoter that allows glial specific expression of proteins encoded by ASLV-A gene vectors. An equal mixture of DF-1 fibroblasts producing RCAS-AP and DF-1 cells producing RCAS-TROY were implanted into the brains of newborn G-tva transgenic mice. As a positive control, mice were implanted with an equal mixture of DF-1 fibroblasts producing RCAS-AP and DF-1 cells producing RCAS-HGF. Mice implanted only with DF-1 fibroblasts producing RCAS-AP served as the negative control. Analysis of brain sections of mice 10 weeks after implantation showed that astrocytes infected with AP alone did not show any distal migration from the injection site (fig. 2). Co-injection of DF-1 fibroblasts producing TROY and the AP marker gene induced astrocytic migration distal to the injection site of the infected astrocytic cells similar to the distal migration of astrocytes following infection with the AP marker and HGF that is a known inducer of cell motility (25) (fig. 2). These data indicate that abnormal expression of TROY stimulates cell migration in situ.
Figure 2.

Aberrant TROY expression increases migration in situ. DF-1 fibroblasts producing RCAS-AP (A,D), an equal mixture of DF-1 cells producing RCAS-AP and DF-1 cells producing RCAS-TROY (B,E) or an equal mixture of DF-1 cells producing RCAS-AP and DF-1 cells producing RCAS-HGF (C,F ) were injected into the frontal lobe of newborn G-tva transgenic mice. 10 weeks after injection, mice were sacrificed and brain sections were analyzed for AP activity. Distal migration from the injection site (asterisk) is indicated by positive AP staining (black arrows).
TROY expression increases therapeutic resistance
Previously, we demonstrated that increased expression of TROY stimulated glioma cell migration and invasion while knockdown of TROY expression inhibited glioma cell migration and invasion (9). Notably, increased expression of TROY did not increase the proliferation of cultured glioma cells relative to control cells (fig. 3A). Current standard of care treatment for GBM patients includes surgical resection followed by radiation with concurrent and adjuvant temozolomide (TMZ). To determine the effect of TROY expression on therapeutic resistance, we examined the effect TROY expression on cell viability following treatment with ionizing radiation or TMZ. At radiation doses of 2 Gy - 6 Gy, increased expression of TROY increased the resistance of glioma cells to treatment with ionizing radiation (fig. 3B). Similarly, increased expression of TROY significantly increased glioma cell resistance to TMZ induced apoptosis (fig. 3C). Conversely, knockdown of TROY expression increased glioma cell sensitivity to TMZ (fig. 3D). TMZ treatment of glioma cells transfected with a non-targeting siRNA significantly decreased cell viability relative to glioma cells treated with vehicle alone. Glioma cells transfected with a siRNA targeting TROY showed increased sensitivity to TMZ treatment with a significant decrease in cell viability relative to the glioma cells transfected with a siRNA targeting TROY treated with vehicle. Moreover, TMZ treatment significantly reduced viability of T98G cells transfected with siRNA targeting TROY relative to T98G cells transfected with a control non-targeting siRNA (p< 0.01). Together, these data indicate that TROY stimulated cell migration and invasion is associated with signaling that increases cell resistance to therapeutic treatment.
Figure 3.

TROY expression increases therapeutic resistance. A, The proliferation of T98G cells expressing TROY relative to control transduced T98G cells expressing copGFP alone was determined at the indicated time points by Alamar Blue assay. B, T98G glioma cells (Ctrl) or T98G cells expressing TROY were irradiated with 2 Gy – 6 Gy and survival was determined after 12 days by clonogenic assay (**p < 0.05). C, T98G glioma cells or T98G cells expressing TROY were treated with vehicle (DMSO) or 250 μM TMZ for 48 hours. The percentage of cellular apoptosis was determined by annexin V staining followed by flow cytometry (**, p<0.001). D, U118 glioma cells transfected with non-targeting siRNA or siRNA targeting TROY were treated with DMSO or 250 μM TMZ for 48 hours. The percent cell viability was measured by Alamar Blue assay and normalized to the control siRNA untreated with TMZ (*, p <0.01, **, p<0.001). Data represents the mean and S.D. from three independent experiments with each experiment conducted in triplicate.
