

Abstract
Aims
Myocardial development is dependent on concomitant growth of cardiomyocytes and a supporting vascular network. The coupling of myocardial and coronary vascular development is partly mediated by vascular endothelial growth factor (VEGFA) signalling and additional unknown mechanisms. We examined the cardiomyocyte specific role of the transcriptional co-activator Cited2 on myocardial microstructure and vessel growth, in relation to Vegfa expression.
Methods and results
A cardiomyocyte-specific knockout of mouse Cited2 (Cited2Nkx) was analysed using magnetic resonance imaging and histology. Ventricular septal defects and significant compact layer thinning (P< 0.02 at right ventricular apex, P< 0.009 at the left ventricular apex in Cited2Nkx vs. controls, n= 11 vs. n= 7, respectively) were found. This was associated with a significant decrease in the number of capillaries to larger vessels (ratio 1.56 ± 0.56 vs. 3.25 ± 1.63, P= 2.7 × 10−6 Cited2Nkx vs. controls, n= 11 vs. n= 7, respectively) concomitant with a 1.5-fold reduction in Vegfa expression (P< 0.02, Cited2Nkx vs. controls, n= 12 vs. n= 12, respectively). CITED2 was subsequently found at the Vegfa promoter in mouse embryonic hearts using chromatin immunoprecipitation, and moreover found to stimulate human VEGFA promoter activity in cooperation with TFAP2 transcription factors in transient transfection assays. There was no change in the myocardial expression of the left-right patterning gene Pitx2c, a previously known target of CITED2.
Conclusions
This study delineates a novel cell-autonomous role of Cited2 in regulating VEGFA transcription and the development of myocardium and coronary vasculature in the mouse. We suggest that coupling of myocardial and coronary growth in the developing heart may occur in part through a Cited2→Vegfa pathway.
Keywords: CITED2, VEGFA, Myocardial development, Capillary growth
Introduction
New cardiac myocytes are efficiently created during foetal life, initially from cardiomyocyte progenitors and subsequently by cardiomyocyte division (reviewed in Bhattacharya et al.1). In gross morphological terms, the thin-walled mouse embryonic ventricle begins to develop finger-like projections of trabecular myocardium at the endocardial surface by embryonic day (E) 10.5 (reviewed in Sedmera et al.2). Thickening of the epicardial myocardium begins by E11.5 and results in the formation of the compact myocardium. This process occurs concomitantly with the formation of the coronary vessels (reviewed in Luttun and Carmeliet3 and Reese et al.4). Many zygotic mutations associated with abnormal extra-embryonic tissue development also have abnormal myocardial development as a phenotypic feature,1 and complementation experiments indicate that myocardial development is dependent on normal development of the extra-embryonic tissues.5 Myocardial compact layer growth abnormalities can also be secondary to abnormal vascular development. Endothelial-specific conditional knockout of several genes [e.g. Nf1, Mapk7, Efnb2, and Casp8 (reviewed in Bhattacharya et al.1)], and pro-epicardial organ-specific knockout of Rxra, result in myocardial growth defects.6 Genetic evidence suggests that the coupling of coronary vessel development to myocardial growth occurs, at least in part, through the vascular endothelial growth factor (VEGFA),7 a known target of the hypoxia-activated transcription factor (HIF1A).8 Myocardial deletion of Vegfa results in fewer coronary microvessels and a thinned ventricular wall.7
The ubiquitously expressed transcriptional co-factor CITED29 can inhibit hypoxia-activated transcription by blocking recruitment of the histone acetyltransferase CREBBP/EP300 to HIF1A.10–12 CITED2 also acts as a co-activator for transcription factors, such as TFAP2, LHX2, PPARA, and SMAD2/3,10,13–16 in part by recruiting CREBBP/EP300. Genetic evidence indicates that Cited2 is essential for cardiac left-right patterning via regulation of the left-right patterning Nodal-Pitx2c pathway.17–20 Zygotic and epiblastic deletion of Cited2 results in atrioventricular septation, outflow tract, and aortic arch defects, and also in left-right patterning defects such as right isomerism.18,19 Loss of Cited2 results in lack of expression of the Nodal target genes Pitx2c, Nodal, and Lefty2 in the left lateral plate mesoderm, explaining the left-right patterning defects. Cited2 is also essential for adrenal, neural crest, liver, lung, lens and placental development and is a regulator of adult haematopoietic stem cells.17,21–26 This early requirement of Cited2 in left-right patterning and in placental development makes it difficult to identify a later specific role in myocardial development. In this study, we therefore investigated the role of Cited2 in the myocardium by conditional deletion in cardiomyocyte precursors.
