

Abstract
Marrow Stromal Cells (MSCs) are relatively rare cells difficult to visualize in marrow biopsies or detect in aspirated marrow. Under specific conditions MSC can be expanded in vitro and the population can give rise to several mesenchymal lineages. “MSC” also refers to mesenchymal stem cells which implies that all cells in the population are multipotent. It is generally agreed that while there may be a few multipotent stem cells in an MSC population the majority are not stem cells. In either case MSC do not produce hematopoietic cells. Although MSCs have been isolated and characterized from several tissues, bone marrow is their most common source for research and clinical use. Primary MSC populations can be derived from bone marrow mononuclear cells with relative ease, but it is important to recognize the cellular heterogeneity within a culture and how this may vary from donor to donor. In this chapter, we will describe methodology to derive primary MSCs from bone marrow screens, an otherwise discarded byproduct of bone marrow harvests used for clinical transplantation. We will also describe some useful techniques to characterize and manipulate MSCs – both primary and immortalized cell lines.
Keywords: Marrow Stromal Cells (MSCs), bone marrow-screen, reverse-transfection, FACS, AutoMACS, CD146, siRNA, miRNA, LTC (Long Term Culture)
1. INTRODUCTION
Stromal cells as an integral component of the the microenvironment (ME)
Maintenance of normal hematopoiesis at all stages of ontogeny requires a complementary microenvironment (ME), which in adult vertebrates resides within the bone marrow(1, 2). Early evidence for the importance of the ME came from experiments in naturally occurring mutant mice. The compound heterozygote SL/SLd mouse could be rescued from effects of low dose radiation by the transplantation of a wild-type spleen but not by wild-type hematopoietic cells suggesting that the defect lies in the ME (the soil) rather than the the hematopoietic stem/progenitor cell (HSPC, or the seed)(3). The SL locus was subsequently discovered to encode for Kit ligand ( KITL or stem cell factor, SCF), a cytokine that is produced by the ME with non-redundant regulatory functions for HSPC maintenance(4, 5). Despite early enthusiasm that KITL might be the critical ME-derived gene product that defines the hematopoietic ME, it has since then become abundantly clear that hematopoietic regulation by the ME is enormously complex with contribution of several cell types and dozens of secreted or surface-bound factors(6). Some of these factors such as CXCL12, SCF and ANGPT1 have non-redundant functions while others such as the Notch and Wnt ligands are redundant, as evidenced by murine gene knock-out models (7–14). These factors are typically not restricted in origin to a single cell type within the ME, further complicating attempts to precisely define the cellular and anatomical location of specific functional niches within the ME(6, 15, 16).
It is now generally accepted that cells of mesenchymal origin such as the osteoblast, endothelium, fibroblast-like stromal cells, and adipocytes as well as cells of hematopoietic origin such as macrophages, osteoclasts and megakaryocytes functionally contribute to the regulation of the HSPC and its subsequent progeny within the ME. The terms “stroma” or “stromal cells” have been historically used to denote the fibroblast-like cells of mesenchymal origin found in primary bone marrow long term cultures (LTCs as detailed later). Precise demarcation of stroma vs. other cells of mesenchymal origin (such as osteoblasts) is problematic with immune-phenotypic techniques given overlap of surface markers and incomplete understanding of different stages of their differentiation from a putative common precursor in vivo. While fibroblast-like stromal cells are best appreciated in in vitro cultures where they proliferate luxuriantly in serum-rich media to form adherent layers, they are more difficult to define in vivo due to (1) their much smaller numerical proportion in comparison to the rapidly proliferating hematopoietic cells and (2) their thin and pleomorphic morphology that renders direct visualization of cells in bone marrow sections challenging, but not impossible with specific stains. Consequently, most studies of stromal cells until recently have been in the in vitro system. Use of genetically modified mouse models using tissue-specific promoters (such as osterix and nestin promoters) and surface markers such as CD146 ( in human primary samples) have been reported in the past few years and have significantly accelerated our understanding of stromal cells and their function in vivo(17–19).
MSC: Misleading Misnomer
Most of the initial interest in these cells after their initial description by Dexter centered around the mechanistic basis of their interaction between hematopoietic cells and how they support hematopoiesis (1, 20). The mid-1990s, however, witnessed a significant interest in stromal cells, which came to be denoted inappropriately as mesenchymal stem cells (MSC), for a wide spectrum of clinical uses ranging from regeneration of damaged tissues like heart and liver to immune modulation of allogeneic graft versus host disease(21–24). Most of these anecdotal observations failed to translate to tangible benefits in larger trials which has dampened enthusiasm of this mode of cellular therapy. Nevertheless some investigators are committed to understanding how infusion of cultured stromal cells, which usually are completely mismatched to the recipient, and rapidly filtered out of systemic circulation by the lungs, could influence tissue regeneration or allo-immune reactions in select clinical situations (25–28). In 2001, a report describing the existence of rare multi-potent adult progenitor cells ( MAPCs) with embryonic stem cell-like potential in MSC populations (29–31). The MAPC had the ability to “transdifferentiate” into multiple tissues but also to revert back to embryonic potential and contribute to all three germ layers. This obviously caused frenzied excitement as the existence of MAPC could eliminate the need for harvesting blastocysts. The existence of MAPCs have since then been largely disavowed and there is general consensus that stromal precursors are not capable of differentiation to tissues outside traditionally recognized mesenchymal lineages, unless deliberately reprogrammed by the forced expression of embryonic transcription factors. (32–37). A comprehensive review of the history and controversies surrounding clinical applications of MSCs and MAPCs is outside the scope of this manuscript and the reader is referred to several papers for further details (25–27, 29, 31, 32, 36, 38).
Defining MSC
MSCs are typically defined as cells that possess the ability to form Colony Forming Unit -Fibroblast (CFU-F) and differentiate into multiple mesenchymal cell types (including, osteoblasts, chondrocytes, adipocytes, and bone marrow stromal cells) under appropriate in vitro culture conditions(39, 40). Although it is generally accepted that these mesenchymal lineages likely originate from common precursors ( much in the same way that a single self-renewing hematopoietic stem cell can self-renew and give rise to mature hematopoietic lineages), unequivocal evidence for such a single common precursor that repopulate all mesenchymal lineages has been lacking (38). This is due to both the obvious difficulty in depleting all mesenchymal cells from a host and the difficulty in reliably transplanting a single stromal cell into such a depleted host. Although only a small portion of the stromal cells from fresh bone marrow isolates or early bone marrow cultures would be considered true MSCs, the term is often inappropriately applied to all cells in a marrow-derived stromal culture.
