

Abstract
Heterogeneity among life traits in mammals has resulted in considerable phylogenetic conflict, particularly concerning the position of the placental root. Layered upon this are gene- and lineage-specific variation in amino acid substitution rates and compositional biases. Life trait variations that may impact upon mutational rates are longevity, metabolic rate, body size, and germ line generation time. Over the past 12 years, three main conflicting hypotheses have emerged for the placement of the placental root. These hypotheses place the Atlantogenata (common ancestor of Xenarthra plus Afrotheria), the Afrotheria, or the Xenarthra as the sister group to all other placental mammals. Model adequacy is critical for accurate tree reconstruction and by failing to account for these compositional and character exchange heterogeneities across the tree and data set, previous studies have not provided a strongly supported hypothesis for the placental root. For the first time, models that accommodate both tree and data set heterogeneity have been applied to mammal data. Here, we show the impact of accurate model assignment and the importance of data sets in accommodating model parameters while maintaining the power to reject competing hypotheses. Through these sophisticated methods, we demonstrate the importance of model adequacy, data set power and provide strong support for the Atlantogenata over other competing hypotheses for the position of the placental root.
Keywords: mammal phylogeny, phylogenetic reconstruction, evolutionary models, placental root, heterogeneous modeling
Introduction
There are conflicting views over the root of the placental mammal phylogeny (Meredith et al. 2011; McCormack et al. 2012; Song et al. 2012; O’Leary et al. 2013). The conflict is largely due to the impact of compressed cladogenesis, a phenomenon shown to have a major negative impact on the resolution of the metazoan phylogeny for example (Rokas et al. 2005). In the case of the diversification of placental mammals, the compressed cladogenesis is so severe that the phylogenetic signature has proven very difficult to decipher. Following a large number of studies using different data sets, data types, and approaches, we now have a small number of areas of conflict remaining in the mammal tree, they concern 1) the position of the root of the placental Superorders (Murphy et al. 2001a; Meredith et al. 2011; McCormack et al. 2012; Song et al. 2012; O’Leary et al. 2013), 2) the branching of extant placental orders (Stanhope et al. 1998; Waddell et al. 1999), and 3) the precise placement of many individual placental species (Janecka et al. 2007). The three major competing hypotheses for the position of the root of the placental phylogeny are illustrated in figure 1. In summary, these alternative hypotheses place the following as the sister group of all remaining placentals: 1) the Afrotheria, for example, elephants and manatees (Murphy et al. 2001b; McCormack et al. 2012), 2) the Xenarthra, for example, armadillos and sloths (O’Leary et al. 2013), and 3) the common ancestor of Afrotheria and Xenarthra (Atlantogenata hypothesis) (Meredith et al. 2011; Song et al. 2012). Some of the most recent contributions to the field of mammal phylogeny have used ultraconserved elements (McCormack et al. 2012) and morphological data (specifically more than 4,500 morphological measurements across 40 extinct and 86 extant placental mammals) (O’Leary et al. 2013), resolving the Afrotheria and Xenarthra, respectively. Interestingly, the work by O’Leary et al. (2013) concluded, among other things, that the Afrotheria did not originate in Africa but in the Americas. Consilience across these different data types would be most desirable, but to achieve this, it would be necessary to have a full understanding of the evolutionary dynamics of all of these data to adequately model them.
Fig. 1.

Three major rooting hypotheses for placental mammals. The three major competing hypotheses for the position of the root are depicted. (A) The Afrotheria root, (B) the Xenarthra root, and (C) the Atlantogenata root.
Retroposons are attractive alternative data type that is less likely to be subject to model misspecification. However, a previous analysis of these rare genomic events by Nishihara et al. (2009) showed similar levels of support for all three competing hypotheses for the position of the placental root (Nishihara et al. 2009). There is some evidence that retroposons are not entirely devoid of homoplasy (Han et al. 2011), which may contribute to signal conflict. Here, we show how adequate modeling can be achieved for protein-coding molecular data.
The current debate over the position of the placental root, and indeed the branching patterns within certain Superorders (e.g., Laurasiatheria), is due in no small part to incorrect ortholog identification (Philippe et al. 2011), long branch attraction (LBA) (Heath et al. 2008), saturation of signal (Moreira and Philippe 2000), compositional bias (Foster and Hickey 1999), evolutionary rate variation between lineages and between different characters in a data set (Li et al. 1987), and model misspecification (Philippe et al. 2011). Each of these factors (alone or in combination) can have a major effect on the resulting topology as outlined later.
Comparisons of protein-coding genes in mammalian genomes have shown that there are substantial variations in both mutational rate and nucleotide composition (Li et al. 1987; Romiguier et al. 2010). The observed heterogeneity can be influenced by many factors including diet (Yang 1998), disease (Usanga and Luzzatto 1985), and intense sexual selection (Dorus et al. 2004). Variations in body size, longevity, metabolic rate, and germ-line generation time across species have also been shown to affect the rate of mutation (Peto et al. 1975; Martin and Palumbi 1993; Bleiweiss 1998; Leroi et al. 2003; de Magalhaes and Costa 2009; Caulin and Maley 2011). Smaller, shorter-lived mammals with high metabolic rates and short germ-line generation times accumulate mutations at a higher rate (Li et al. 1996). With such variation in mutational rate across lineages, these data are also likely to suffer the effects of LBA. Holton and Pisani (2010) have shown that LBA has led to the incorrect resolution of Coelomata as a clade; the influence of this systematic bias is also a concern for mammal phylogeny reconstruction. GC-biased gene conversion in the DNA repair machinery (Galtier 2003) results in a higher GC content over time for those species with higher mutational rates, and this has resulted in mammal genome data manifesting compositional biases across species (Romiguier et al. 2010). By disregarding compositional heterogeneity in a data set (i.e., by not modeling it adequately), we can consistently identify the wrong tree as optimal (Foster 2004). Foster gave examples where homogeneous models gave an incorrect topology, but using a tree-heterogeneous composition model obtained the correct topology and was also shown to fit the data adequately (Foster 2004; Foster et al. 2009). The importance of modeling both compositional heterogeneity across the tree and heterogeneity in exchange rates over the data set has been shown by Cox et al. (2008) where strong support for the incorrect topology was obtained if one employed models that did not account for these types of heterogeneity in the data.
In this article, we employ two major classes of heterogeneous modeling: first those that accommodate compositional heterogeneity and exchange rate heterogeneity across the phylogeny. As previously described by Foster et al. (2009), these are node-discrete composition heterogeneous (NDCH) and node-discrete rate matrix heterogeneous (NDRH) models. The second class of heterogeneous models applied are those that allow for heterogeneity across the data set (Lartillot and Philippe 2004). All previous attempts to reconstruct the mammal phylogeny using molecular data have used homogeneous models that do not account for variation in composition or exchange rates across the tree or the data set (Murphy et al. 2001b; Bininda-Emonds et al. 2007; Meredith et al. 2011). In contrast, here we will use models that adequately describe the complex reality of the evolutionary processes. However, more sophisticated models demand more data to discriminate among competing hypotheses. For example, alignment lengths of greater than 1,000 positions (i.e., concatenated data sets) have been shown to be necessary for profile mixture models to reliably calculate the shapes of the profiles of the data (Quang et al. 2008). Therefore, if heterogeneous models are to be employed then the size and the quality of the data set are critical.