Methods
Mice
Cited2+/− mice (Cited2tm1Bha),17 Nkx2.5Cre mice (Nkx2-5tm(cre)Rjs),27 and Cited2flox/flox females (Cited2tm2Bha)19 were crossed to create Cited2−/− and wild-type control embryos, and Cited2+/flox;Nkx2.5Cre (control) and Cited2−/flox;Nkx2.5Cre (tissue-specific deletion, referred to as Cited2Nkx) embryos. Embryos were collected at the indicated time-point after detection of a vaginal plug [embryonic day (E) 0.5] and genotyped using allele-specific polymerase chain reaction (PCR; details of primers available on request). All studies were performed in accordance with UK Home Office Animals (Scientific Procedures) Act 1986.
Imaging
Magnetic resonance imaging (MRI) was performed on a 9.4T MR system (Varian, USA) as described previously on embryos with littermate controls at E15.5,28 n= 7 control and n= 11 Cited2Nkx. Embryos were subsequently sectioned and stained with haematoxylin and eosin.
Measurements
All measurements were taken using transverse sections featuring the mitral and tricuspid valve in a four-chamber view, blind to the embryo genotype. For vessel counts, heart sections were analysed at the same positions taken for myocardial measurements. High-resolution photographs at ×10 magnification were taken of each heart, and capillaries (defined as vessels not >1 red cell in width) and large vessels (larger than two red cells in diameter) counted. The left ventricular (LV) apex, septum, and right ventricular (RV) apex were photographed and the number and type of vessel in the endocardial, myocardial, and subepicardial areas at these sites counted. The septum included all the septal myocardium viewable between the epicardial aspect of the septum to the atrioventricular valves, and the LV apex included all the LV myocardium between the left atrioventricular groove and the LV side of the septum, with the same criteria applied to the right.
Quantitative reverse transcriptase-polymerase chain reaction, chromatin immunoprecipitation, and transient transfection assays
Quantitative reverse transcriptase polymerase chain reaction (qPCR) was performed from dissected embryo hearts (E13.5) as described.19 Primer details are stated in the Supplementary material online. Chromatin immunoprecipitation assays from embryonic hearts were performed as described in the Supplementary material online, followed by qPCR for detection. Transient transfection assays were performed as described (Supplementary material online, methods).
Statistical and data analysis
Genotype deviation from expected Mendelian ratios was analysed using the χ2 test. The calculated probability of a type 1 error was calculated using the CHIDIST function in Excel (Microsoft Office 2007, Microsoft, Redmond, WA, USA).
For measurements of the heart size, the value of each analysed heart was plotted and graphed according to genotype with the mean shown and error bars demonstrating the standard error of the mean. Statistical significance was estimated using a two-tailed t-test assuming equal variance, taking a value of P< 0.05 to be significant. For qPCR, all reactions were performed in triplicate for each sample and the mean of each data triplicate was taken to be the value of the sample and exploratory data analysis used to visualize these graphically to see the data distribution, plotting each mean and the standard error of the mean for each particular population.
As a quality control, only samples for which the threshold cycle (Ct) replicate values were within 1 Ct value of each other were used for subsequent analysis.29 In graphs detailing the qPCR results, each dot in each column represents data from a single embryo heart of the relevant genotype. Overall means were compared using a two-tailed t-test, assuming equal variation. For hypothesis driven experiments, the criterion for statistical significance was P< 0.05 if one, or P< 0.025 if two hypotheses were tested (using the Bonferroni correction).30 The statistical package in Excel (Microsoft Office 2007, Microsoft, Redmond, WA, USA) was used for all tests.
Results
Cardiomyocyte deletion of Cited2 causes myocardial compact layer thinning and ventricular septal defects
To investigate a role for Cited2 within the developing heart, a cardiac-specific deletion of Cited2 (Cited2−/flox;Nkx2.5Cre, hereafter referred to as Cited2Nkx) was generated by using the Cited2flox allele19 and the Nkx2.5Cre strain27 that expresses Cre recombinase in cardiomyocytes from E7.5. Cited2Nkx mice were viable, born at a normal Mendelian frequency, and survived at least to weaning (Cited2+/f= 15, Cited2−/f= 16, Cited2−/flox;Nkx2.5Cre= 17, Cited2−/flox;Nkx2.5Cre= 11, total 59 mice) (Supplementary material online, Table S1). Examination of embryo genotypes revealed that there was no loss of Cited2Nkx embryos in late gestation at E15.5 (Cited2+/f= 10, Cited2−/f= 17, Cited2−/flox;Nkx2.5Cre= 7, Cited2−/flox;Nkx2.5Cre= 12, total 46 E15.5 embryos) (Supplementary material online, Table S1). Examination of hearts from E9.5 embryos bearing a Nkx2-5Cre recombined Cited2flox allele (where the Cited2 promoter drives expression of lacZ) showed that recombination was highly efficient in myocardial cells, appearing complete in the common atrium, atrioventricular canal, primitive ventricle, bulbus cordis, and outflow tract (Figure 1).
Figure 1.