Given the heterogeneity of the stromal population, several groups have attempted to enrich for the CFU-F population using cell surface markers. The first effort reported was the generation of the murine monoclonal antibody STRO-1, an IgM antibody specific for an undefined polysaccharide motif. STRO-1 binds to approximately 10% of bone marrow mononuclear cells (BMMNC), most of which are nucleated erythroid cells(41). When nucleated erythroids are excluded by Glycophorin A positivity, STRO-1 positive cells define a population that is 100 fold enriched for CFU-F. Furthermore, STRO-1 can be easily identified in the stromal cell layers of the long term culture. Despite widespread use of this antibody to enrich for CFU-Fs, an obvious limitation is its presence on hematopoietic cells, preventing use in bone marrow sections or mixed primary long term cultures. Over the past two decades, several groups have tried to extend on this work by attempting to isolate MSC using various cell surface markers including CD49a, CD63, CD90, NGFR, CD105, CD106, CD140b, CD146 (17, 42–47). Of these markers, only cells that express high levels of CD146 have been shown to transfer a functional hematopoietic ME when transplanted into immunodeficient mice(17). This antibody can also be used to identify progenitors in-vivo by immune histochemistry (see figure) and our group has found this to be a useful marker to enrich for MSC (details of protocol explained later)(48).
MSC Cultures
Although culture conditions can be varied to promote stromal growth over hematopoietic growth resulting in further enrichment of fibroblast-like cells, they neverhteless remain highly heterogeneous and contaminated with cells of hematopoietic origin, most commonly macrophages. These cultures are typically referred to as MSC cultures. Setting up primary MSC cultures is relatively straight forward, but one must expect wide variation among donors and between cultures from the same donor. Differences in growth kinetics and relative proportions of different cell types are common, as is the ability to support different hematopoietic stages and lineages. MSC quality is also influenced by viral infections like Cytomegaloviruses (CMV), and technical issues which are often unavoidable, including a delay in processing, or a significant contamination by erythroid cells necessitating hypotonic hemolysis of samples(49). Even adjusting for these factors, the phenotypic and functional characteristics of these cells are difficult to predict. Cloned stromal cell lines have circumvented many of these problems; however, results from cell lines must be reproduced in primary cells to be considered valid. While cell lines are readily available, descriptions of current techniques to isolate primary cells and manipulate them in vitro are not as available.
Most tissues that have discernible connective tissue components have mesenchymal progenitors that can be grown in cultures that resemble MSC.(38). Bone Marrow has remained the most commonly used and best studied source of MSCs due to relative ease of access by bone marrow aspiration. Although normal bone marrow aspirates can be obtained by volunteer donations or remunerated study participants, this is often problematic for obvious reasons. Although relatively safe, bone marrow aspirates are painful and invasive and major complications like severe hemorrhage from a severed aberrant blood vessel or damage to internal organs have occurred. An alternate strategy that our group and others has successfully used for several years is to salvage cells from bone marrow screens used to prepare marrow for clinical transplantation. The shift away from bone marrow mononuclear cells to peripheral blood mononuclear cells (PBMC) as the preferred source of stem cells for clinical use has meant that the number of bone marrow harvests has reduced drastically in the past two decades. Bone marrow harvests however continue to be performed for specific indications ( like pediatric donors or for donor conditions like sickle cell trait or as part of study protocols that require bone marrow as a source for the stem cells) and hence result in a respectable number of marrow screens available for extraction of Bone Marrow Mononuclear Cells (BMMNC) in most larger centers. The methods described in this manuscript will focus on the use of bone marrow screens for MSCs; but can obviously be used for BMMNCs from a fresh marrow aspirate should that be available.
2. MATERIALS
2.1 IRB approval and personnel training
As with any other work involving biological materials, appropriate training of personnel, and up-to-date institutional approval of protocols is essential. Institutional Review Board (IRB) approval has to be in place before the work commences. Depending on the particular institution, the investigators may be eligible for expedited review and human subjects exclusion given that the screens may be provided after removing personal identifiers and that the material is otherwise discarded. Proper training and certification of personnel in handling of bio-hazardous materials is similarly critical given that the marrow screens could be a source of infection although donors are typically tested extensively prior to marrow harvest. (See Note 1)
2.2 Cell and Tissue Culture Set up
Proper cell and tissue culture techniques and equipment is of critical importance in any tissue culture methodology, but especially so for primary MSC cultures given that they could be contaminated at multiple steps of their set up and maintenance, and the relatively long periods (weeks to months) that they may need to be maintained. Most laboratories which perform tissue culture are set up with biosafety laminar flow cabinets ( or TC hoods), incubators that can maintain temperature, CO2 content and humidity (TC incubator), bench-top centrifuges to spin down cells, vacuum traps, water baths, standard inverted microscopes ( for cell counting and monitoring of cell growth) etc. The set up and maintenance of these are hence not discussed in detail here.
2.3 Bone Marrow Screens
Bone Marrow Screens are filters used to remove particulate materials including marrow spicules, fat globules and small clots from the bone marrow aspirated from donors for the purpose of transplantation (Figure 1). They can be procured from the cell processing or apheresis laboratories of clinical centers where bone marrow aspirates are processed and can be provided without any patient identifiers other than age, sex and other important pertinent clinical parameters like cytomegalovirus (CMV) infection status. If personal identifying information is excluded, this will typically allow for the samples to be considered exempt from human subject research allowing for less stringent IRB protocols). Given that the screens are clinical grade products, they are typically sterile and contamination of its contents unlikely at the time of processing in the clinical laboratory. General aseptic precautions at the time of transfer to the research laboratory would suffice. Typically two screens of different pore size ( 500 and 200 microns) are serially connected to filter the marrow. These can be attached to each other to form a sterile loop at the time of transfer avoiding spillage of contents and contamination (Figure 2A) attached to each other. If your laboratory is in the process of setting up a working relationship with the clinical cell processing laboratory for purposes of procuring these screens, it would be worthwhile to explain in detail to the facility supervisor and the staff, the scientific basis of your studies and the practical details of what you intend to do with the screens as this will allow for expeditious transfer. (See Note 2)
Figure 1. Schematic of Bone Marrow Harvest and Screens.
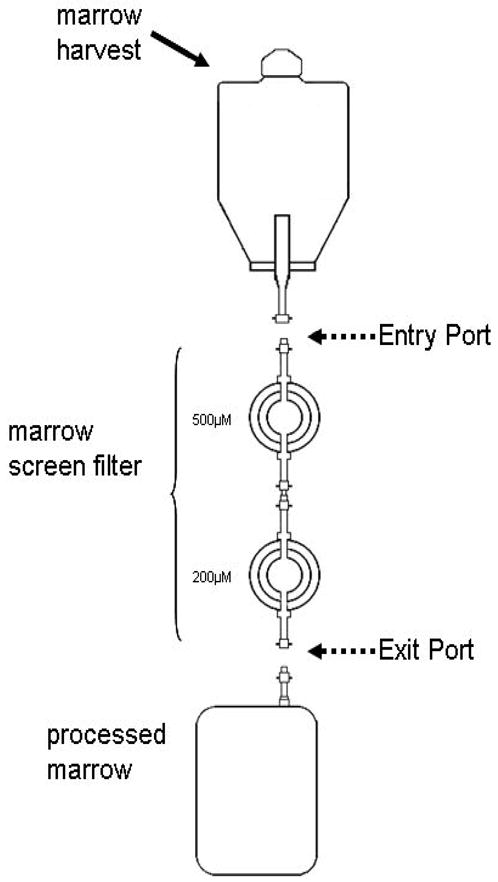
The bone marrow aspirate collected from donors is hooked up typically to two filters called screens connected in series ( 500 μM first followed by 200 μM) to filter out particulate matter including spicules, clots and tissue fragments. The filtrate is collected for infusion ( processed marrow). The marrow screens are then made available to research laboratories with the entry port and exit connected to each other.