To date, there have been many studies that used concatenated data sets to investigate the position of the placental root (Murphy et al. 2001b, 2007; Kriegs et al. 2006; Asher 2007; Hallstrom and Janke 2010). These data sets have included nuclear and mitochondrial genes (with protein-coding genes treated either as nucleotide or amino acid sequences), noncoding DNA, morphological data, and rare genomic events (Murphy et al. 2001b, 2007; Kriegs et al. 2006; Hallstrom and Janke 2010). It is now clear, however, that not all data sets are equally suitable for all phylogenetic questions at all phylogenetic depths, as shown most recently by Song et al. (2012). The work of Brown et al. (1982) showed that mitochondrial genes and noncoding DNA sequences accumulate mutations at a faster rate than nuclear protein-coding genes, making them less appropriate for resolving deep nodes on a phylogeny. In addition, DNA sequences are more prone to mutational saturation than amino acid data sets (Kosiol et al. 2007) and might be more strongly affected by biases related to lineage-specific codon usage preferences (Rota-Stabelli et al. 2013).
To fully address the root of the placental mammals, evolutionary models that account for heterogeneity in both composition and the rate of mutational change (i.e., exchange rate) across the tree and the data must be employed (Foster et al. 2009). Along with modeling these features, it is essential for the data set to have the power to reject alternative hypotheses and to be sufficiently large to meet the requirements of the increase in parameterization. Here, we have exploited recent advances in modeling heterogeneity of composition and exchange rates across the tree and the data in a Bayesian framework, and using a data set generated from completed genomes, we address the problem of the root of the placental mammals.
Results and Discussion
First, we set out to determine whether there was a significant improvement in fit with the use of heterogeneous models over homogeneous models for mammal data sets. To perform this test, we used two sources of data: 1) a previously published data set containing 66 taxa and a maximum alignment length of 9,789 base pairs referred to as “66TaxonSet” (see Materials and Methods) (4) and 2) a data set assembled specifically for this analysis containing 39 taxa and an alignment length of 23,900 amino acids (aa) referred to throughout as “39TaxonSet.” We tested the suitability of these data sets for resolving the root of the mammal phylogeny. The treatment of the data sets and their naming system are described in table 1.
Table 1.
Summary of Data Sets Analyzed.
| Data Sets | Data Set Type | Alignment Length |
|---|---|---|
| 66TaxonSet_nuc | Nucleotide | 9,789 bp |
| 66TaxonSet_aa | Amino acid | 2,190 aa |
| 66TaxonSet_day | Dayhoff recoded 6 | 2,190 characters |
| 39TaxonSet_aa | Amino acid | 23,900 aa |
| 39TaxonSet_day | Dayhoff | 23,900 |
Note.—The name of each data set as per main text, the data type and corresponding alignment lengths are shown.
Heterogeneous Models Fit “66TaxonSet” Better than Homogeneous Models
In the first instance, we wished to determine whether the homogeneous models were adequate for 66TaxonSet. To determine which model describes 66TaxonSet, we analyzed the data at three levels in total, the first two were the nucleotide level (66TaxonSet_nuc) and the amino acid level (66TaxonSet_aa). Recoding into Dayhoff categories tends to ameliorate compositional heterogeneity and affords us the opportunity to apply GTR-like rate matrices to these resized data sets using a relatively small number of free parameters (Hrdy et al. 2004). Therefore, NDRH models were applied to the Dayhoff recoded data set (66TaxonSet_day). These data were recoded into a total of 6 Dayhoff categories as follows: Category 1 [C]; Category 2 [S, T, P, A, and G]; Category 3 [N, D, E, and Q]; Category 4 [H, R, and K]; Category 5 [M, I, L, and V], and Category 6 [F, Y, and W] (Hrdy et al. 2004). The 66TaxonSet_nuc and 66TaxonSet_aa showed compositional heterogeneity where 13 out of 66 had P values of approximately 0.002, and 47 out of 66 taxa had P values of 0.000, respectively. Therefore, neither 66TaxonSet_nuc nor _aa fit the homogenous model (P values were not corrected for multiple comparisons), whereas the 66TaxonSet_day was compositionally homogeneous (supplementary table S1, Supplementary Material online). These results support the thesis that the nucleotide and amino acid data sets from 66TaxonSet require models that accommodate compositional heterogeneity over the tree and the data.
We use two approaches to model heterogeneity: one is to model heterogeneity across the phylogeny using the NDCH/NDRH models (Foster 2004), and the second is to model heterogeneity across the data using the CAT model (Lartillot and Philippe 2004). The NDRH model allows for character state exchange rates to vary over the topology, and the NDCH model allows for the composition to vary over the topology, independently of each other. The CAT model, developed by Lartillot and Philippe (2004), models heterogeneity over the data using a mixture of site profiles. Here, we will define homogeneous models as those that do not allow for compositional variation or exchange rate variation across the tree or data set, that is, they do not incorporate either NDRH/NDCH or CAT models described earlier. A homogeneous model with a single exchange rate matrix, a single composition vector, a proportion of invariable sites, and four categories of gamma distributed associated rate variation is denoted as 1GTR + 1C + I + 4Γ, this shorthand notation is used throughout the article.
Our strategy was to investigate whether models that account for heterogeneity over the phylogeny and data are a better fit to mammal data than previously employed homogeneous models. The best-fitting homogeneous model was identified using ModelGenerator v85 (Keane et al. 2006), and the models accommodating heterogeneity across the phylogeny were evaluated by progressively increasing the number of exchange rate matrices and composition vectors (or both) until there was no further improvement from increased parameterization. Bayes factor (BF) comparisons were used to determine when the addition of parameters no longer significantly improved the model fit to the data. The difference between the fit of the two models to the data set must have 2(ln BF) >6 to be deemed significant (see table 2 for summary of models used in 66TaxonSet and supplementary table S2, Supplementary Material online, for complete list).
Table 2.
Models Applied Using P4.
| Model | Number of GTR Rate Matrices | Number of Composition Vectors | Parameters |
|---|---|---|---|
| 1GTR + 1C + I + 4Γ | 1 | 1 | 12 |
| 2GTR + 1C + I + 4Γ | 2 | 1 | 18 |
| 3GTR + 1C + I + 4Γ | 3 | 1 | 24 |
| 4GTR + 1C + I + 4Γ | 4 | 1 | 30 |
| 5GTR + 1C + I + 4Γ | 5 | 1 | 36 |
| 6GTR + 1C + I + 4Γ | 6 | 1 | 42 |
| 1GTR + 2C + I + 4Γ | 1 | 2 | 16 |
| 1GTR + 3C + I + 4Γ | 1 | 3 | 20 |
| 1GTR + 4C + I + 4Γ | 1 | 4 | 24 |
| 1GTR + 5C + I + 4Γ | 1 | 5 | 28 |
| 1GTR + 6C + I + 4Γ | 1 | 6 | 32 |
| 1GTR + 7C + I + 4Γ | 1 | 7 | 36 |
| 2GTR + 5C + I + 4Γ | 2 | 5 | 34 |
| 3GTR + 5C + I + 4Γ | 3 | 5 | 40 |
| 4GTR + 5C + I + 4Γ | 4 | 5 | 46 |
Note.—The model codes used in this study; the number of GTR rate matrices applied to the data; the number of composition vectors estimated; and the number of free parameters estimated for each model. All parameters were free to vary. In all cases, the proportion of invariable sites (+I) = 0.3 and the gamma distributed associated rate variation (+Γ) = 0.7.