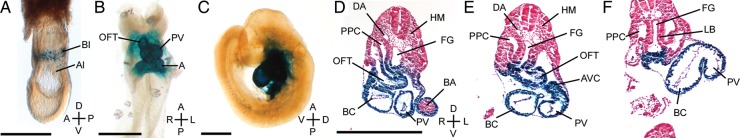
lacZ reporter activity following Nkx2-5Cre mediated recombination of the Cited2flox allele. Embryos of the Cited2+/f;Nkx2.5Cre genotype were examined for LacZ expression at E7.5 (A), E8.5 (B) and E9.5 (C). LacZ activity was first observed in developing blood islands at E7.5 with the primitive heart tube staining at the heart looping stage (E8.5). (D–F) The E9.5 embryo was sectioned to demonstrate that recombination was highly efficient in myocardial cells. A, primitive atrium; Al, allantois; AVC, atrioventricular canal; BA, branchial arch; BC, bulbus cordis; BI, blood islands; DA, dorsal aorta; FG, foregut; HM, head mesenchyme; LB, lung bud; OFT, outflow tract; PPC, pleuroperitoneal canal; PV, primitive ventricle. Scale bars, 1 mm. Axes: D, dorsal; V, ventral; A, anterior; P, posterior.
Analysis of E15.5 embryos by MRI and by histology, however, revealed a spectrum of cardiac defects affecting septation (Figure 2, Figure 3A–C). From 11 Cited2Nkx embryos examined, 6 showed ventricular septal defects (VSD), of which 2 also had atrioventricular septal defects. None of the mutant embryos had left-right patterning defects, seen when Cited2 is deleted earlier in development.19 Furthermore, no outflow tract or aortic arch patterning defects were observed. No cardiac defects were seen in control littermates (n= 7).
Figure 2.
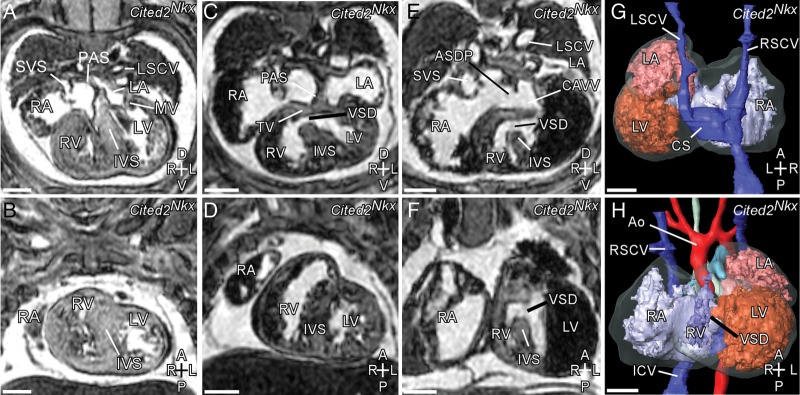
Spectrum of cardiac defects in Cited2Nkx embryos visible by MRI. (A, C, E, transverse sections; B, D, F, coronal sections) (A) and (B) apparently normal Cited2Nkx embryo heart with pectinated right atrium (RA), systemic venous sinus (SVS), mitral valve (MV), left and right ventricle (LV, RV), and interventricular septum (IVS). (C and D) Cited2Nkx embryo heart with ventricular septal defect (VSD). (E and F) Cited2Nkx embryo with ventricular septal defect, septum primum atrial septal defect (ASDP), common atrioventricular valve (CAVV). (G and H) 3D reconstruction of Cited2Nkx embryo shown in (E) and (F). RSCV, right superior caval vein; LSCV, left superior caval vein; ICV, inferior caval vein; Ao, aortic arch; CS, coronary sinus. Scale bar, 500 µm. Axes: D, dorsal; V, ventral; A, anterior; P, posterior; R, right; L, left.
Figure 3.

Cardiac defects and measurements in Cited2Nkx embryo hearts. Haematoxylin and eosin stained transverse sections from control (A) (n= 7) and Cited2Nkx (B and C) embryos (n= 11). The right and left ventricles (RV, LV), interventricular septum (IVS), right and left atria (RA, LA), ventricular septal defects (VSD), and compact (thick line) and trabecular (thin line) zones are indicated. The compact layer of the myocardium in both ventricles is visibly thicker in the control than in the Cited2Nkx hearts. Scale bar, 500 µm; axes D, dorsal; V, ventral; R, right; L, left. (D–F) Thoracic and transventricular diameters in Cited2Nkx embryos. (D) Maximum thoracic diameter, (E) maximum transventricular diameter, (F) cardiothoracic ratio, (G) corrected compact layer thickness (mm) for right ventricle and for the left ventricle in control and Cited2Nkx hearts, respectively. (H) Trabecular layer thickness (mm) for right ventricle and for the left ventricle in control and Cited2Nkx hearts, respectively. (I) Ratio of compact to trabecular layer thickness for right ventricle and for the left ventricle in control and Cited2Nkx hearts, respectively. Each point represents data from an individual embryo and lines indicate the mean and standard error of the mean.