Figure 2. Extraction of bone marrow mononuclear cells (BMMNC) from marrow screens.
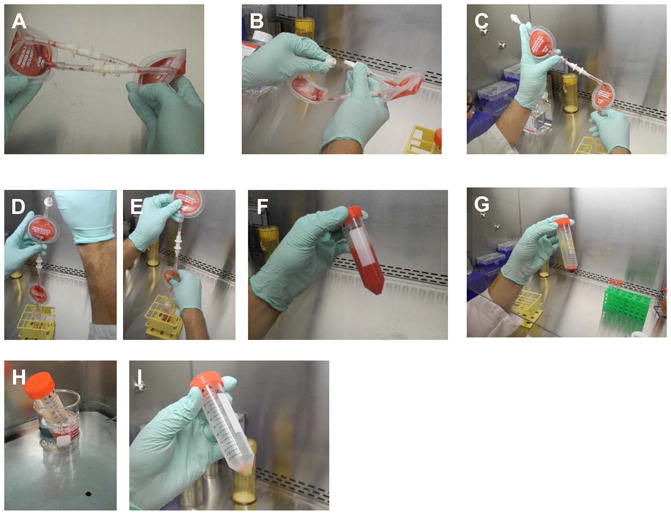
A: Screens are typically provided with the two filters (500 μM and 200 μM) connected in series as a sterile loop.
B. Carefully disengage the entrance port of the 500 μM filter from the exit port of the 200 μM filter.
C: Pour any liquid contents left in the screen to a 50 cc conical tube.
D: Inject 25 cc of PBS-EDTA into the entrance port of the 500 μM filter and collect the run through carefully.
E: Try to dislodge any particulate material in the filters into the PBS-EDTA solution
F: Approximately 50 cc of bone marrow particular material is eluted into a 50 cc conical tube.
G: The cell pellet after the first wash is rich with erythroid cells which need to be mostly removed before plating in culture.
H: Hemolysis of collected cells in hemolysis buffer at 37 degrees.
I: Post-hemolysis and a further wash in PBS-EDTA, cell pellet is pale and has little visible erythroid contamination.
2.4 Human Stromal Cell lines
Various groups have reported on immortalized stromal cell lines, typically generated using retroviral vectors. Our group has previously reported on the use of human stromal cell lines isolated after immortalization with retrovirally expressed human papilloma virus (HPV) E6/E7 proteins; two cell lines termed HS5 and HS27a have been used to represent functionally distinct stromal niches that exist within the larger context of the adult vertebrate marrow microenvironment(50). HS5 represents a stromal phenotype that secretes large quantities of cytokines thus driving differentiation of hematopoietic stem and progenitor cells (HSPC) to mature myeloid lineages. HS27a in contrast produces high levels of HSPC-niche associated genes (such as CXCL12, Jagged1 and Angiopoietin1) that help maintain HSPC in their primitive undifferentiated states. (See Note 3)
2.5 Flow Cytometry and sorting of stromal cells based on CD146 expression: Flow sorters and MACS sorters
Flow-based sorting as described here for CD146 expression is typically available in large research centers where well-trained staff perform the sorting using shared instruments. (See Note 4)For magnetic Bead sorting for enrichment of stromal precursors by removal of CD14 and CD45 positive cells, we have utilized proprietary magnet-conjugated antibodies from Miltenyi Biotec (see Note 5).
2.6 Media for MSC growth and LTC growth
Human MSC can be grown in a variety of media formulations as long as they are replete with serum. These include alpha alpha Minimal Essential Media, Dulbecco’s Modified Eagle’s Media (DMEM) and RPMI-1640. DMEM commonly comes in low glucose or high glucose variants and low glucose formulations are routinely used by most groups for propagating MSC. Although the other base media formulations have differences in components, in our anecdotal experience, they can be used interchangeably as long as they are serum supplemented. Fetal Bovine Serum (FBS) supplemented at 10 to 20% final volume is considered essential for good proliferation of all fibroblast cultures including bone marrow derived stromal cells. FBS is known to have high concentrations of several fibroblast mitogens (from the PDGF and FGF family). (See Note 6) Serum free formulations with defined chemical additives such as cytokines and small molecules, as have been developed for hematopoietic colony growth, would be an important technical advancement for the field; however at this time FBS remains a necessary supplement for MSC cultures. Addition of antibiotics such as Penicillin-Streptomycin and antifungal agents such as amphotericin B should be considered, the latter especially in climates where mold contamination is known to occur in cultures that have to be maintained for several weeks.
Media for primary Long Term Culture (LTCs) is typically composed of Iscoves’s media supplemented with screened horse serum (HS) in addition to FBS and several other additives (See Note 7). Although the human stromal cell lines Hs27a and Hs5 were initiated in LTC media they can be grown in RPMI-1640 supplemented with 10% FBS.
2.7 Media Formulations and other solutions
Fetal bovine serum (FBS) and Horse Serum (HS)
Thaw frozen FBS/HS at 4°C overnight. Heat inactivate FBS/HS by incubating at 56°C for 30 minutes to inactivate complement. Filter sterilize using 0.45 u bottle filter ( make sure that to agitate the serum while in the filter chamber to prevent clogging of filter pores by clots. Dispense into appropriate size aliquots (50ml conicals or 100 mm bottles) working aliquots and freeze at −20°C.
Bovine Serum Albumin, 10% solution
Dissolve 100g fraction V BSA in 1 liter of deionized water. Filter sterilize using a low-protein-binding 0.45-um filter. Store in 50 to 100ml aliquots at −20°C.
MSC Medium (DMEM based; components purchased from standard suppliers as GIBCO or Fisher Scientific)
50 ml of pre-screened FBS
5 ml of 100X Penicillin/Streptomycin
5 ml of 100X Amphotericin B (optional)
DMEM (low glucose, replete with pyruvate and glutamate ) to a total of 500 ml.
Mix components and filter sterilize with 0.45 uM bottle filter, store at 4°C for use in less than 3 months.
LTC Medium
62.5 ml pre-screened FBS (screened to support LTC growth) to a final concentration of 12.5%
62.5 ml pre-screened HS (screened to support LTC growth) to a final concentration of 12.5 %
5 ml of 100 X Penicillin-Streptomycin
5 ml of 100X Amphotericin B (optional)
5 ml of L-glutamine
5 ml of Pyruvate
250 ml of 2X Isocoves Modified Dulbecco’s Medium (IMDM) prepared from powder (GIBCO).
500 ul of 103 M hydrocortisone stock (Final concentration 106 M)
Distilled tissue culture grade water (endotoxin free) - add to total of 500 ml.
Mix all components and filter sterile with 0.45 micron filter and store at 4°C for up to 3 months. Can also be frozen at −20°C for future use if larger volumes are made.
Stromal Cell Line Medium (HS5 and HS27a)
50 ml of pre-screened FBS
5 ml of 100X Penicillin/Streptomycin
445 ml of RPMI-1640 (replete with pyruvate and glutamate )
Mix components and filter sterilize with 0.45 micron bottle filter, store at 4°C for use in less than 3 months.