BF analysis indicated that there is strong evidence in favor of the heterogeneous model containing three GTR matrices, five composition vectors, an invariable sites category, and four categories of gamma distributed associated rate variation, that is, 3GTR + 5C + I + 4Γ, over the homogeneous model (1GTR + 1C + I + 4Γ), 2(ln BF) = 1097.1. The largest change in BF scores is seen in the shift between the compositionally homogeneous model (1GTR + 1C + I + 4Γ) and the model that allows for compositional heterogeneity across the tree (1GTR + 2C + I + 4Γ) with 2(ln BF) = 727.7. All BF comparisons are detailed in supplementary table S3, Supplementary Material online. Posterior predictive simulations on 66TaxonSet_nuc showed that homogeneous models do not fit the data, see figure 2A, whereas the best fitting model that accounts for heterogeneity across the phylogeny (3GTR + 5C + I + 4Γ) does fit the data, see figure 2B. This result indicates that the model employed in the original publication of the 66TaxonSet was not adequate to describe the data (Murphy et al. 2001a).
Fig. 2.

Fit of alternative models applied to 66TaxonSet_nuc, and posterior predictive simulations for the homogeneous model and heterogeneous model of “best-fit” for 66TaxonSet_nuc. Models are shown on x axis. The specifications of each model, that is, the number of rate matrices and composition vectors, are given inside the bar that represents that model. The y axis shows the ln L score, the higher the bar the worse the fit of the model. Inset (A) shows posterior predictive simulations for the homogeneous model (1GTR + 1C + I + 4Γ). Inset (B) shows posterior predictive simulation for the best-fit heterogeneous model (3GTR + 5C + I + 4Γ). The black bar graphs in insets (A) and (B) represent the posterior predictive simulations of these models on 66TaxonSet_nuc, and the arrow in each inset represents the χ2 position in the simulation for the real data.
To evaluate the improvement of fit on increasing composition vectors over amino acid data sets, the number of composition vectors was increased incrementally over the 66TaxonSet_aa. It was found that models 1JTT + 5C + I + 4Γ, 1JTT + 6C + I + 4Γ, and 1JTT + 7C + I + 4Γ did not reach convergence during the Markov Chain Monte Carlo (MCMC) run, this is not surprising given each of these models contain over 100 free parameters. BF comparisons indicated that the compositionally heterogeneous model 1JTT + 4C + I + 4Γ provided best fit to the data and is better than the compositionally homogeneous model 1JTT + 1C + I + 4Γ by 2(ln BF) = 369.4. However, posterior predictive simulations showed that both homogeneous (1JTT + 1C + I + 4Γ) and the best fit model allowing for compositional heterogeneity over the phylogeny (1JTT + 4C + I + 4Γ) fit the data with tail area probabilities of 0.79 and 0.97, respectively (supplementary fig. S1, Supplementary Material online). In summary, the posterior predictive simulation results show that the compositionally homogeneous model was satisfactory in describing the 66TaxonSet_aa; however, BF comparisons show that the model that accounts for compositional heterogeneity across the phylogeny describes the data better.
To evaluate the effect of multiple rate matrices on our amino acid data set and remove a layer of compositional heterogeneity, the 66TaxonSet_aa was recoded to 6 Dayhoff categories (66TaxonSet_day). It was found that increasing the number of rate matrices improved the model fit, as shown in comparison of model 1GTR + 1C + I + 4Γ with model 2GTR + 1C + I + 4Γ where 2(ln BF) = 67.0. This is compared with the impact of increasing the composition vectors, where model 1GTR + 1C + I + 4Γ compared with model 1GTR + 2C + I + 4Γ, 2(ln BF) = 16.4. BF comparisons identified model 5GTR + 2C + I + 4Γ as the best fitting model, and when compared with the homogeneous model (1GTR + 1C + I + 4Γ), 2(ln BF) = 169.3. Posterior predictive simulations show that both the homogeneous model 1GTR + 1C + I + 4Γ and the heterogeneous model of best-fit 5GTR + 2C + I + 4Γ were both adequate in describing the data.
Next, we wished to pursue the impact of models that accommodate heterogeneity across the data set, and so we applied profile mixture model CAT to the data. BF comparisons were not employed to test the fit of the CAT model as they were not implemented in PhyloBayes v3.2 (Lartillot et al. 2009), instead we used 10-fold Bayesian cross validation. The fit of data set heterogeneous models CAT and CAT-GTR were compared against the fit of homogeneous models 1GTR + 1C + I + 4Γ and 1JTT + I + 4Γ. Results show that for both 66TaxonSet_nuc and 66TaxonSet_day, a 1GTR + I + 4Γ model fits the data best (supplementary table S7, Supplementary Material online). For 66TaxonSet_aa, CAT is the best fitting model. The results of cross validation combined with the BF analyses show that the model using the GTR rate matrix and accommodating compositional heterogeneity describe the data better than compositionally homogeneous models. Therefore, these heterogeneous models were used to derive optimal phylogenies for 66TaxonSet_nuc and 66TaxonSet_day, whereas the CAT model was used to find the optimal tree for 66TaxonSet_aa, see summary table 3.
Table 3.
Summary of Data Quality and Phylogenetic Tests Applied to 66TaxonSet and 39TaxonSet.
| Data Sets | Compositional Heterogeneity Detected | P4 | PhyloBayes | Root Position (P4/PhyloBayes) | Reject Competing Hypotheses 2Ln(BF) = 6–10 | Single strongly Supported Hypothesis |
|---|---|---|---|---|---|---|
| 66TaxonSet_nuc | Yes | 3GTR + 5C + I + 4Γ | 1GTR + 1C + 4Γ | Atlantogenataa/Atlantogenata | Afrotheria and Xenarthra | None |
| 66TaxonSet_aa | Yes | 1JTT + 4C + I + 4Γ | CAT | Xenarthraa/Atlantogenata | Afrotheria and Atlantogenata | None |
| 66TaxonSet_day | No | 5GTR + 2C + I + 4Γ | 1GTR + 1C + 4Γ | Xenarthra/Xenarthraa | None | None |
| 39TaxonSet_nuc | Yes | 2GTR + 4C + 4Γ | CAT | Atlantogenata/Atlantogenataa | None | None |
| 39TaxonSet_aa | Yes | 1JTT + 5C + 4Γb | CAT_GTR | Atlantogenata/Atlantogenata | — | Atlantogenata |
| 39TaxonSet _day | Yes | 2GTR + 4C + 4Γ | CAT_GTR | Atlantogenata/Atlantogenata | Afrotheria and Xenarthra | Atlantogenata |
Note.—The results of the χ2 test of compositional homogeneity are shown. The model of best fit from P4 and PhyloBayes analyses are given. The placental root supported by each data set is indicated for both the P4 and the PhyloBayes analyses. The ability of each data set to reject alternative rooting hypotheses is given (2(ln BF) > 6 indicates alternative rooting hypotheses can be strongly rejected).
aRooting hypotheses that have a PP support value of <0.50, that is, poor support. A hypothesis is denoted as being the “single strongly supported hypothesis” if it has both 1) a high PP at the placental root and 2) all other hypotheses could be rejected.
bThis model did not fit the data.