Cited2Nkx embryos had smaller thoracic cavities (P< 0.009) (n= 7) than their littermate controls (n= 11), and smaller transventricular diameters (P< 0.02; Figure 3D and E). However, correcting the heart size for thoracic diameter, using the cardiothoracic ratio (defined as the maximum transverse diameter of the heart across the ventricles divided by the maximum diameter of the thorax in the section), the hearts were of the same size relative to the embryo size (Figure 3F). For all subsequent measurements, the value obtained was divided by the thoracic diameter to correct for this.
The compact layer of myocardium was significantly thinner in Cited2Nkx embryo hearts than their littermate controls at both the RV and LV apex (P < 0.02 and P < 0.009, respectively; Figure 3G). The trabecular layer, however, was the same thickness (Figure 3h). When the myocardial thickness was examined as a ratio of compact to trabecular layers, it was significantly reduced in Cited2Nkx hearts at both the RV (P < 0.024) and LV (P < 0.007) apex (Figure 3I), indicating a selective developmental defect in the compact layer compared with the trabecular layer in both ventricles.
Cardiomyocyte deletion of Cited2 causes abnormal microvessel development
Abnormal myocardial compact layer development is often secondary to abnormal vascular development. To examine possible causes of defective compact layer development, the number of vessels was counted in each ventricle at the LV and RV apex and septum. There was a significant decrease in the number of total vessels seen (mean 56 ± 24 in control vs. 41.5 ± 20 in Cited2Nkx, P= 0.018; Figure 4C) (control n= 7, Cited2Nkx= 11 hearts examined). When actual vessel types were examined, it was found the number of large vessels was unchanged but there was almost half the number of capillaries present (large vessels, 15.7 ± 11.0 vs. 17.4 ± 9.9, P= 0.56 and capillaries, 40.7 ± 18.4 vs. 24.1 ± 12.3, P= 0.002; control vs. Cited2Nkx, respectively for each; Figure 4D). The ratio of capillaries to large vessels was also examined, and found to be significantly decreased in the Cited2Nkx vs. control hearts (1.56 ± 0.56 vs. 3.25 ± 1.63, P= 2.7 × 10−6; Figure 4E). This demonstrates that there is a reduced presence of capillaries relative to larger vessels.
Figure 4.
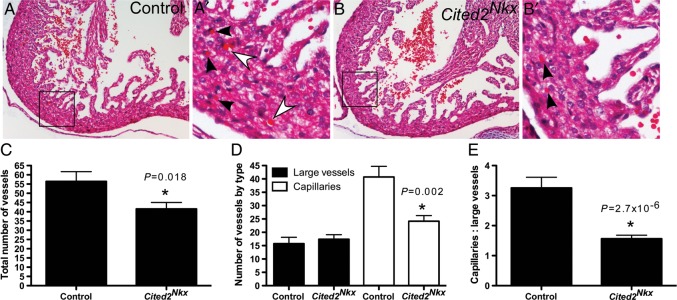
Blood vessel analysis in Cited2Nkx hearts. (A and B), Haematoxylin and eosin-stained transverse sections of control and Cited2Nkx hearts at ×10 magnification. (A′ and B′) Enlargement of boxed areas in (A) and (B). Capillaries (vessels 1 red cell in width) are indicated by black arrowheads. Large vessels (>1 red cell in width) are indicated by white arrowheads. (C) Total number of vessels counted in control (n= 7) and in Cited2Nkx hearts (n= 11). (D) Large vessel and capillary counts in control and in Cited2Nkx hearts. (E) Ratio of large vessels to capillary counts in control and Cited2Nkx hearts. Bars indicate the mean and lines standard error of the mean.
Cited2 positively regulates Vegfa expression in the developing heart
As deletion of Cited2 in cardiomyocytes caused VSDs, abnormal myocardium and abnormal vessels, a candidate gene approach was taken to identify potential Cited2 target genes. We tested two candidates that have previously been linked to Cited2. These are Pitx2c18,19 and Vegfa.12 Embryo hearts were isolated at E13.5, just prior to completion of ventricular septation. To ensure Cited2 knockout in our conditionally knocked out samples, we first performed a qPCR analysis for Cited2. There was a 4.7-fold decrease in Cited2 expression between control (n= 12) and Cited2Nkx (n= 12) hearts, (Figure 5A) validating our samples for further analysis. We next performed qPCR for Pitx2c and for Vegfa. There was no significant difference seen in Pitx2c expression (control= 10 and Cited2Nkx n= 11), (Figure 5B). A significant 1.5-fold decrease in Vegfa expression levels was found in the Cited2Nkx hearts compared with controls (P < 0.02, n= 12 for each genotype) (Figure 6A).