Solutions and Buffers
10 X PBS
| NaCl | 800 g |
| KCl | 20g |
| Na2HPO4 *2H2O | 144 g |
| KH2PO4 | 24g |
Dissolve in 10 liters of Deionized water, filter sterilize and store at 4°C. The pH is approximately 6.8 at this time. To make working 1X solution, take 100ml of 10x stock and add 900ml of deionized water. The pH should adjust to 7.4 with dilution and this should be verified with pH meter.
10x HBSS (Hanks’ balanced salt solution)
| NaCl | 80g |
| KCl | 4g |
| Na2HPO4·7H2O | 0.9g |
| KH2PO4 | 0.6g |
Add deionized water to 1 liter and filter sterilize. Store at 4°C.
To make 1x working stock, mix 100ml of 10x stock with 800 ml of deionized water. Add 0.35 g NaHCO3 and 1grma of D-glucose. Bring volume upto 1 liter and adjust pH to 7.4. Store at 4°C.
EDTA (ethylenediaminetetraacetic acid), 0.5 M (pH 8.0)
Dissolve 186.1 g disodium EDTA dihydrate in 700 ml water.
Adjust pH to 8.0 with 10 M NaOH (~50 ml; add slowly).
Add water to 1 liter and Autoclave. To make 2mm working stock, add 1ml to of 0.5M EDTA to 250mL of PBS.
AUTOMACS/FACS BUFFER (PBS with 2mM EDTA, 0.5% BSA)
To 473 ml of 1x PBS, add 2 ml of 0.5 M EDTA, and 25ml 10% BSA solution.
PBS-EDTA
To 500 ml 1X PBS, add 2 ml of 0.5M EDTA
Citrate Saline Solution (For non-enzymatic cellular dissociation)
50.3 gm of KCl
22.06 gm of sodium citrate
Dissolve in tissue culture grade water to total of 500 ml ( for a 10X solution). Filter sterilize or autoclave and store at 4°C.
Make 1X solution (final concentrations of KCl is 135 mM and sodium citrate is 15 mM) as needed for use with tissue culture grade water.
** Various commercial non-enzymatic preparations (from vendors such as millipore and sigma-aldrich) and also available
Hemolytic Buffer
10X stock solution (1000 ml)
| NH4Cl8 | 3.0 g |
| NaHCO3 | 10.0 g |
| EDTA | 0.4 g |
Dissolve salts in approximately 500 ml sterile water and after all the salts have dissolved, make up the volume to a total of 1000 ml. Filter sterile with 0.45 micron filter and store at 4°C. Reconstitute 1X solution (typically not more than 50 ml at one time) as needed.
Other Solutions and Buffers
Trypsin-EDTA (0.25 % or 0.05 % trypsin and 1 mM EDTA) available from various vendors
Phosphate Buffered Saline (PBS)
PBS-EDTA (addition of EDTA to 2 mM final concentration)
Ficoll Solution (1.073 or 1.077 gm/ml) from various vendors.
Antibodies
CD45 microbeads, Miltenyi Biotech, catalog #130-045-801
Isotype: Mouse IgG2a
CD14 microbeads, Miltenyi Biotech, catalog #130-050-201
Isotype: Mouse IgG2a
CD146 microbeads, Miltenyi Biotech, catalog #130-093-596
Isotype: Mouse IgG1
FITC Mouse anti-Human CD146, BDBiosciences, catalog # 560846
Clone: P1H12, Isotype: Mouse IgG1, κ ( same clone is also available from other vendors including ebiosciences)
Reverse Transfection reagents
OptiMEM Media ( Life/GIBCO catalogue # 31985-070 )
Lipfectamine 2000 Reagent ( Life/GIBCO catalogue # 11668019)
RPMI-1640 with 10% FBS without antibiotics.
3. METHODS
3.1 Isolating bone marrow cells from marrow screens
As mentioned above, it is absolutely critical to maintain a sterile working environment and minimize contamination of the cell preparation at each step of this protocol given that the cultures need to be maintained for several weeks in nutrient rich media and hence antibiotics/antifungal supplementation of culture media alone will be insufficient to prevent contamination by microbes.
-
1
In the Aliquot approximately 50 cc of PBS with EDTA in a 50 ml conical tube.
-
2
Using a 18 guage needle connected to a 50 cc syringe, aspirate the PBS-EDTA to the syringe. Keep by the side for later use.
-
3
Spray down the Screen with 70% ethanol in the TC hood and as mentioned above, screens are provided typically attached to each other as a loop (Figure 2A), this helps to keep the contents sterile and preventing spillage of contents. Disengage the screens from each other by opening the connectors (Figure 2B) taking care not to spill the contents of the screen. Carefully open the connector such that the liquid contents can be poured out of the inlet of the larger pore sized filter into a 50 cc conical tube. (for example, if a 500 micron and 200 micron filter are connected in series, pour out of the inlet of the 500 micron filter, Figure 2C). This way the larger trapped particles of bone spicules could be harvested.
-
4
Attach the 50 cc syringe directly to the connector of the screens or through the 18 gauge and inject 25 ml of PBS-EDTA taking extreme care not to let the PBS-marrow mix to spill out. Collect the flow through in the 50 cc conical tube ( Figure 2D and E)). Alternatively, one can reconnect the two screens and swish the contents to dislodge of the filters into the PBS-EDTA solution ( for about 1–2 minutes) and pour the contents into the 50 cc conical tube.
-
5
Repeat the wash process ( step 4) once for a total of about 50 cc wash out (Figure 2F).
-
7
Centrifuge the wash out in an appropriate table-top centrifuge for 5 minutes at 4 degree Celsius to obtain a cell pellet (Figure 2G). Carefully aspirate the supernatant and immediately re-suspend in either PBS-EDTA or in hemolytic buffer (to remove erythroid cells as described in 3.2 or 3.3). (See Note 8) Further processing of the cell pellets can be done by either direct hemolysis or density gradient separation. Getting rid of the erythroid cells is necessary as lysis of erythroid cells in culture is known to inhibit adequate stromal growth. In our experience, direct hemolysis is usually sufficient for isolation of bone marrow cells for MSC/LTC cultures since the hemolyzed sample can be directly plated to culture without further delay. If the isolated bone marrow cells are to be used for other purposes such as flow-sorting or there is a delay in further use of the cells, the polymorphonuclear cells (PMNs or mature neutrophils) could cause clumping of cells. Both protocols are hence described below.
3.2 Hemolysis of Bone Marrow Washout
Add approximately 5 ml of 1X Hemolytic buffer to the bone marrow pellet after the pellet is loosened by gentle “racking” ( running across a plastic rack used for holding 15 cc conical tubes).
Incubate in a 37 degree water bath. Gently agitate every 30 seconds by swirling the contents of the tube monitoring for a change in color of the contents from “blood red” to “wine red” and relative clearing of contents (Figure 2H). Limit total hemolysis time to not more than 5 minutes to avoid cell clumping and lysis of non-erythroid cells.
Bring tube back to the TC hood, add 45 cc of PBS-EDTA to the tube to make the volume up to 50 cc. Spin at 400 g force for 5 minutes.