Phylogenetic Results of Best Fitting Models on 66TaxonSet
In contrast to the previously published topology for 66TaxonSet, there are significant topological differences resulting from the application of models that account for heterogeneity over the phylogeny and the data. However, the posterior probability (PP) support values for placement of the placental root using the best fitting models remains low: 66TaxonSet_nuc and 66TaxonSet_aa support the Atlantogenata hypothesis with a PP of 0.46 (3GTR + 5C + I + 4Γ) and 0.51 (CAT), respectively. The 66TaxonSet_day supports the Xenarthra rooting with a high PP score of 0.98 (5GTR + 2C + I + 4Γ). However, comparison of BF scores showed that 66TaxonSet_day is unable to reject the Atlantogenata hypothesis, 2(ln BF) = 1.96 or the Afrotheria rooting, 2(ln BF) = 5.24. All BF comparison scores are given in supplementary table S4, Supplementary Material online. The results indicate that although models that accommodate heterogeneity across the phylogeny and the data described all treatments of 66TaxonSet better (66TaxonSet_ -nuc, -aa, and -day), this data set does not have sufficient power to support a single topology and reject all other competing phylogenetic hypotheses for the placement of the placental mammal root.
Heterogeneous Modeling of the 39TaxonSet
Given the current availability of many complete mammal genomes, we moved forward to assemble a new data set referred to as the 39TaxonSet. The data set consists of single copy orthologous families and contains 71,700 aligned nucleotide positions across 39 taxa. Overall, this data set contains fewer taxa but is four times the length of 66TaxonSet_nuc. Single gene orthologs were assembled using a strict best reciprocal Basic Local Alignment Search Tool (Blast) hit approach (see Materials and Methods). These data were analyzed at the 1) nucleotide level (39TaxonSet_nuc), 2) amino acid level (39TaxonSet_aa), and 3) these data were recoded into six Dayhoff categories (39TaxonSet_day) in the same way as for the 66TaxonSet.
The 27 genes comprising the 39TaxonSet had an overall GC content in the range 30.36–45.44% across all 39 taxa. The model fit test showed that 26/27 genes tested in the 39TaxonSet_nuc were compositionally heterogeneous. The level of heterogeneity varied on a gene-by-gene basis, overall 17/27 of the genes tested had at least 25/39 compositionally heterogeneous taxa (P values < 0.05). The results of the composition test have been provided in supplementary table S5(D), Supplementary Material online.
A total of 25 different models were applied to the 39TaxonSet and these ranged from the simplest model, that is, the homogeneous model with one composition vector and one exchange rate matrix, to the more parameter-rich heterogeneous models, including for example. Six GTR rate exchange matrices and six compositions vectors (for 39TaxonSet_nuc), one JTT rate exchange matrix and seven composition vectors (for 39TaxonSet_aa), and finally up to five GTR exchange rate matrices and four estimated composition vectors (39TaxonSet_day) (see supplementary table S2, Supplementary Material online, for complete set of models tested). The 39TaxonSet_nuc failed the χ2 tests of compositional homogeneity (P value = 0.00), whereas the model fit test (Foster 2004) showed that 17/39 taxa were not modeled adequately by the homogeneous model 1GTR + 1C + 4Γ· Amino acid data are known to ameliorate problems with compositional biases, possible codon usage biases and, in addition, they become saturated for superimposed substitutions more slowly (Philippe et al. 2011), therefore we constructed the 39TaxonSet_aa data set. Although the 39TaxonSet_aa data set passed the χ2 tests of compositional homogeneity (P value = 1.00), there were 35 out of 39 taxa that failed to fit the expectation of the homogeneous model 1JTT + 4Γ. Finally, for the 39TaxonSet_day, there were 21 out of 39 taxa that failed to fit the homogeneous model GTR + 1C + 4Γ (supplementary table S1, Supplementary Material online).
BF analyses were employed to determine whether there was strong evidence for the application of more parameter rich models over less parameter rich models for 39TaxonSet_nuc. It was found that homogeneous model 1GTR + 1C + 4Γ had ln L = − 623,990.33, whereas the heterogeneous model of best fit 2GTR + 4C + 4Γ had ln L = −620,944.12. The BF analysis shows very strong support for the use of the heterogeneous model with 2(ln BF) = 6,092.42. The posterior predictive simulations showed that the heterogeneous model 2GTR + 4C + 4Γ was able to accommodate the 39TaxonSet_nuc data (P = 0.79), whereas the homogeneous model 1GTR + 1C + 4Γ was not (P = 0.00) (supplementary fig. S1, Supplementary Material online). We also found strong support for the application of heterogeneous models to the amino acid data set, as 1JTT + 4Γ had ln L = −225,184.25 and the heterogeneous model of best-fit 1JTT + 5C + 4Γ scored ln L = −223,954.76. This provided strong support for the heterogeneous model with 2(ln BF) = 2,459.0 (supplementary table S3, Supplementary Material online). Overall, these BF analyses show very strong support for the application of heterogeneous models. However, posterior predictive simulations showed that for 39TaxonSet_aa models, allowing for compositional heterogeneity across the phylogeny did not adequately describe the data (P = 0.000) (supplementary fig. S1, Supplementary Material online).
At this point in our analysis, we wished to examine models that vary across the phylogeny (NDRH and NDCH) to determine whether they could further improve the fit of the model to the data. The application of GTR-like protein models in a phylogeny heterogeneous context is problematic (the 20 character states implies too many parameters with each additional composition vector or exchange rate matrix). It was therefore necessary to test the fit of NDRH-based models to the Dayhoff recoded data set, 39TaxonSet_day. BF comparisons between models that accommodate both compositional and exchange rate heterogeneity across the phylogeny 2GTR + 4C + 4Γ (ln L − 103,791.31) and the homogeneous model 1GTR + 1C + 4Γ (ln L − 103,967.31) show strong support for the heterogeneous model with 2(ln BF) = 352.0 (supplementary table S3, Supplementary Material online). The fit of the model (2GTR + 4C + 4Γ) to these data are supported by posterior predictive simulations with a tail-area probability of 0.004 (supplementary fig. S1, Supplementary Material online). Although this heterogeneous model does not fit the data within the 95% confidence interval, it is a far better descriptor of the data than the homogeneous model 1GTR + 1C + 4Γ that 1) has a tail-area probability of 0.00 and 2) lies well outside the posterior distribution of the simulated data set.