Figure 5.

Cited2 and Pitx2c expression in E13.5 Cited2Nkx hearts. Quantitative reverse transcriptase polymerase chain reaction was used to assess Cited2 and Pitx2c RNA expression in control and in Cited2Nkx hearts. (A) A 4.7-fold decrease in Cited2 expression in Cited2Nkx hearts compared with controls was found (n= 12 control, n= 12 Cited2Nkx, P= 1.07 × 10−5). (B) No significant difference was seen in Pitx2c expression levels (n= 10 control, n= 11 Cited2Nkx). For each panel, each dot represents data from a single E13.5 heart of the relevant genotype. All expression levels are relative to 18s RNA expression levels. Lines indicate mean and standard error of the mean.
Figure 6.

Vegfa and Hif1a expression in E13.5 Cited2Nkx and Cited−/−2 hearts. Quantitative reverse transcriptase polymerase chain reaction was used to assess RNA levels. (A) Vegfa expression in control and in Cited2Nkx hearts showing a 1.5-fold reduction in Cited2Nkx hearts (P< 0.02) (n= 12 control, n= 12 Cited2Nkx). (B) Hif1a expression in control and in Cited2Nkx hearts (n= 12 control, n= 12 Cited2Nkx). (C) Vegfa in wild-type control and in congenic Cited2−/− hearts (n= 12 control, n= 12 Cited2−/−). (D) Hif1a in wild-type control and in Cited2−/− hearts (n= 12 control, n= 12 Cited2−/−). For each panel, each dot represents data from a single E13.5 heart of the relevant genotype. All expression levels are relative to 18s RNA expression levels. Lines indicate mean and standard error of the mean.
Next, we tried to ascertain if this decrease in Vegfa could have arisen from a decrease in Hif1a expression, this being a major regulator of Vegfa expression.8 To ensure consistency, we performed these experiments on the same conditionally deleted Cited2Nkx hearts, but observed no difference in Hif1a expression (Figure 6B).
To determine whether Vegfa expression was affected in zygotic deletion of Cited2, we analysed by qPCR a separate series of C57Bl6/J congenic Cited2−/− hearts and wild-type littermate controls (n= 12 for each genotype). Here, there was a non-significant increase in Vegfa expression, compared with control (Figure 6C). We also performed a qPCR for Hif1a in these samples; there was no significant change observed between congenic Cited2−/− hearts and wild-type littermate controls (Figure 6D).
CITED2 positively regulates Vegfa expression via TFAP2 transcription factors
Our previous work established that CITED2 and TFAP2 family members physically interact, and that CITED2 acts as a co-activator of TFAP2 transcription factors.13,17,18 Vascular endothelial growth factor expression has been shown to be positively31–35 or negatively36 modulated by TFAP2 factors in other cellular systems. Analysis of the human VEGFA and mouse Vegfa promoters using the transcription factor database (http://www-bimas.cit.nih.gov/) showed that both human and mouse promoters contain multiple AP2 consensus-binding sites (Figure 7A), consistent with previous observations.37 Using chromatin immunoprecipitation, CITED2 could be detected at the Vegfa promoter in developing wild-type mouse embryonic hearts (Figure 7B). We next tested, by transient transfection assays, whether CITED2 and TFAP2 could modulate the transcriptional activity of the human VEGF promoter. These co-transfection experiments showed that CITED2 cooperates with TFAP2C to co-activate the human VEGF promoter by ∼2.5-fold in comparison with the promoter transfected with the control vectors (Figure 7C). These results indicate that endogenous CITED2, a transcriptional co-activator, is present at the Vegfa promoter, and that CITED2 is a positive regulator of Vegfa transcription, working at least in part through TFAP2 sites in the Vegfa promoter.
Figure 7.

CITED2 is present at the Vegfa promoter in mouse embryonic hearts and cooperates with TFAP2 to co-activate the human VEGFA promoter. (A) Diagram of the human and mouse Vegfa promoters. The positions of putative TFAP2-binding sites are indicated (grey arrowheads). The black arrow indicates the transcriptional start site. The positions of DNA fragments in the mouse promoter amplified by PCR for chromatin immunoprecipitation assay are indicated with blue arrowheads. (B), chromatin immunoprecipitation from Cited2+/+ and Cited2−/− embryonic hearts (E13.5) with anti-Cited2 antibody. The enrichment of the immunoprecipitated DNA in Cited2+/+ vs. Cited2−/− hearts was analysed by quantitative reverse transcriptase polymerase chain reaction using primers (VGF1 to VGF4) spanning the Vegfa promoter region [shown in (A)]. Results (bars indicating mean and lines standard error of the mean) from three independent experiments are shown as enrichment of immunoprecipitated DNA over the input chromatin. (C) Hep3B cells were transiently transfected with the pVEGF-luc reporter (40 ng), with CMV-lacZ (100 ng), CMV-CITED2 or the control vector (80 ng), CMV-TFAP2A, CMV-TFAP2C, and the control vector (40 ng). Results (bars indicating mean and lines standard error of the mean, two independent experiments performed in quadruplicate) are presented as relative luciferase units, corrected for β-galactosidase activity. The control transfection value (at the extreme left) is set at 1.