Examine the tube contents for the adequacy of hemolysis (both supernatant and cell pellet). (See Note 9)The final pellet after adequate hemolysis would be pale and devoid of frank erythroid presence (Figure 2I)
3.3 Ficoll Density gradient Separation to Yield BONE MARROW mononuclear cells (BMMNC)
Density gradient separation based on the polysaccharide Ficoll to separate whole blood or bone marrow to different components is a useful technique for isolation of MSCs and stromal precursors since they are contained in the mononuclear fraction at the interphase between ficoll and diluted bone marrow. Layering of blood or marrow on ficoll is a delicate technique and it is recommended that it is learnt from those proficient with the technique and practiced on more easily obtainable samples like peripheral blood before attempting on limited resources like bone marrow aspirate. Ficoll density (g/cm3) 1.077 is classically used for BMMNC preparation from human bone marrow. (see Note 10) Also important to remember is that the gradient separation is not absolute and does result in the loss of BMMNC (including the stromal precursors) to other fractions. Hence if the sample is limited ( such as part of a clinical procurement) and to be used for direct plating for MSC cultures, it is recommended that one consider direct hemolysis instead of ficoll gradient separation.
-
1
Move Ficoll from 4 degree fridge to TC hood at least 30 minute before separation since Ficoll density is sensitive to temperature. Do not warm in a 37 degree water bath. To expedite equilibration to room temperature, one can aliquot the Ficoll to the Falcon tubes about 30 minute before separation (Step 3 of this protocol).
-
2
Dilute bone marrow with three volumes of medium HBSS. Diluting the blood or BM increases the yield of mononuclear cells and decreases the hang-up of RBCs.
-
2
Place 3.0 ml ficoll into each clear 15 ml round bottom tubes (Falcon 2057). As noted above, it is critical to have ficoll should be at room temperature (RT) since the density is temperature dependent.
-
3
Tilt the tube to wet the side with ficoll then gently layer 6–8 ml of the diluted bone marrow on top (Figures 3A–D). Pre-wetting the side of the tube helps the bone marrow flow smoothly down the side and prevents breaking the interface.
-
4
Centrifuge tubes at 400g force for 30 minutes at RT. Mononuclear cells will appear as a white cloud at the interface and red cells will be pelleted at the bottom of the tube (Figure 3E).
-
5
Collect the interface (mononuclear cells) on top of the ficoll with a cotton-stuffed pasteur pipet (Figures 3F–J). Single use sterile plastic pipettes could also be used. Pool the interface cells from two layers into a labeled 15 ml V tube, fill tube up with HBSS to dilute the ficoll and wash it off the cells by centrifuging at 400 g force for 10 minutes, RT to pellet cells. (This is the first wash).
-
6
Aspirate off supernatant and resuspend the pellet by tapping the bottom of the tube, or by gently dragging the tube across the top of a test tube rack (“racking”). Wash the cells with 10–15 ml HBSS or;
-
7
If there is visible RBC contamination of the pellet, lyse the RBCs by following the protocol as detailed in 3.2
-
8
Then fill up tube with HBSS and centrifuge tube at 1200 rpm, 5 minutes. This slow spin keeps platelets from pelleting.
-
9
Consolidate cells to a single 15 ml tube, get a cell count and wash again as in step 8.
-
10
Cells can now be counted prior to plating in culture.
Figure 3. Extraction of bone marrow mononuclear cells (BMMNC) by ficoll gradient separation.

A: 3ml of ficoll at room temperature is aliquoted in to 15 cc Falcon tubes
B–D: 6–8 mf of dilute bone marrow is carefully layered on the ficoll layer taking care not to mix the two layers up.
E: After centrifugation at 400 g for 30 minutes at room temperature, different layers become apparent. The mononuclear cells are trapped in the layer between ficoll and serum.
F–J: With a sterile glass pipetted (stuffed with cotton at proximal end for seal), the mononuclear layer is carefully collected minimizing suction of both the ficoll layer between and the serum layer above.
3.4 Setting up Human Marrow Long Term Cultures (LTCs)
Primary Long Term Cultures ( LTCs or Dexter cultures) as originally described by Dexter in 1977 is a marrow derived primary culture that supports both hematopoietic and stromal growth and has been used as an in vitro approximation of the complex cellular interactions in vivo for decades (20). The culture media is hence optimized for hematopoietic growth in addition to stromal growth. In the course of days to several weeks after setting up LTCs, stromal elements become confluent and sustain primitive hematopoietic precursors within stromal layers (seen as stacked phase-dark cells in phase contrast microscopy and are referred to as cobble stone areas or CSAs (long arrows in Figure 4A and B)). As these progenitors divide and mature, they are released to the media, seen as phase-light cells (short arrows in Figures 4A and B). The cultures can be sustained by “demi-depletion” by which half the volume of media is removed every week or so allowing to remove floating mature hematopoietic precursors, replenishing nutrients while not completely depleting the cytokine milieu created by the stromal cells. In contrast to MSC cultures, LTCs are typically not split at confluence since the complex cellular interactions between hematopoietic and stromal elements would be perturbed and not easily reconstituted in the next passage.
Figure 4. Primary Long Term Cultures (LTCs or Dexter Cultures).
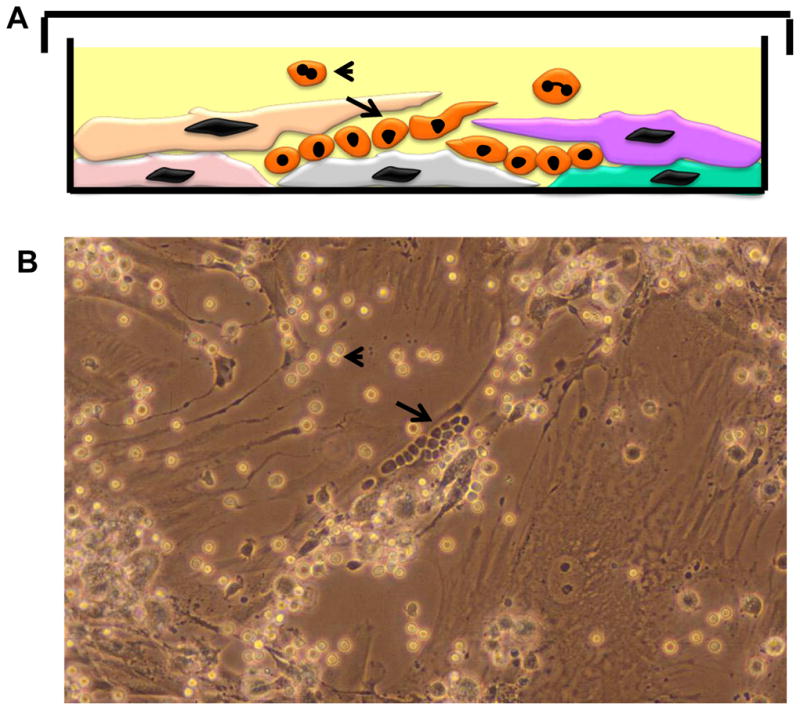
A: Cartoon depicting vertical layer of an LTC with stromal and hematopoietic elements.
B: Phase contrast micrograph of an LTC.