We wished to determine which rooting position was supported by the 39TaxonSet_nuc using the NDCH and NDRH models and the CAT-based models. The 39TaxonSet_nuc was assembled for this purpose and following cross validation we identified CAT as a better fit to the data set; however, there was very little support in favor of the Atlantogenata rooting (PP = 0.44). Posterior predictive simulations also showed that this data set is compositionally heterogeneous, across the entire alignment (Z score = 14.15) and across 20/39 taxa (supplementary table S7, Supplementary Material online). We therefore tested the NDCH and NDRH models and identified the 2GTR + 4C + 4Γ as the best fit. Under the 2GTR + 4C + 4Γ, we found support for the Atlantogenata rooting; however, the 39TaxonSet_nuc data were unable to reject the competing hypotheses for the position of the root.
The CAT and CAT-GTR models were applied to the 39TaxonSet_aa to determine whether profile mixture models fit the data better than the 1GTR + 1C + 4Γ and 1JTT + 4Γ-based models. The fit was again assessed using 10-fold Bayesian cross validation. The CAT-GTR model was identified through cross validation as a better fit to the 39TaxonSet_aa. At the amino acid level, there were less compositionally deviating taxa than the nucleotide level with only 9/39 taxa having a Z score > 2. These nine compositionally heterogeneous taxa (Z score > 2) are denoted in figure 3. To determine the effect of the observed compositional heterogeneity on the phylogeny we 1) removed the offending taxa and repeated the phylogenetic analysis and 2) analyzed the data at the Dayhoff category level (39TaxonSet_day). With the compositionally heterogeneous taxa removed the CAT-GTR model on the reduced 39TaxonSet_aa (amino acids), data set now passed the global test of compositional homogeneity. Analysis of the 39TaxonSet_day determined that CAT-GTR was the best fit to the 39TaxonSet_day when compared with CAT and 1GTR + 1C + 4Γ models. Posterior predictive simulations of 39TaxaSet_day show that although the CAT-GTR model globally fits the data (Z score = 1.89), 10 taxa are compositionally heterogeneous (Z score > 2). As recoding the 39TaxaSet_day data to Dayhoff categories did not ameliorate compositional heterogeneity under the CAT-GTR model, it was determined that the model that accounts for compositional and exchange rate heterogeneity over the phylogeny 2GTR + 4C + 4Γ better described the 39TaxonSet_day data set and the CAT-GTR model better described the 39TaxonSet_aa data set, see summary table 3.
Fig. 3.

Mammal phylogeny reconstructed using two independent approaches. Phylogeny reconstruction carried out using CAT-GTR model on the 39TaxonSet_aa in PhlyoBayes v3.2 and on 39TaxonSet_day using the heterogeneous model 2GTR + 4C + 4Γ in P4. The support values for both methods are given at each node: the numerator is the Bayesian support value for nodes based on CAT-GTR in PhyloBayes; the denominator is the support value based on the 2GTR + 4C + 4Γ model in P4. Support values for the 2GTR + 4C + 4Γ model are shown where they are in agreement with the CAT-GTR model topology. The color scheme represents the major groups of mammals: red, Euarchontoglires; blue, Laurasiatheria; green, Afrotheria; purple, Xenarthra; gray, Marsupials (* represents compositionally heterogeneous taxa). Gallus gallus and Ornithorhynchus anatinus were both compositionally heterogeneous but were excluded in this figure to retain clarity of placental mammal branch lengths.
Phylogenetic Results of Best Fitting Models on 39TaxonSet
Support is found for the Atlantogenata position for the root using a model that accounts for compositional and exchange rate heterogeneity across the tree (2GTR + 4C + 4Γ) on the 39TaxonSet_nuc and 39TaxonSet_day data sets as well as a model that accounts for heterogeneity across the data set (CAT-GTR) on 39Taxonset_day and 39Taxonset_aa data sets. The branch leading to the common ancestor of Xenarthra and Afrotheria is the most strongly supported position for the sister node to all other placental mammals, see figure 3. All topologies obtained are available for download from http://dx.doi.org/10.5061/dryad.vh49j. The removal of compositionally heterogeneous taxa (identified in 39TaxonSet_aa through Posterior Predictive simulations and highlighted in fig. 3) does not alter the topology and the Atlantogenata root remains the preferred position compared with the two alternative positions considered.
We wished to assess whether the 39TaxonSet had the power to reject alternative positions for the root. Although CAT-GTR is the best fitting model overall, it is not feasible to compare alternative topologies under CAT-based models employed in PhyloBayes (Lartillot et al. 2009). Therefore, we used the best fitting P4 heterogeneous model 2GTR + 4C + 4Γ (supports the Atlantogenata hypothesis with a PP value of 1.00 for 39TaxonSet_nuc and PP value of 0.96 for 39TaxonSet_day). Using the 2GTR + 4C + 4Γmodel, the BF analyses indicated that 39TaxonSet_day can discriminate between alternative phylogenetic hypotheses with BF scores as follows: Afrotheria rooting 2ln (BF) = 16.08 and Xenarthra rooting 2(ln BF) = 6.54, whereas the 39TaxonSet_nuc is unable to do so. A recent study has shown that nucleotide data are more susceptible to saturation at deeper nodes compared with amino acid data (Rota-Stabelli et al. 2013), and this may account for the lack of power in the nucleotide data set to distinguish between alternative hypotheses.
The analysis of the 39TaxonSet_day data set shows that although an improvement in fit of the model to the data set is observed with the addition of compositional vectors and exchange rate matrices, support for Atlantogenata as the sister group of all the other placental mammals is not eroded with more complex models.
To test whether the NDCH and NDRH models are robust to poor taxon sampling, we systematically reduced the number of representatives in each of the Afrotheria, Xenarthra, Laurasiatheria, and Euarchontoglires Superorders to a single taxon and we reran the phylogenetic reconstruction analyses using the heterogeneous models. In the case of the Euarchontoglires, we included the human and mouse independently as the representative taxon sampled. In each of these reduced taxon sets, the heterogeneous models (2GTR + 4C + 4Γ and CAT-GTR) recovered the Atlantogenata rooting hypothesis (see data online at the Dryad digital repository: doi:10.5061/dryad.vh49j).
The phylogeny produced using the CAT-GTR model on the 39TaxonSet_aa is almost completely congruent with that obtained through the analysis of the 39TaxonSet_day using the 2GTR + 4C + 4Γ model (See fig. 3). The only disagreement between these two topologies lies within the Laurasiatheria and relates to the placement of the five orders within this clade. Our focus in this analysis was the resolution of the interordinal relationships within the placental mammal phylogeny, to address the intraorder placement of the Laurasiatheria, denser taxon sampling through sequencing of currently poorly represented orders is necessary.
Conclusions
We set out to determine whether more sophisticated models that account for compositional heterogeneity and exchange rate heterogeneity across both the phylogeny and the data are required for the resolution of the placental root. Analyzing previously published data and methods, we have found that homogeneous models and shorter alignments cannot fully resolve this phylogenetic problem as they are not capable of rejecting all other competing hypotheses.