Discussion
In this study, we have identified, by conditional deletion of Cited2 from the developing myocardium, a cell autonomous role for Cited2 in mouse cardiac myocytes. We show that Cited2 is required for normal myocardial thickening, ventricular septation, and coronary vascular development. We also show that conditional deletion of Cited2 in the mouse myocardium results in deficiency of the mouse Vegfa transcript, that CITED2 is found at the mouse Vegfa promoter in vivo, and that CITED2 transactivates a VEGFA promoter in cells. The defect in myocardial compact layer formation observed at E15.5 is at a time in which myocyte proliferation in this layer is the predominant type of growth seen,38–40 and we hypothesize that defective myocardial proliferative growth is the most likely mechanism for this observation. This may be secondary to the lack of a supporting vascular network required to support the rapid increase in cell number.
This is distinct from the mechanisms of cardiac malformation and left-right patterning defects reported previously in Cited2 deficiency18 as there is no deficiency in Pitx2c expression. Vegfa is essential for normal coronary development, and normal coronary development is required for normal myocardial development. The levels of VEGFA are tightly regulated during normal development, as evidenced by the fact that loss or gain of even a single allele results in cardiac malformation.41–43 The phenotype described here is similar to that found in a mycocyte-specific deletion of VEGFA, where the myocardium was found to be the main source of VEGFA. The conditional knockout mice were viable but subsequently showed impaired function and reduced weight, with thin ventricular myocardium and reduced capillary number.7 We, therefore, suggest that one mechanism by which Cited2 deficiency affects myocardial and coronary development is, at least in part, via deficiency of VEGFA.
Our results, using a cardiac-specific conditional knockout, are the opposite of the previously reported increased cardiac Vegfa expression observed in zygotic deletion of Cited2.12 Based on this we interpret the results of zygotic Cited2 deletion on Vegfa as the effect of severe global embryonic hypoxia resulting from complex cardiac malformations or the abnormal placental development seen with zygotic deletion,25 and activation of Vegfa expression by HIF1A.8 In keeping with this re-interpretation, we too found an increase in Vegfa expression in the zygotic deletion of Cited2 (although this was not statistically significant). Tissue-specific deletion of Cited2 here permitted more focused study at a tissue-specific level, eliminating the gross effects of abnormal placental development and severe cardiac malformation and the further non-tissue-specific gene responses this would entail.
CITED2 is a key regulator of HIF1A, blocking its interaction with CREBBP/EP300, and hence HIF1A-mediated transcription. Our results would suggest that mechanisms regulating downstream target genes of HIF1A may be more complex. CITED2 directly blocks HIF1A-mediated transcription10–12 but, by allowing transcription of a downstream target such as Vegfa, it may be protective in severe hypoxia, allowing some Vegfa transcription and vessel development but not at too high a level, preventing disorganized vessel growth and ensuring that not all Vegfa transcription is lost. Another model of cardiomyocyte-specific Cited2 deletion was reported recently, in which no septation, myocardial, or coronary defects were noted.44 The reasons for the differences from our study are not clear but could result from reduced efficacy of Cited2 deletion in cardiomyocytes or from the use of a mixed genetic background, either of which could result in reduced penetrance of phenotype.
There may be alternative mechanisms for the abnormalities in myocardial and coronary development observed in the myocardial-specific knockout of Cited2. Trp53 is an additional key regulator, coordinating myocardial and coronary growth via the HIF1 and VEGF pathway, and could be an important intermediary particularly as it has been shown to be a target of Cited2.23,45 However, previous complex mutants of Cited2 and p53 have been created without much effect visible on the cardiac phenotype,28 suggesting its role in the developing myocardium is unclear. The role of other regulators of embryonic myocardial and coronary development—such as GATA4 (reviewed in Bhattacharya et al.1), and VEGFB46—needs to be addressed in future studies.
Taken together, our results suggest that Cited2 controls the coupling of embryonic myocardial and coronary vascular development, at least in part via regulation of Vegfa transcription. We speculate that activation of the Cited2→Vegfa pathway may be beneficial in heart failure or ischaemia, where such coupled myocardial and coronary growth may be desirable. Moreover, we speculate that Cited2 may also play a necessary role in tumour angiogenesis by ensuring tumour growth proceeds with a concomitant vascular network.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by Wellcome Trust Program Grant [083228/Z/07/Z]; Wellcome Trust Core Award [090532/Z/09/Z]; Wellcome Trust Clinical Fellowship [076257/Z/04/Z]; and British Heart Foundation Chair Award [CH/09/003]. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Conflict of interest: none declared.