TROY expression increases survival signaling
We examined whether TROY mediated increased therapeutic resistance was linked to increased survival signaling. T98G or SNB19 glioma cells were transfected with epitope-tagged TROY or empty vector, serum starved for 16 hours, then lysed and immunoblotted with antibodies to Akt and IkBα (fig. 4A, 4B). In both cell lines, expression of TROY resulted in increased phosphorylation of both Akt and IkBα but, did not alter total expression levels. As phosphorylation of IkBα is an indicator of NF-κB activation, we examined the effect of TROY expression on activation of NF-κB. TROY expression increased phosphorylation of NF-κB (fig. 4C). Treatment of cells with the Akt inhibitor LY294002 significantly reduced the TROY mediated NF-κB phosphorylation while treatment of cells with the NF-κB inhibitor BAY117082 did not alter TROY mediated Akt phosphorylation (fig. 4C) indicating that Akt lies upstream of NF-κB in the TROY signaling pathway. Notably, TROY mediated resistance to TMZ treatment is dependent upon Akt and NF-κB activity. As shown in figure 5, TMZ treatment of control cells but not TROY expressing cells significantly increased apoptosis as assayed by immunoblot analysis for cleaved PARP (fig. 5A). Resistance of TROY expressing cells to TMZ induced apoptosis was abrogated following inhibition of Akt or NF-κB (fig. 5B).
Figure 4.
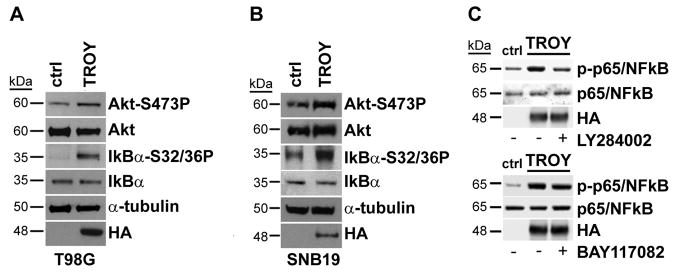
TROY expression increases survival signaling. A, T98G glioma cells and T98G cells expressing TROY or B, SNB19 glioma cells and SNB19 cells expressing TROY were serum starved for 24 hours, the cells lysed, and the lysates immunoblotted with the indicated antibodies. C, T98G cells or T98G cells expressing TROY were treated with 20 μM LY29402 (upper panel) or 20 μM BAY117082 (lower panel) for 30 min. Whole cell lysates were immunoblotted with the indicated antibodies.
Figure 5.

TROY induced resistance to TMZ is dependent upon Akt and NF-κB. A, T98G glioma cells expressing TROY or control T98G cells were serum deprived for 24 hr then treated with 250 μM TMZ for 48 hr. TMZ induced apoptosis was assayed by immunoblot analysis of cell lysates with an antibody to cleaved PARP. B, T98G glioma cells expressing TROY were left untreated, treated with 250 μM TMZ for 48 hr, or treated with 250 μM TMZ for 48 hr in combination with 20 μM LY294002 or 20 μM BAY-11-709. Apoptosis was assayed by immunoblot analysis of cleaved PARP.
To directly link TROY signaling to Akt and NF-κB activation, we employed a regulated homodimerization system (24, 31) to mimic the oligomerization observed in members of the TNFR superfamily. Glioma cells were transduced with a recombinant lentivirus encoding a chimeric protein containing the TROY cytoplasmic domain fused to a FK506 binding protein domain. Addition of a synthetic cell-permeant dimerizer (AP20187) induced homodimerization of the TROY cytoplasmic domain and phosphorylation of Akt and NF-κB (fig. 6A). The TROY membrane proximal domain (MPD) is a potential signaling module consisting of the 14 juxtamembrane amino acids that is enriched in basic residues. The MPD of the related TNFRSF6 (CD95) receptor has been reported to mediate the activation of Rac1 (32). Interestingly, regulated homodimerization of a TROY cytoplasmic domain containing a deletion of the membrane proximal domain (ΔMPD) did not induce phosphorylation of Akt or NF-κB. Similarly, expression of full length TROY with a deletion of the MPD did not induce phosphorylation of Akt of NF-κB in serum starved glioma cells (fig. 6B) indicating an important role for the juxtamembrane domain in TROY signaling. Indeed, unlike expression of wild type TROY, expression of TROY ΔMPD appeared to function as a dominant negative and significantly inhibited glioma cell migration (fig. 6C) and cell survival following irradiation (fig. 6D). Expression of WT TROY and TROY ΔMPD was equivalent in transduced cells by immunoblotting (fig. 6E).