In our 39TaxonSet, we show that models that allow heterogeneity across the data outperform those tested that allow for heterogeneity across the phylogeny. We illustrate that our single copy orthologous data set (39TaxonSet) is sufficiently large to accommodate these parameter rich models. Significantly, these heterogeneous models achieve adequacy in modeling the evolutionary history of these sequences and they are robust to taxonomic sampling. We demonstrate that even though heterogeneous NDRH and NDCH models fit nucleotide data better than homogeneous modes and give PP support values of 1.00 to the Atlantogenata hypothesis, CAT-based models only support the Atlantogenata hypothesis with a PP = 0.44, and it is still not possible to distinguish between competing hypotheses using nucleotide data. The inability of the 39TaxonSet_nuc data set to reject the competing hypotheses for the position of the placental root is not surprising. Codon bias has been demonstrated at deep phylogenetic positions (Rota-Stabelli et al. 2013), and nucleotide data will tend to saturate more than amino acid data. Overall, these characteristics of the data provide a strong argument for the use of amino acids for deep-level phylogeny. Strong statistical support for the Atlantogenata hypothesis is achieved with the 39TaxonSet_day data set and heterogeneous models, and all major alternative hypotheses for the placement of the root of placental mammals can be fully rejected.
As with any phylogenetic question, congruence across multiple independent data sets and independent analyses is highly desirable. The Atlantogenata hypothesis is being recovered with greater frequency in mammal phylogenetic analyses (Madsen et al. 2001; Scally 2001; Hudelot et al. 2003; van Rheede et al. 2006; Nishihara et al. 2007; Hallstrom et al. 2007; Wildman et al. 2007; Hallstrom and Janke 2008; Prasad et al. 2008; Song et al. 2012). Here, we have shown that when better fitting models are used support for the Atlantogenata hypothesis is increased. The work presented here demonstrates that the mammal phylogeny is knowable, and that better phylogenetic modeling and the associated larger data requirements are of paramount importance to improving our understanding of mammalian phylogenetics.
Materials and Methods
Gene and Taxon Sampling
The first data set analyzed in the article was taken from Murphy et al. (2001b) and received no additional treatment or editing of the alignment, it is referred to as 66TaxonSet. This data set contains 66 species and 15 loci composed of 11 coding, 1 mtDNA, and 3 3′-UTR sequences totaling an alignment length of 9,789 bp. The protein-coding portion of these data consists of 11 genes representing 2,190 aligned amino acid positions.
The second data set was assembled specifically for this analysis using species available on the Ensembl server (version 60) and the Polar Bear (Ursus maritimus) genome, kindly supplied freely online by the Beijing Genomics Institute, China (Li et al. 2011), this data set is referred to as the 39TaxonSet. The longest canonical transcripts for the 39 taxa (38 mammals plus chicken) were analyzed using the best reciprocal Blast hit approach (Hubbard et al. 2005). One-to-one orthologs were identified using standalone BlastP (Altschul et al. 1990) with e−6, and genes were assigned to the category of one-to-one ortholog following best reciprocal Blast hit analysis and visual inspection of all alignments for all families. Only those single gene orthologs with high sequence quality across the entire gene length were retained. This resulted in a total of 27 single gene orthologous families for further analysis. The careful curation of the data set ensured accurate single gene ortholog identification despite the low genome coverage (≤2×) in 19/39 of the species used in this analysis. The taxon coverage was between 67.11% and 95.55% of the total concatenated nucleotide alignment (79,884 nucleotides). Details of the 27 genes in the 39TaxonSet used in this study, including gene names, accession numbers, alignment lengths, GC content, and composition homogeneity on a gene and species level are given in supplementary table S5, Supplementary Material online.
The 39TaxonSet alignment was manually edited using the Se-Al software (Rambaut 2001), sequencing errors and misaligned regions were identified and removed. The edited amino acid alignment was 27,200 aa in length. On removal of invariant sites, the total alignment length for the 39TaxonSet was 39,275 bp or 11,039 aa. The Ensembl unique identifiers for all 27 genes are available in supplementary table S5, Supplementary Material online, and the complete set of multiple sequence alignments are available at http://dx.doi.org/10.5061/dryad.vh49j.
Test for Compositional Homogeneity
The 66TaxonSet and the 39TaxonSet were tested for compositional homogeneity by comparing the compositions of the taxa with the mean composition of the data using the χ2 test for compositional homogeneity. This test does not take the topology of the taxa into account and as such cannot accommodate individual lineages that may deviate from the average compositional distribution (Rzhetsky and Nei 1995). Therefore, we applied a second test for compositional homogeneity that is a tree and model-based composition test (Foster 2004). We generated 100 simulated data sets based on the given model, phylogenetic tree and sample 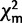 (where m is the expected value from the model). The goodness of fit measure, or χ2-value, from the actual data was then compared with the simulated data (
(where m is the expected value from the model). The goodness of fit measure, or χ2-value, from the actual data was then compared with the simulated data (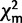 ). With the topology fixed, the model parameters and branch lengths were optimized in a maximum likelihood framework, and these data were considered homogeneous if the calculated value is greater than 95% of the null distribution (Foster 2004).
). With the topology fixed, the model parameters and branch lengths were optimized in a maximum likelihood framework, and these data were considered homogeneous if the calculated value is greater than 95% of the null distribution (Foster 2004).
Model Selection
Substitution models were originally selected following model testing using ModelGenerator v85 (Keane et al. 2006). Resultant models from this test were used as a starting point for the generation of tree heterogeneous models. The logarithm of the marginal likelihood was calculated using the Newton Raftery equation 16 (Newton et al. 1994) in P4 (Foster 2004). Through comparison of BFs, we can identify the model that fits the data best (while simultaneously not over fitting the model to the data). Twice the difference in logarithms was calculated and then compared with the Kass and Raftery table (Kass and Raftery 1995).
The CAT and CAT-GTR were tested against homogeneous models in PhyloBayes v3.2 (Lartillot et al. 2009) using Bayesian Cross Validation (Stone 1974), which is implemented in PhyloBayes v3.2 (Lartillot et al. 2009). Cross Validation was calculated by splitting the data set into two unequal parts, the learning set and the test set. The parameters of the model were estimated on the learning set, given the tree obtained from the MCMC run. The parameters were then used to calculate the likelihood of the test set. This was repeated 10 times for each model and the average of the overall likelihood score was taken. The scores from each model were then compared and the top scoring model was chosen, see supplementary table S7, Supplementary Material online, for cross validation calculations.
P4 Phylogenetic Analysis
The 66TaxonSet was analyzed at three levels: 1) the nucleotide data set (66TaxonSet_nuc), 2) amino acid data set (66TaxonSet_aa), and 3) the recoded Dayhoff categories (66TaxonSet_day). For the 39TaxonSet, heterogeneous models were applied at two levels: 1) amino acids (39TaxonSet_aa) and 2) the recoded Dayhoff categories (39TaxonSet_day). The models applied to the data set are shown in supplementary table S2, Supplementary Material online. For the 66TaxonSet, the proportion of invariable sites (+I) and Gamma distributed associated rate variation (+Γ) varied throughout the MCMC run to determine the optimal values. For the 39TaxonSet, +I were removed and the +Γ value was optimized in P4 (Foster 2004). This estimate for Γ was then fixed for the remainder of the analyses. This removed the computational load required for optimizing unnecessary parameters and therefore improved the speed of the analyses. For each of the heterogeneous models tested, 10 independent MCMC runs were used and these were permitted to run long after the likelihood values of the chain converged. Only runs that had converged were used in further analyses, this was determined by the agreement between topologies from the 10 independent MCMC runs and parameters. Topologies obtained from heterogeneous models in P4 are available for download from http://dx.doi.org/10.5061/dryad.vh49j.