Supplementary Material
References
- 1.Bhattacharya S, Macdonald ST, Farthing CR. Molecular mechanisms controlling the coupled development of myocardium and coronary vasculature. Clin Sci (Lond) 2006;111:35–46. doi: 10.1042/CS20060003. [DOI] [PubMed] [Google Scholar]
- 2.Sedmera D, Pexieder T, Vuillemin M, Thompson RP, Anderson RH. Developmental patterning of the myocardium. Anat Rec. 2000;258:319–337. doi: 10.1002/(SICI)1097-0185(20000401)258:4<319::AID-AR1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 3.Luttun A, Carmeliet P. De novo vasculogenesis in the heart. Cardiovasc Res. 2003;58:378–389. doi: 10.1016/s0008-6363(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 4.Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circ Res. 2002;91:761–768. doi: 10.1161/01.res.0000038961.53759.3c. [DOI] [PubMed] [Google Scholar]
- 5.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 6.Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giordano FJ, Gerber H-P, Williams S-P, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci. 2001;98:5780–5785. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 9.Dunwoodie SL, Rodriguez TA, Beddington RSP. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech Dev. 1998;72:27–40. doi: 10.1016/s0925-4773(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin DH, Li SH, Chun YS, Huang LE, Kim MS, Park JW. CITED2 mediates the paradoxical responses of HIF-1alpha to proteasome inhibition. Oncogene. 2008;27:1939–1944. doi: 10.1038/sj.onc.1210826. [DOI] [PubMed] [Google Scholar]
- 12.Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, Yuan S, Prabhakar NR, Herrup K, Conlon RA, Hoit BD, Watanabe M, Yang YC. The essential role of Cited2, a negative regulator for HIF-1alpha, in heart development and neurulation. Proc Natl Acad Sci USA. 2002;99:10488–10493. doi: 10.1073/pnas.162371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braganca J, Eloranta JJ, Bamforth SD, Ibbitt JC, Hurst HC, Bhattacharya S. Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding Protein, and CITED2. J Biol Chem. 2003;278:16021–16029. doi: 10.1074/jbc.M208144200. [DOI] [PubMed] [Google Scholar]
- 14.Chou YT, Wang H, Chen Y, Danielpour D, Yang YC. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–5560. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- 15.Glenn DJ, Maurer RA. MRG1 binds to the LIM domain of Lhx2 and may function as a coactivator to stimulate glycoprotein hormone alpha-subunit gene expression. J Biol Chem. 1999;274:36159–36167. doi: 10.1074/jbc.274.51.36159. [DOI] [PubMed] [Google Scholar]
- 16.Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J Biol Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 17.Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- 18.Bamforth SD, Braganca J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat Genet. 2004;36:1189–1196. doi: 10.1038/ng1446. [DOI] [PubMed] [Google Scholar]
- 19.Macdonald ST, Bamforth SD, Chen CM, Farthing CR, Franklyn A, Broadbent C, Schneider JE, Saga Y, Lewandoski M, Bhattacharya S. Epiblastic Cited2 deficiency results in cardiac phenotypic heterogeneity and provides a mechanism for haploinsufficiency. Cardiovasc Res. 2008;79:448–457. doi: 10.1093/cvr/cvn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weninger WJ, Floro KL, Bennett MB, Withington SL, Preis JI, Barbera JP, Mohun TJ, Dunwoodie SL. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development. 2005;132:1337–1348. doi: 10.1242/dev.01696. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Doughman YQ, Gu S, Jarrell A, Aota S, Cvekl A, Watanabe M, Dunwoodie SL, Johnson RS, van Heyningen V, Kleinjan DA, Beebe DC, Yang YC. Cited2 is required for the proper formation of the hyaloid vasculature and for lens morphogenesis. Development. 2008;135:2939–2948. doi: 10.1242/dev.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Haviernik P, Bunting KD, Yang Y-C. Cited2 is required for normal hematopoiesis in the murine fetal liver. Blood. 2007;110:2889–2898. doi: 10.1182/blood-2007-01-066316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kranc KR, Schepers H, Rodrigues N, Bamforth S, Villadsen E, Ferry H, Bouriez-Jones T, Sigvardsson M, Bhattacharya S, Jacobsen SE, Enver T. Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell. 2009;5:659–665. doi: 10.1016/j.stem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu X, Lam E, Doughman Y-Q, Chen Y, Chou Y-T, Lam M, Turakhia M, Dunwoodie SL, Watanabe M, Xu B, Duncan SA, Yang Y-C. Cited2, a coactivator of HNF4[alpha], is essential for liver development. Embo J. 2007;26:4445–4456. doi: 10.1038/sj.emboj.7601883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Withington SL, Scott AN, Saunders DN, Lopes Floro K, Preis JI, Michalicek J, Maclean K, Sparrow DB, Barbera JP, Dunwoodie SL. Loss of Cited2 affects trophoblast formation and vascularization of the mouse placenta. Dev Biol. 2006;294:67–82. doi: 10.1016/j.ydbio.