Figure 6.

Requirement for the MPD in TROY signaling. A, SNB19 glioma cells were transduced with an inducible homodimerization chimera encoding the WT TROY cytoplasmic domain (left) or a chimera encoding the TROY cytoplasmic domain with a deletion of the MPD (right). Serum starved cells were treated with the synthetic dimerizer AP20817 (50 nM) for varying time points, lysed, and immunoblotted with the indicated antibodies. B, T98G glioma cells expressing full length TROY (WT), TROY containing a deletion of the MPD (ΔMPD), or GFP (ctrl), were serum starved, lysed, and immunoblotted with the indicated antibodies. C, Radial cell migration assay T98G glioma cells expressing GFP (ctrl), T98G cells expressing wild type TROY (WT), or T98G expressing TROY containing a deletion of the MPD (ΔMPD). Migration was assayed over 24 hours on 10 μg/ml laminin (*, p < 0.05). D, T98G cells expressing GFP (ctrl), wild type TROY (WT), or the TROY ΔMPD variant were subjected to irradiation (2 Gy) and the surviving fraction was measured by colony formation 12 days later (**, p < 0.05). E, Cell lysates of T98G cells expressing GFP (ctrl), wild type TROY (WT), or the TROY ΔMPD variant were immunoblotted with the indicated antibodies.
Knockdown of TROY expression increases survival of orthotopic xenograft mice
To determine the effect of TROY expression on glioma tumor progression in vivo, we examined the effect of silencing TROY expression on the survival of mice with intracranial xenografts established with the primary glioblastoma cell line GBM10. GBM10 is from a panel of serially passaged GBM xenografts established from patient tumors that maintain characteristic morphological and molecular properties of the original tumor (17, 18). GBM10 cells were stably transduced with a lentiviral construct encoding a shRNA targeting TROY or with a non-targeting control shRNA and intracranially implanted into nude mice. Immunoblots of GBM10 cells transduced with a shRNA targeting TROY demonstrated greater than 90% reduction in TROY expression relative to GBM10 cells transduced with a control non-targeting shRNA (fig. 7). Mice with intracranial xenografts established with GBM10 cells transduced with the shRNA targeting TROY exhibited significantly increased survival relative to mice with xenografts established with GBM10 cells transduced with the control non-silencing shRNA substantiating a role for TROY expression in GBM tumor progression.
Figure 7.

Knockdown of TROY expression increases xenograft survival. A, Kaplan-Meier survival curves of athymic nude mice with intracranial xenografts of GBM10 transduced with a control non-targeting shRNA or a shRNA targeting TROY. Curves show a significant survival benefit for mice with xenografts with knockdown of TROY expression (p = 0.004). B, lysates of transduced GBM10 cells used for intracranial xenografts were immunoblotted 48 hours after injection with the indicated antibodies.
Discussion
In previous studies, we demonstrated that the TNFRSF family member TROY is overexpressed in advanced glial tumors and promotes glioblastoma invasion through Pyk2-Rac1 signaling. Moreover, we demonstrated that TROY expression negatively correlates with overall patient survival. Therefore, TROY may represent a heretofore-unappreciated therapeutic target to improve clinical outcomes in glioblastoma. In the current study, we further investigated the potential role of TROY in glioblastoma survival and resistance. The major findings of this report are as follows: (1) TROY expression was significantly increased in patient GBM tumor samples with TROY mRNA exhibiting increased expression in the invasive cell population, (2) aberrant expression of TROY in mouse astrocytes in vivo using glial-specific gene transfer in transgenic mice induced astrocyte migration within the brain, (3) increased TROY expression did not increase cell proliferation but increased resistance of glioma cells to both IR- and TMZ- induced apoptosis while knockdown of TROY increased TMZ sensitivity, (4) TROY increased survival signaling through activation of Akt and NF-κB, (5) TROY induced resistance to TMZ was dependent upon Akt and NF-κB, (6) TROY induced NF-κB phosphorylation and stimulation of migration required the membrane proximal region of the cytoplasmic domain, (7) knockdown of TROY increased xenograft survival. The current results further support a role for TROY in GBM and suggest that targeting TROY and its signaling pathway represents a potential approach to increase tumor vulnerability and improve the therapeutic response of glioblastoma.