Long arrows in both panels depict the cobble stone areas (CSAs) comprised of primitive hematopoietic precursors trapped within the stromal layers and appear as “phase-dark” cells resembling a cobble-stone. Mature myeloid cells are released into the supernatant when the more primitive precursor cells in the CSAs divide and mature ( short arrow in both panels) and appear as “phase-light cells” in phase-contrast micrographs.
Prepare BMMNC with techniques described in sections 3.2 (typically adequate) or 3.3
- Count cells and set up in TC flasks/plates in LTC medium per the following scheme:
T75 Flask (or 100 mm dish): 15–20 × 106 cells in 12 ml T25Flask: 5–10 × 106 cells in 5 ml 6 well plate: 3 × 106 cells in 2–3 ml/well 24 well plate: 1 × 106 in 1 ml/well Culture cells at 37°C, 5 % CO2, feeding every 7 days by demi-depleting the media ( removing about half of the spent medium which contains non-adherent monocytes and myeloid cells) and replacing with equal volume of fresh LTC medium. A confluent culture of fibroblasts, macrophages, and adipocytes should be formed in 10–14 days and cobblestone areas of developing myeloid cells within the layer may also be visible. Non-adherent cells can be assayed at feeding time (by counting with hematocyometer of flow-cytometry for particular hematopoietic markers). LTCs can typically be maintained for several weeks to months before they decline.
3.5 Setting up and passaging of human MSC (hMSC) cultures
Protocol for hMSC cultures are similar to that of LTCs, except that the media used is is DMEM replete with 10% FCS.
Plate cells in densities as described above for LTCs.
Fibroblast-appearing MSCs should be evident 24–48 hours after plating although the preponderance of non-adherent hematopoietic cells might make initial visualization difficult.
About 48 hours after plating, remove supernatant cells by suctioning (with sterile precautions) in the tissue culture hood. The cultures may be rinsed with HBSS if the hematopoietic cells form clumps. Refeed with equal amount of fresh media.
CFU-Fs become rapidly evident in the next 3–5 days (Figure 5A) and cultures become confluent in about 2 weeks from when they were plated (Figure 5B).
Since primary cells have contact inhibition, MSCs could be maintained for several weeks after reaching confluence without splitting cultures; but to expand their numbers, the MSC cultures can be split at a 1:3 ratio after they reach 75 to 80% confluence.
To split, rinse cultures 2–3 times with PBS-EDTA ( half the volume of tissue culture media) after the media is suctioned off. Then add Trypsin EDTA ( 1 ml of 0.25% or 4 ml of 0.05% Trypsin) and return to 37°C.
After 5 minutes, determine how dislodgeable the cells are in response to initial Trypsin-EDTA treatment. If needed, the initial Trypsin-EDTA can be suctioned off and an equal volume added and the plate returned to 37°C ( this will allow for the initial Trypsin to presumably loosen up the extracellular matrix and the second Trypsin to loosen the cell to cell adhesions).
After another 5 minutes, add 2 ml of FBS (or bovine calf serum which is adequate to inactive trypsin but is much cheaper than FBS) to inactivate the trypsin. Scrape the cells off the plate with a sterile cell scraper and transfer cells to a 50 cc conical.
Wash plate with 5–10 ml of PBS-EDTA to collect cells that may have been left behind.
Spin cells at 400 g force for 5 minutes. Remove supernatant by suctioning.
Resuspend pellet by “racking”. Add requisite amount of media ( 3 fold if splitting 1–3) and plate to adherent plastic plates.
MSCs will maintain exponential growth typically for at least 4–5 passages and slow down after that. The characteristics of MSCs will become more homogeneous after each passage and closer to fibroblasts derived from any other tissue site after a few passages.
Figure 5. Morphology of MSC cultures.
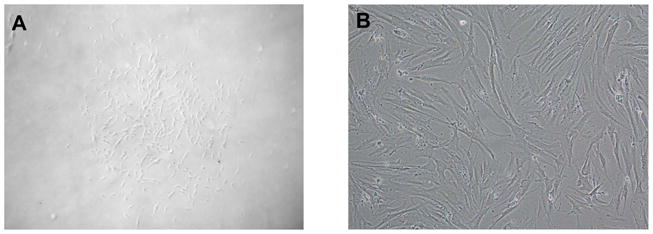
A: a Typical CFU-F that arises out of a single stromal precursor after 5–7 days of culture.
B: Confluent cultures that develop after 2 weeks or more of culture.
3.6 Flow-based ISOLATION OF MSC BY CD146high expression
Although a variety of immune-phenotypic markers have been used to define functional subtypes within marrow stromal cells, we have found CD146 as initially described by Sacchetti et al to be particularly useful (17). Commercially available CD146 antibodies are robust reagents that can be used for immune-histochemistry (Figure 6A–B) and flow-cytometry (Figure 6C). In our experience, sorting cells based on CD146 positivity does correlate to expression of other stem cell niche associated genes such as CXCL12 and Angiopoietin1 as described by Sacchetti et al in their original description of CD146+ self-renewing osteoprogentors(17, 48). In contrast to Sacchetti et al (who performed flow-based sorting on whole bone marrow mononuclear cells), we performed flow-based sorting on hMSC expanded for 1–2 passages (48). This was done to increase the proportion of stromal cells being sorted (allowing for most hematopoietic cells to be washed after the stromal precursors attach to the plastic dishes) and consequently decrease the total of number of cells that needed to be sorted and reagents used. Although stromal cells have likely changed their function and behavior even by this short culture period, our group has reported that the CD146+ fraction of cells continue to express HSPC niche-associated genes (CXCL12, Angiopoietin1) at a higher level when compared to CD146 negative fraction (48). CD146 has also been found to be strongly expressed in the human stromal cell line Hs27a which is functionally similar to the CD146+ osteoprogenitor fraction as defined by Sacchetti et al; Hs5 in comparison is CD146 negative as would be consistent with its function(17, 48).
Figure 6. CD146 expression in primary stromal cells and its use in sorting stromal cells.

A and B: Immune Histochemistry (IHC) for CD146 expression of normal human bone marrow ( Panel A is an isotype antibody control. CD146 positive cells are present in a perivascular distribution, a location consistent with other models of where the HSPC niche might reside.
C: Flow-cytometry analysis of MSC cultures after 1 passage ( 10–14 days of culture). A variable proportion of cells are CD146 positive and they inhabit of continuum of antigen expression. Two MSCs set up from separate donors are shown.
D: Typical flow-sorting results from CD146-based sorting of MSCs. Approximately 35% of cells are deemed CD146 positive ( pre-sort, top histogram), and sorted to negative ( middle histogram) and CD146 negative ( bottom histogram) using a FACS-ARIA sorter ( Beckton Dickinson and Company) with a 100 μM nozzle.