PhyloBayes Phylogenetic Analysis
Both the 66TaxonSet and the 39TaxonSet were analyzed using CAT and CAT-GTR models along with homogeneous JTT + 4Γ and GTR + 1C + 4Γ (+I parameter added for 66TaxonSet) in PhyloBayes v3.2 (Lartillot et al. 2009). For each model tested, two independent runs using four MCMC chains were applied, these were sampled every 100 trees until convergence was reached, see download http://dx.doi.org/10.5061/dryad.vh49j.
Posterior Predicative Simulations
Each model was tested for goodness-of-fit using posterior predictive simulations (Foster 2004). These tests were applied to models that accommodate compositional heterogeneity across the phylogeny in P4 (Foster 2004) and models that accommodate heterogeneity across the data in PhyloBayes v3.2 (Lartillot et al. 2009). Using the given model for a data set and the parameters estimated from the MCMC run for that data set, simulated data sets were created. The fit of the model is estimated by the tail-area probability (Gelman et al. 1995). If the real data falls within the distribution of the simulated data set then the composition of the data is well described by the given model. Using posterior predictive simulations in PhyloBayes v3.2 (Lartillot et al. 2009), the compositional deviation for each taxon along with the overall fit of the model to composition were assessed. The output for posterior predictive simulations in PhyloBayes v3.2 (Lartillot et al. 2009) were computed and given in the form of Z scores. A Z score > 2 indicated that the model failed to fit the data, see supplementary figure S1 and table S6, Supplementary Material online, for the 66TaxonSet and 39TaxonSet scores, respectively.
Supplementary Material
Supplementary tables S1–S7 and figure S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
C.C.M. and A.E.W. carried out single gene ortholog identification. C.C.M. carried out all data assembly, phylogenetic, and statistical analyses. C.C.M., M.J.O’C., P.G.F., D.P., and J.O.McI. contributed to methodology, phylogenetic tests, and statistical analyses. M.J.O’C. conceived of the study, its design, and coordination. All authors participated in writing the manuscript. The authors thank the Irish Research Council for Science, Engineering and Technology (Embark Initiative Postgraduate Scholarship: RS2000172 to C.C.M.). M.J.O’C. and A.E.W. are funded by Science Foundation Ireland Research Frontiers Program: EOB2673, and MJO’C by the associated SFI STTF-11. M.J.O’C. acknowledge the Fulbright Commission for the Fulbright Scholar Award 2012-2013. C.C.M. thanks the DCU Orla Benson Award 2011 for funding and Roberto Feuda for assistance with PhyloBayes analyses. The authors thank the SFI/HEA Irish Centre for High-End Computing (ICHEC) and the SCI-SYM centre DCU for processor time and technical support. The authors also thank three anonymous reviewers for their comments that served to greatly improve the quality and clarity of the manuscript.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Asher RJ. A web-database of mammalian morphology and a reanalysis of placental phylogeny. BMC Evol Biol. 2007;7:108. doi: 10.1186/1471-2148-7-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda-Emonds OR, Cardillo M, Jones KE, et al. (10 co-authors) The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- Bleiweiss R. Slow rate of molecular evolution in high-elevation hummingbirds. Proc Natl Acad Sci U S A. 1998;95:612–616. doi: 10.1073/pnas.95.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WM, Prager EM, Wang A, Wilson AC. Mitochondrial DNA sequences of primates: tempo and mode of evolution. J Mol Evol. 1982;18:225–239. doi: 10.1007/BF01734101. [DOI] [PubMed] [Google Scholar]
- Caulin AF, Maley CC. Peto’s paradox: evolution’s prescription for cancer prevention. Trends Ecol Evol. 2011;26:175–182. doi: 10.1016/j.tree.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A. 2008;105:20356–20361. doi: 10.1073/pnas.0810647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhaes JP, Costa J. A database of vertebrate longevity records and their relation to other life-history traits. J Evol Biol. 2009;22:1770–1774. doi: 10.1111/j.1420-9101.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- Dorus S, Evans PD, Wyckoff GJ, Choi SS, Lahn BT. Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat Genet. 2004;36:1326–1329. doi: 10.1038/ng1471. [DOI] [PubMed] [Google Scholar]
- Foster PG. Modeling compositional heterogeneity. Syst Biol. 2004;53:485–495. doi: 10.1080/10635150490445779. [DOI] [PubMed] [Google Scholar]
- Foster PG, Cox CJ, Embley TM. The primary divisions of life: a phylogenomic approach employing composition-heterogeneous methods. Philos Trans R Soc Lond B Biol Sci. 2009;364:2197–2207. doi: 10.1098/rstb.2009.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PG, Hickey DA. Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J Mol Evol. 1999;48:284–290. doi: 10.1007/pl00006471. [DOI] [PubMed] [Google Scholar]
- Galtier N. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 2003;19:65–68. doi: 10.1016/s0168-9525(02)00002-1. [DOI] [PubMed] [Google Scholar]
- Gelman AB, Carlin JS, Stern HS, Rubin DB. London: Chapman & Hall; 1995. Bayesian data analysis. [Google Scholar]
- Hallstrom BM, Janke A. Resolution among major placental mammal interordinal relationships with genome data imply that speciation influenced their earliest radiations. BMC Evol Biol. 2008;8:162. doi: 10.1186/1471-2148-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom BM, Janke A. Mammalian evolution may not be strictly bifurcating. Mol Biol Evol. 2010;27:2804–2816. doi: 10.1093/molbev/msq166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom BM, Kullberg M, Nilsson MA, Janke A. Phylogenomic data analyses provide evidence that Xenarthra and Afrotheria are sister groups. Mol Biol Evol. 2007;24:2059–2068. doi: 10.1093/molbev/msm136. [DOI] [PubMed] [Google Scholar]
- Han KL, Braun EL, Kimball RT, et al. (17 co-authors) Are transposable element insertions homoplasy free?: an examination using the avian tree of life. Syst Biol. 2011;60:375–386. doi: 10.1093/sysbio/syq100. [DOI] [PubMed] [Google Scholar]
- Heath TA, Zwickl DJ, Kim J, Hillis DM. Taxon sampling affects inferences of macroevolutionary processes from phylogenetic trees. Syst Biol. 2008;57:160–166. doi: 10.1080/10635150701884640. [DOI] [PubMed] [Google Scholar]
- Holton TA, Pisani D. Deep genomic-scale analyses of the metazoa reject Coelomata: evidence from single- and multigene families analyzed under a supertree and supermatrix paradigm. Genome Biol Evol. 2010;2:310–324. doi: 10.1093/gbe/evq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdy I, Hirt RP, Dolezal P, Bardonova L, Foster PG, Tachezy J, Embley TM. Trichomonas hydrogenosomes contain the NADH dehydrogenase module of mitochondrial complex I. Nature. 2004;432:618–622. doi: 10.1038/nature03149. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Andrews D, Caccamo M, et al. (52 co-authors) Ensembl 2005. Nucleic Acids Res. 2005;33:D447–D453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudelot C, Gowri-Shankar V, Jow H, Rattray M, Higgs PG. RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol Phylogenet Evol. 2003;28:241–252. doi: 10.1016/s1055-7903(03)00061-7. [DOI] [PubMed] [Google Scholar]