TROY is an orphan member of the TNFR superfamily. Two recent genome-wide association studies have identified TROY as a susceptibility factor in nasopharyngeal carcinoma and lung cancer (33, 34). TROY is widely expressed in embryonic tissues, including progenitor cells in the ventricular and sub-ventricular zones of the brain, but its postnatal expression is highly restricted. The tightly regulated expression of TROY suggests that alterations in TROY expression may have undesirable effects on cell proliferation and migration. This suggestion is supported by the demonstration that TROY is highly expressed in primary and metastatic melanoma cells but not in normal melanocytes (35). A recent study demonstrated that TROY expression is significantly upregulated in tumor infiltrating microglia in GBM and is involved in the regulation of their migration (36). We have previously reported that the levels of expression of TROY in glial tumors correlate with tumor grade and that increased TROY expression was an indicator of poor outcome (9). Here we showed that TROY protein expression was significantly increased in patient GBM tumor samples and GBM tumors in situ on a tumor microarray. Notably, laser capture microdissection demonstrated that TROY mRNA was significantly increased in GBM cells at the invasive tumor edge. Moreover, using astrocyte-specific gene transfer in situ we showed that altering TROY expression resulted in migration of astrocytes. This data indicates that abnormal expression of TROY can be an inducer of glioma cell migration in situ. Therefore, the aberrant re-expression of TROY observed in GBM supports an important role for TROY in GBM progression and possibly serves as a good predictor of survival outcome to be utilized in complimenting other prognostic indicators.
Invasion not only makes surgical resection impossible it also carries with it the potential for increased resistance to chemotherapeutic agents. Studies have demonstrated that there is an inverse relationship between invasion and sensitivity to apoptosis induced by chemotherapeutic agents (15). In addition, expression profiling of invasive glioma cells has demonstrated a significant decrease in pro-apoptotic genes (14, 16). Our previous demonstration that increased TROY expression stimulated glioma cell migration and invasion (9) suggested that increased TROY expression could be accompanied by increased resistance to therapeutic treatment. Consistent with this hypothesis, our current studies demonstrate that increased TROY expression significantly increased the resistance of glioma cells to both ionizing radiation and TMZ treatment. Conversely, knockdown of TROY expression both inhibited cell migration and increased the sensitivity of glioma cells to TMZ linking TROY expression to therapeutic resistance. The observed increased resistance was independent of an effect on proliferation. Although TROY expression did not alter cell proliferation, increased TROY expression increased survival signaling through activation of Akt and NF-κB. The activation of Akt and NF-κB has been strongly implicated in the resistance to apoptosis (37-39). The activation of Akt and NF-κB in serum starved glioma cells could be induced by dimerization of the TROY cytoplasmic domain alone directly linking their phosphorylation to TROY signaling. Moreover, the TROY mediated resistance to TMZ induced apoptosis was blocked by inhibition of Akt or NF-κB substantiating their role in TROY survival signaling. Interestingly, knockdown of TROY expression significantly increased the survival of mice with intracranial xenografts suggesting that reducing TROY signaling in the GBM cells slowed tumor progression even in the absence of additional therapeutic challenge. Since we observed that silencing TROY expression significantly increased glioma cell sensitivity to TMZ in vitro, we anticipate that knockdown of TROY expression and survival signaling will likely increase the benefit of TMZ therapy. This is an area that is currently under investigation using intracranial xenografts.
The ligands that mediate TROY signaling and the important functional domains of TROY remain to be fully defined. It has been reported that lymphotoxin-α is a ligand for TROY and that lymphotoxin-α binding to TROY activated NF-κB reporter activity (40) although the biological significance of this interaction is unknown. The cytoplasmic domain of TROY contains a conserved protein module designated the membrane proximal domain. The MPD was first characterized in Fas (CD95, TNFRSF6). The MPD of Fas consists of the 14 amino acid residues immediately adjacent to the transmembrane domain, is enriched in basic amino acids, is conserved among TNFRSF members, and is required for Fas induced activation of Rac1 (32). Our data indicated that the MPD of TROY was required for the capacity of TROY to activate Akt and NF-κB. Increased expression of TROY but not TROY ΔMPD stimulated Akt and NF-κB phosphorylation. Similarly, induced dimerization of the TROY cytoplasmic domain but, not dimerization of the TROY ΔMPD cytoplasmic domain stimulated Akt and NF-κB phosphorylation. Furthermore, expression of TROY ΔMPD also abrogated the capacity of TROY to stimulate glioma cell migration supporting a central role for the MPD in TROY signal transduction.