Flow-cytometry and flow-based sorting of adherent cells such as fibroblasts and stromal cells is not straightforward and certain important distinctions between hematopoietic cells need to be kept in mind. Stromal cells, consistent with their fixed state in vivo and adherent state in vitro are not physiologically meant to flow through narrow capillaries without blocking them and consequently are difficult to easily sort into different populations while preserving viability. Since these cells are typically larger, the use of a larger nozzle (100 or 130 microns) for the sorting machine would be ideal and most modern flow-sorters are equipped to do this. While preparing the cells, it is important to avoid using trypsin to dislodge the cells since CD146 is known to digest the CD146 epitope. (see Note 11) Finally, viability of cells after sorting is typically about 50% at best ( again likely reflecting the non-physiological nature of single cell capillary flow for these cells) and if these cells were to be re-plated for culture, the loss of viability needs to be considered.
-
1
Wash cultures initially with PBS-EDTA 3 times. Typically 10 to 20 100 mm dishes are used for one sorting experiment.
-
2
Add enough citrate saline solution to cover the bottom culture ( for 100 mm plates, 5 ml of solution). Return plates to 37 degree incubator for 5 minutes.
-
3
Scrape cells off the plate with a sterile cell scraper. Move the cells to a 50 ml conical. Wash the plate with an additional 5 ml of PBS-EDTA to collect any remaining cells and add to the conical tube.
-
4
Spin cells down at 400 g force for 5 minutes. Remove supernatant.
-
5
Gently resuspend cells by “racking” the pellet. Add 5 ml of PBS-EDTA and mix thoroughly with the pipetteman several times.
-
6
Wash a second time with PBS-EDTA and resuspend cells as described above.
-
7
Determine if cells are significantly clumped together (primary MSC cultures are often clumped together) and you may opt to repeat the PBS-EDTA resuspension step once more. If not, filter through a sterile 100 micron filter and proceed to counting and antibody labeling.
-
8
Determine viable cell number with a hemocytometer count.
-
9
Resuspend up to 10 million cells in a clear 5 ml Falcon tube (#2058) in sterile FACS buffer in a total volume not to exceed 1 ml. (see Note 12)
-
10
Add 10 ul of anti-CD146 antibody per 1 million cells. Incubate at 4°Cfor 30 minutes.
-
11
Wash three times by adding 4 ml of FACS buffer and spinning at 800 g force for 5 minutes and resuspending the cell pellet after each wash.
-
12
After final wash, filter cells once more through a 100 micron filter and resuspend in a final cell concentration of no more than 1 million cells per ml ( to prevent cellular clumping).
-
13
Perform FACS-based sorting of cells on sorting machine, preferably with a 100 or 130 micron nozzle. Ensure that the cells are agitated often to prevent clumping. Typical results for 14. FACS-based sorting are shown in Figure 6D.
-
15
After sort, spin down cells at 800 g for 30 minutes, count with hemocytometer and proceed with specific experiment.
3.7 Isolation of MSC by Depletion Of CD45 and CD14
An alternate method to enrich for MSCs is by depleting the bone marrow of hematopoietic cells. Most mature cells of hematopoietic origin are CD45 ( pan leucocyte antigen) positive; some monocytes and macrophages may not be removed by CD45 depletion alone and depleting CD14 positive cells in addition further enriches for stromal precursors. As in the CD146 protocol above (section 3.6), enriching for adherent cells only by plating cells in plastic adherent plates for 24 hours can reduce the total reagents that would need to be used if unselected fresh BMMNC is used. Availability of magnetic conjugated antibodies for CD45 and CD14 make this protocol easy to perform with magnetic separators such as Auto-MACS ( described earlier) which we have typically utilized for this purpose.
Isolate BMMNC per ficoll gradient separation protocol above
Plate BMMNC at a density of 50 ×10^6 cells per 100 mm plate in MSC Media ( DMEM with 10% FCS).
Next day, wash off the non-adherent cells 3 times with HBSS
Add 5ml of 0.25% trypsin-EDTA and incubate at 37°C for 5 minutes to remove adherent cells. Visualize cells under microscope to ensure adequate trypsinization. Dislodge with cell scraper if needed.
Rinse flask with 5 ml media to inactivate trypsin and collect cells in 15 ml conical
Spin cells down at 1200 RPM for 5 minutes
Resuspend cells in AutoMACS buffer (80uL for upto 10^7 cells). Add 20uL each of CD45 and CD14 microbeads.
Incubate on ice for 20 minutes
Wash with 4mL of Automacs buffer after incubation
Perform Automacs with “Deplete” protocol, once only
Spin down the negative fraction at 400 g and resuspend in MSC media.
Plate in 100 mm dishes or T-75 flasks with 0.1 to 0.5 × 10^6 cells per flask, if the yield is low, plate at lower densities as well.
Isolated fibroblasts colonies should be visible starting day 2–3. To remove all the non-stromal components, change media every 2–3 days for the first 2 weeks as described for MSC cultures.
3.8 Stromal Cell lines
Stromal cell lines HS5 and HS27are stable and well characterized lines that can be used to approximate various tissue microenvironments. They are easy to culture and can be manipulated with relative ease (at least when compared to primary MSC).
-
1
Pre-warm media ( RPMI-1640 supplemented with 10% FBS) in a 37 degree water bath. Aliquot 45 ml of media to a 50 cc conical tube.
-
1
Thaw cryo-preserved cell vial ( typically containing 5 ×10^6 cells) in a 37 degree water bath. As with other cryo-preserved cells, the rule is to “freeze slowly and thaw quickly” to avoid cryogenic damage to cells.
-
2
Remove vials to tissue culture hood once the frozen media is loosening from the side of the vial. Open vial, Add about 1 ml of pre warmed media to vial to complete the thaw.
-
3
Add the thawed cell-media mixture to the 50 cc conical containing warm media. Mix well and spin down cells at 400 g for 5 minutes.
-
4
Suction off supernatant and plate cells to one T75 flask ( or 100 mm plate) with 10 ml media. Transfer to incubator
-
5
Determine viability of cells the next day – all viable cells should be attached and exhibiting fibroblast-like pleomorphic morphology. Cells can be split in about 48 hours ( 1:3) if they are nearly confluent.
-
6
Cells can be maintained by replacing media without splitting for upto a week after attaining full confluence.
-
7
To freeze stromal cell lines, use RPMI-1640 supplemented with 30% FBS. Resuspend about 5×10^6 cells per ml of this media and aliquot to cryovials. Add tissue culture grade DMSO to a final volume of approximately 10% ( 100 ml to a 1 ml suspension). Move to cryo-freezing containers (such as “Mr Frosty” that allows for graded temperature drop) and place in −80 freezer. Move frozen vials to liquid nitrogen in 24 to 48 hours for long term storage.
3.9 Reverse transfection of stromal cell line with siRNA/miRNA
While stromal cells are not particularly difficult to grow and propagate, they can be challenging to transfect transiently with plasmids or small RNAs. When working with siRNAs to inhibit specific transcripts or miRNA, low efficiency transfection ( less than 25%) is almost guaranteed to result in a failed experiment, since the downstream tests of changes in RNA/protein levels or function is likely not be reflected in those cells which were not transfected. Commercially available lipid preparations such as lipfectamine 2000 from Invitrogen have been successfully adapted to the high efficiency transfection of several adherent cell-types (such as HEK-293Ts, HeLa). Typical protocols for liposomal transfections for cells add the lipid-RNA mix to already adherent cells. Stromal cells are typically difficult to transfect with this methodology presumably due to extra-cellular matrix that is laid down immediately by freshly plated cells that make lipid-based transfection difficult. One way around this difficulty is by “reverse transfection”. In reverse transfection cells are added to the media that already has the lipid-RNA mixes; higher efficiency likely results from the cells being exposed to liposomal mixture before they adhere and secrete matrix proteins (48). We use siRNA or miRNA at 5–10 nmol final concentration, but can be optimized based on knock-down achieved. The following are for a 12 well plate, but can be scaled up or down as needed. All reagents need to be brought to room temperature before beginning the protocol. (See Note 13)
In a 5 ml clear sterile Falcon tube (#2058), Mix 100 ul OPTIMEM with 2 ul lipofectamine 2000.