- Janecka JE, Miller W, Pringle TH, Wiens F, Zitzmann A, Helgen KM, Springer MS, Murphy WJ. Molecular and genomic data identify the closest living relative of primates. Science. 2007;318:792–794. doi: 10.1126/science.1147555. [DOI] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–795. [Google Scholar]
- Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McInerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C, Holmes I, Goldman N. An empirical codon model for protein sequence evolution. Mol Biol Evol. 2007;24:1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- Kriegs JO, Churakov G, Kiefmann M, Jordan U, Brosius J, Schmitz J. Retroposed elements as archives for the evolutionary history of placental mammals. PLoS Biol. 2006;4:e91. doi: 10.1371/journal.pbio.0040091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Leroi AM, Koufopanou V, Burt A. Cancer selection. Nat Rev Cancer. 2003;3:226–231. doi: 10.1038/nrc1016. [DOI] [PubMed] [Google Scholar]
- Li B, Zhang G, Willersleve E, Wang J, Wang J. [Internet]. GigaScience; 2011. Genomic data from the polar bear (Ursus maritimus) [cited 2013 July 23]; Available from: http://dx.doi.org/10.5524/100008. [Google Scholar]
- Li WH, Ellsworth DL, Krushkal J, Chang BH, Hewett-Emmett D. Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Mol Phylogenet Evol. 1996;5:182–187. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- Li WH, Tanimura M, Sharp PM. An evaluation of the molecular clock hypothesis using mammalian DNA sequences. J Mol Evol. 1987;25:330–342. doi: 10.1007/BF02603118. [DOI] [PubMed] [Google Scholar]
- Madsen O, Scally M, Douady CJ, et al. (10 co-authors) Parallel adaptive radiations in two major clades of placental mammals. Nature. 2001;409:610–614. doi: 10.1038/35054544. [DOI] [PubMed] [Google Scholar]
- Martin AP, Palumbi SR. Body size, metabolic rate, generation time, and the molecular clock. Proc Natl Acad Sci U S A. 1993;90:4087–4091. doi: 10.1073/pnas.90.9.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Faircloth BC, Crawford NG, Gowaty PA, Brumfield RT, Glenn TC. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 2012;22:746–754. doi: 10.1101/gr.125864.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, Janecka JE, Gatesy J, et al. (22 co-authors) Impacts of the cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- Moreira D, Philippe H. Molecular phylogeny: pitfalls and progress. Int Microbiol. 2000;3:9–16. [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O’Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001a;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Eizirik E, O’Brien SJ, et al. (11 co-authors) Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001b;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17:413–421. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, Raftery AE, Davison AC, et al. (34 co-authors) Approximate Bayesian-inference with the weighted likelihood bootstrap. J R Stat Soc B Methodol. 1994;56:3–48. [Google Scholar]
- Nishihara H, Maruyama S, Okada N. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc Natl Acad Sci U S A. 2009;106:5235–5240. doi: 10.1073/pnas.0809297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Okada N, Hasegawa M. Rooting the eutherian tree: the power and pitfalls of phylogenomics. Genome Biol. 2007;8:R199. doi: 10.1186/gb-2007-8-9-r199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary MA, Bloch JI, Flynn JJ, et al. (23 co-authors) The placental mammal ancestor and the post-K-Pg radiation of placentals. Science. 2013;339:662–667. doi: 10.1126/science.1229237. [DOI] [PubMed] [Google Scholar]
- Peto R, Roe FJ, Lee PN, Levy L, Clack J. Cancer and ageing in mice and men. Br J Cancer. 1975;32:411–426. doi: 10.1038/bjc.1975.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Worheide G, Baurain D. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 2011;9:e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad AB, Allard MW, Green ED. Confirming the phylogeny of mammals by use of large comparative sequence data sets. Mol Biol Evol. 2008;25:1795–1808. doi: 10.1093/molbev/msn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang LS, Gascuel O, Lartillot N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics. 2008;24:2317–2323. doi: 10.1093/bioinformatics/btn445. [DOI] [PubMed] [Google Scholar]
- Rambaut A. Se-AL. 2001 [Internet]. [cited 2009 Dec 2]. Available from: http://tree.bio.ed.ac.uk/software/seal/ [Google Scholar]
- Rokas A, Kruger D, Carroll SB. Animal evolution and the molecular signature of radiations compressed in time. Science. 2005;310:1933–1938. doi: 10.1126/science.1116759. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJ, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20:1001–1009. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota-Stabelli O, Lartillot N, Philippe H, Pisani D. Serine codon usage bias in deep phylogenomics: pancrustacean relationships as a case study. Syst Biol. 2013;62:121–133. doi: 10.1093/sysbio/sys077. [DOI] [PubMed] [Google Scholar]
- Rzhetsky A, Nei M. Tests of applicability of several substitution models for DNA sequence data. Mol Biol Evol. 1995;12:131–151. doi: 10.1093/oxfordjournals.molbev.a040182. [DOI] [PubMed] [Google Scholar]
- Scally M. Molecular evidence for the major clades of placental mammals. J Mammal Evol. 2001;8:239–277. [Google Scholar]
- Song S, Liu L, Edwards SV, Wu S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc Natl Acad Sci U S A. 2012;109:14942–14947. doi: 10.1073/pnas.1211733109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhope MJ, Waddell VG, Madsen O, de Jong W, Hedges SB, Cleven GC, Kao D, Springer MS. Molecular evidence for multiple origins of Insectivora and for a new order of endemic African insectivore mammals. Proc Natl Acad Sci U S A. 1998;95:9967–9972. doi: 10.1073/pnas.95.17.9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone M. Cross-validatory choice and assessment of statistical predictions. J R Stat Soc. 1974;36:111–147. [Google Scholar]
- Usanga EA, Luzzatto L. Adaptation of Plasmodium falciparum to glucose 6-phosphate dehydrogenase-deficient host red cells by production of parasite-encoded enzyme. Nature. 1985;313:793–795. doi: 10.1038/313793a0. [DOI] [PubMed] [Google Scholar]
- van Rheede T, Bastiaans T, Boone DN, Hedges SB, de Jong WW, Madsen O. The platypus is in its place: nuclear genes and indels confirm the sister group relation of monotremes and Therians. Mol Biol Evol. 2006;23:587–597. doi: 10.1093/molbev/msj064. [DOI] [PubMed] [Google Scholar]
- Waddell PJ, Cao Y, Hauf J, Hasegawa M. Using novel phylogenetic methods to evaluate mammalian mtDNA, including amino acid-invariant sites-LogDet plus site stripping, to detect internal conflicts in the data, with special reference to the positions of hedgehog, armadillo, and elephant. Syst Biol. 1999;48:31–53. doi: 10.1080/106351599260427. [DOI] [PubMed] [Google Scholar]
- Wildman DE, Uddin M, Opazo JC, et al. (10 co-authors) Genomics, biogeography, and the diversification of placental mammals. Proc Natl Acad Sci U S A. 2007;104:14395–14400. doi: 10.1073/pnas.0704342104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.