In summary, the current data substantiate an important role for TROY in glioblastoma pathobiology. Previously, we have shown that TROY expression stimulates glioma cell migration and invasion in vitro. The results of the current study show that TROY expression is significantly increased in GBM, increased TROY expression stimulates survival signaling, and increased TROY expression increases therapeutic resistance. In addition, aberrant expression of TROY stimulated cell migration in situ positioning TROY as a contributor, and possibly a driver, of the malignant dispersion of glioma cells. Together, these observations support TROY or its signaling effectors as novel targets to inhibit glioma cell survival, limit cell invasion, and decrease therapeutic resistance.
Acknowledgments
Grant Support
This works was supported in part by NIH grants R01 CA130940 (NLT), R01 CA103956 (J.C.L.), and P50 CA108961 (NLT and JCL).
Footnotes
The authors declare they have no conflicts of interest
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu S, Tamada K, Ni J, Vincenz C, Chen L. Characterization of TNFRSF19, a novel member of the tumor necrosis factor receptor superfamily. Genomics. 1999;62:103–7. doi: 10.1006/geno.1999.5979. [DOI] [PubMed] [Google Scholar]
- 4.Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, et al. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–51. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 5.Pispa J, Mikkola ML, Mustonen T, Thesleff I. Ectodysplasin, Edar and TNFRSF19 are expressed in complementary and overlapping patterns during mouse embryogenesis. Gene Expr Patterns. 2003;3:675–9. doi: 10.1016/s1567-133x(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 6.Hisaoka T, Morikawa Y, Kitamura T, Senba E. Expression of a member of tumor necrosis factor receptor superfamily, TROY, in the developing olfactory system. Glia. 2004;45:313–24. doi: 10.1002/glia.10323. [DOI] [PubMed] [Google Scholar]
- 7.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–9. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 8.Hisaoka T, Morikawa Y, Kitamura T, Senba E. Expression of a member of tumor necrosis factor receptor superfamily, TROY, in the developing mouse brain. Brain Res Dev Brain Res. 2003;143:105–9. doi: 10.1016/s0165-3806(03)00101-9. [DOI] [PubMed] [Google Scholar]
- 9.Paulino VM, Yang Z, Kloss J, Ennis MJ, Armstrong BA, Loftus JC, et al. TROY (TNFRSF19) is overexpressed in advanced glial tumors and promotes glioblastoma cell invasion via Pyk2-Rac1 signaling. Mol Cancer Res. 2010;8:1558–67. doi: 10.1158/1541-7786.MCR-10-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baranwal S, Alahari SK. Rho GTPase effector functions in tumor cell invasion and metastasis. Curr Drug Targets. 2011;12:1194–201. doi: 10.2174/138945011795906534. [DOI] [PubMed] [Google Scholar]
- 11.Khalil BD, El-Sibai M. Rho GTPases in primary brain tumor malignancy and invasion. J Neurooncol. 2012;108:333–9. doi: 10.1007/s11060-012-0866-8. [DOI] [PubMed] [Google Scholar]
- 12.Rathinam R, Berrier A, Alahari SK. Role of Rho GTPases and their regulators in cancer progression. Front Biosci. 2011;16:2561–71. doi: 10.2741/3872. [DOI] [PubMed] [Google Scholar]
- 13.Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and Nuclear Factor-κB and correlate with poor patient outcome. Cancer Res. 2006;66:9535–42. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- 14.Demuth T, Rennert JL, Hoelzinger DB, Reavie LB, Nakada M, Beaudry C, et al. Glioma cells on the run - the migratory transcriptome of 10 human glioma cell lines. BMC Genomics. 2008;9:54. doi: 10.1186/1471-2164-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joy AM, Beaudry CE, Tran NL, Ponce FA, Holz DR, Demuth T, et al. Migrating glioma cells activate the PI3-K pathway and display decreased susceptibility to apoptosis. J Cell Sci. 2003;116:4409–17. doi: 10.1242/jcs.00712. [DOI] [PubMed] [Google Scholar]
- 16.Mariani L, Beaudry C, McDonough WS, Hoelzinger DB, Demuth T, Ross KR, et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: a cDNA microarray analysis. J Neurooncol. 2001;53:161–76. doi: 10.1023/a:1012253317934. [DOI] [PubMed] [Google Scholar]