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 26.Xu B, Qu X, Gu S, Doughman Y-Q, Watanabe M, Dunwoodie SL, Yang Y-C. Cited2 is required for fetal lung maturation. Dev Biol. 2008;317:95–105. doi: 10.1016/j.ydbio.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2–5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 28.Schneider JE, Bose J, Bamforth SD, Gruber AD, Broadbent C, Clarke K, Neubauer S, Lengeling A, Bhattacharya S. Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev Biol. 2004;4:16–27. doi: 10.1186/1471-213X-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams PS. Data analysis and reporting. In: Dorak TM, editor. Real-time PCR. New York: Taylor and Francis Group; 2006. pp. 41–62. [Google Scholar]
- 30.Perneger TV. What's wrong with Bonferroni adjustments. BMJ. 1998;316:1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brenneisen P, Blaudschun R, Gille J, Schneider L, Hinrichs R, Wlaschek M, Eming S, Scharffetter-Kochanek K. Essential role of an activator protein-2 (AP-2)/specificity protein 1 (Sp1) cluster in the UVB-mediated induction of the human vascular endothelial growth factor in HaCaT keratinocytes. Biochem J. 2003;369(Pt 2):341–349. doi: 10.1042/BJ20021032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clifford RL, Deacon K, Knox AJ. Novel regulation of vascular endothelial growth factor-A (VEGF-A) by transforming growth factor (beta)1: requirement for Smads, (beta)-CATENIN, AND GSK3(beta) J Biol Chem. 2008;283:35337–35353. doi: 10.1074/jbc.M803342200. [DOI] [PubMed] [Google Scholar]
- 33.Gille J, Reisinger K, Asbe-Vollkopf A, Hardt-Weinelt K, Kaufmann R. Ultraviolet-A-induced transactivation of the vascular endothelial growth factor gene in HaCaT keratinocytes is conveyed by activator protein-2 transcription factor. J Invest Dermatol. 2000;115:30–36. doi: 10.1046/j.1523-1747.2000.00020.x. [DOI] [PubMed] [Google Scholar]
- 34.Gille J, Swerlick RA, Caughman SW. Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2-dependent DNA binding and transactivation. EMBO J. 1997;16:750–759. doi: 10.1093/emboj/16.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopes FL, Desmarais J, Ledoux S, Gevry NY, Lefevre P, Murphy BD. Transcriptional regulation of uterine vascular endothelial growth factor during early gestation in a carnivore model, Mustela vison. J Biol Chem. 2006;281:24602–24611. doi: 10.1074/jbc.M602146200. [DOI] [PubMed] [Google Scholar]
- 36.Ruiz M, Pettaway C, Song R, Stoeltzing O, Ellis L, Bar-Eli M. Activator protein 2alpha inhibits tumorigenicity and represses vascular endothelial growth factor transcription in prostate cancer cells. Cancer Res. 2004;64:631–638. doi: 10.1158/0008-5472.can-03-2751. [DOI] [PubMed] [Google Scholar]
- 37.Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D'Amore PA. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- 38.Toyoda M, Shirato H, Nakajima K, Kojima M, Takahashi M, Kubota M, Suzuki-Migishima R, Motegi Y, Yokoyama M, Takeuchi T. jumonji downregulates cardiac cell proliferation by repressing cyclin D1 expression. Dev Cell. 2003;5:85–97. doi: 10.1016/s1534-5807(03)00189-8. [DOI] [PubMed] [Google Scholar]
- 39.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 40.Erokhina EL. Proliferation dynamics of cellular elements in the differentiating mouse myocardium. Tsitologiia. 1968;10:1391–1409. [PubMed] [Google Scholar]
- 41.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 42.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 43.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 44.Lopes Floro K, Artap ST, Preis JI, Fatkin D, Chapman G, Furtado MB, Harvey RP, Hamada H, Sparrow DB, Dunwoodie SL. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum Mol Gen. 2011;20:1097–1110. doi: 10.1093/hmg/ddq554. [DOI] [PubMed] [Google Scholar]
- 45.Kranc KR, Bamforth SD, Braganca J, Norbury C, Van Lohuizen M, Bhattacharya S. Transcriptional coactivator Cited2 induces Bmi1 and Mel18 and controls fibroblast proliferation via Ink4a/ARF. Mol Cell Biol. 2003;23:7658–7666. doi: 10.1128/MCB.23.21.7658-7666.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bry M, Kivela R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, Carmeliet P, Lemstrom KB, Shibuya M, Yla-Herttuala S, Alhonen L, Mervaala E, Andersson LC, Knuuti J, Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.