- 17.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12:2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 18.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 19.Kislin KL, McDonough WS, Eschbacher JM, Armstrong BA, Berens ME. NHERF-1: modulator of glioblastoma cell migration and invasion. Neoplasia. 2009;11:377–87. doi: 10.1593/neo.81572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, et al. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol. 2003;162:1313–21. doi: 10.1016/S0002-9440(10)63927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 22.Nakada M, Niska JA, Tran NL, McDonough WS, Berens ME. EphB2/R-Ras signaling regulates glioma cell adhesion, growth, and invasion. Am J Pathol. 2005;167:565–76. doi: 10.1016/S0002-9440(10)62998-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fortin SP, Ennis MJ, Savitch BA, Carpentieri D, McDonough WS, Winkles JA, et al. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol Cancer Res. 2009;7:1871–81. doi: 10.1158/1541-7786.MCR-09-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amara JF, Clackson T, Rivera VM, Guo T, Keenan T, Natesan S, et al. A versatile synthetic dimerizer for the regulation of protein-protein interactions. Proc Natl Acad Sci U S A. 1997;94:10618–23. doi: 10.1073/pnas.94.20.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fortin SP, Ennis MJ, Schumacher CA, Zylstra-Diegel CR, Williams BO, Ross JT, et al. Cdc42 and the guanine nucleotide exchange factors Ect2 and Trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol Cancer Res. 2012;10:958–68. doi: 10.1158/1541-7786.MCR-11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci U S A. 1998;95:1218–23. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loftus JC, Yang Z, Tran NL, Kloss J, Viso C, Berens ME, et al. The Pyk2 FERM domain as a target to inhibit glioma migration. Mol Cancer Ther. 2009;8:1505–14. doi: 10.1158/1535-7163.MCT-08-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipinski CA, Tran NL, Bay C, Kloss J, McDonough WS, Beaudry C, et al. Differential role of proline-rich tyrosine kinase 2 and focal adhesion kinase in determining glioblastoma migration and proliferation. Mol Cancer Res. 2003;1:323–32. [PubMed] [Google Scholar]
- 29.Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NF-κB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280:3483–92. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 30.Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, et al. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene. 1999;18:5253–60. doi: 10.1038/sj.onc.1203087. [DOI] [PubMed] [Google Scholar]
- 31.Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–71. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Ruan W, Lee CT, Desbarats J. A novel juxtamembrane domain in Tumor Necrosis Factor Receptor superfamily molecules activates Rac1 and controls neurite growth. Mol Biol Cell. 2008;19:3192–202. doi: 10.1091/mbc.E08-02-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bei J-X, Li Y, Jia W-H, Feng B-J, Zhou G, Chen L-Z, et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat Genet. 2010;42:599–603. doi: 10.1038/ng.601. [DOI] [PubMed] [Google Scholar]
- 34.Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z, et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat Genet. 2011;43:792–6. doi: 10.1038/ng.875. [DOI] [PubMed] [Google Scholar]
- 35.Spanjaard RA, Whren KM, Graves C, Bhawan J. Tumor necrosis factor receptor superfamily member TROY is a novel melanoma biomarker and potential therapeutic target. Int J Cancer. 2007;120:1304–10. doi: 10.1002/ijc.22367. [DOI] [PubMed] [Google Scholar]
- 36.Jacobs VL, Liu Y, De Leo JA. Propentofylline targets TROY, a novel microglial signaling pathway. PLoS One. 2012;7:e37955. doi: 10.1371/journal.pone.0037955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belda-Iniesta C, de Castro Carpeno J, Casado Saenz E, Cejas Guerrero P, Perona R, Gonzalez Baron M. Molecular biology of malignant gliomas. Clin Transl Oncol. 2006;8:635–41. doi: 10.1007/s12094-006-0033-9. [DOI] [PubMed] [Google Scholar]
- 38.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 39.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashimoto T, Schlessinger D, Cui CY. Troy binding to lymphotoxin-α activates NFκB mediated transcription. Cell Cycle. 2008;7:106–11. doi: 10.4161/cc.7.1.5135. [DOI] [PubMed] [Google Scholar]