In a separate tube, mix 100 ul OPTIMEM with 0.625 ul of 5 nmol mirna/sirna.
Let these mixtures stand for 5 minutes, then gently mix together. Keep for 10–15 minutes
In the interim, plate 500 ul of media without antibiotics (usually RPMI with 10 % FCS) in a 12 well plate. Return to TC incubator at 37°C for 10–15 minutes.
Add 200 ul of the lipofectamine-rna-OPTMIMEM mixture to the 12 well plate slowly.
Return to incubator for another 10–15 minutes, total of about 30 minutes.
Suspend stromal cells ( 80,000 to 100,000) in 500 ul of antibiotic free media (RPMI with 10 % FCS)
Add the cells suspension drop by drop to the well containing the lipid mixture/media. Gently tilt spread evenly ( avoid circular swirling as this will allow for cells to congregate at the center of each plate). Return to incubator Change media ( RPMI-1640 with 10% FBS and antibiotics) after 6 hours or overnight incubation.
We typically can get efficiencies up to 80 with above protocol ( when using anti-GFP siRNA or plasmid for controls, Figure 7)
Figure 7. Reverse Transfection of stromal cell lines.
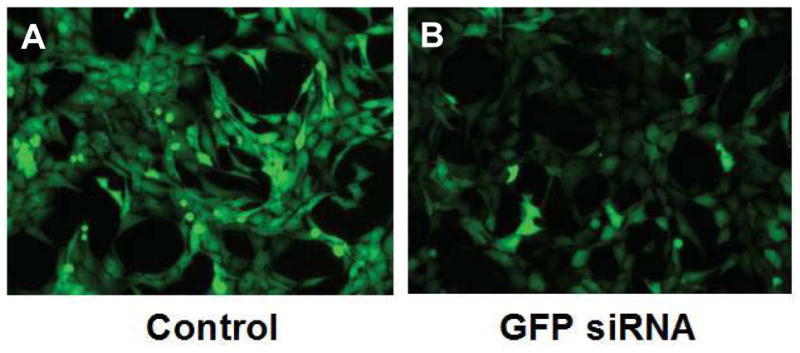
A: Stromal cell line HS27a stably expressing Green Fluorescent Protein (GFP) was transfected with control scrambled siRNA and visualized by inverted fluorescent microscopy after 48 hours.
B: Hs27a-GFP cell lines transfected with anti-GFP siRNA showing marked reduction in GFP expression of most cells.
3.10 Concluding Remarks and Summary
Mesenchymal stromal cells (MSC) can be isolated from bone marrow with relative ease. Bone marrow screens which are typically discarded by-products of bone marrow harvests are an excellent source of marrow cells and should be considered by investigators with access to large clinical centers. The foremost consideration while working with MSCs is to realize the wide variability of almost all biological characteristics amongst MSCs established from various donors. This is partly due to the complex nature of the culture itself: stromal cells inhabit a continuum of differentiation from the earliest stromal precursor capable of multilineage differentiation to mature mesenchymal cells (osteoid, adipocytic chondroid etc). Add to this the presence of macrophages and other hematopoietic cells and the reason for wide variations become clear. Flow-based sorting for CD146 has proven to be a useful technique in sorting for those earlier stromal precursors that both define CFU-Fs as well as support hematopoietic stem and precursor cells. It is important to recognize that stromal cells, consistent with their non-circulating and non-transplantable biology in vivo, dont fare well with flow-based sorting and hence special precautions have to be undertaken while sorting for them. The use of well characterized stromal cell lines can overcome the problems of primary MSC heterogeneity and should be considered when mechanistic studies of the stromal microenvironment are undertaken; although validation with primary MSC would be needed.
Acknowledgments
Supported in part by NIH grants DK073701, DK082757, HL104070, DK082783, HL099993, and DK056465, Bethesda, MD, U.S.A.
Footnotes
Aliquots of frozen marrow mononuclear cells are available for purchase through the NIH-NIDDK-sponsored Core Centers for Excellence in Hematology (CCEH) for investigators in the US at much reduced prices compared to commercial vendors and eliminates most of the regulatory concerns. Please visit the CCEH website at http://cceh.fhcrc.org for further details.
Although immediate processing would be ideal, we have successfully generated robust MSC cultures from screens preserved at 4°C or at room temperature for up to 48 hours (allowing for transport between collaborator’s laboratories).
Both Hs27a and Hs5 are available from American Type Culture Collection (ATCC, Manassas, VA) or can be directly obtained from Beverly Torok-Storb’ laboratory in Seattle, WA.
We have used the FACS Aria sorter (Beckton, Dickinson and Company or BD) for our studies but multi-color sorters from other manufacturers such as Beckman Coulter should work just as well for these applications.
Several varieties of instrumentation are available for separation of cells. The Auto-MACS automated system called is particularly useful for larger cell numbers and can sort cells maintaining aseptic status and is the system we use routinely. This instrument is also typically available through flow cytometry core facilities. The protocol could be optimized for use with other magnetic sorters with relative ease.
Since FBS is a complex biological product mass-produced as a side-product from slaughterhouses, biological variability from batch to batch is inevitable and it is recommended to screen lots of FBS for performance in the particular biological assay of interest. Commercial suppliers often provide small (~ 50 ml) aliquots of FBS for testing to individual laboratories and hold those lots for several weeks to allow for testing. Batches screened by the commercial suppliers (deemed appropriate for MSC growth) is also available directly albeit at typically much higher costs.
The horse serum needs to be specifically screened for the ability to support LTCs and not MSCs.
Avoid delays in anticoagulant-free state given that marrow-cell pellets have powerful pro-coagulants activated and could result in clots and clumps.
With complete or near-complete hemolysis, the supernatant should be wine-red in color and the cell pellet is pale or pinkish but not frank red. If hemolysis is inadequate steps 1–3 can be repeated 1–2 times (further hemolysis is unlikely to be helpful) keeping in mind not to allow exposure to hemolytic buffer for more than 5 minutes each time.
Recently, reports have indicated that ficoll at 1.073 may be more suitable for MSC progenitor isolation although this observation has not been rigorously validated.
Protocols that do not use trypsin tend to leave primary MSC cells in a clumped state, so extra precaution needs to be taken to get the cells in to single cells suspension.
Given the extreme stickiness of primary stromal cells, it would be ideal to limit total cells in the approximately 5 million cells.
Also note that transfection needs to be performed in antibiotic-free media as presence of antibiotics will reduce transfection efficiency and cell viability.
References
- 1.Chabannon C, Torok-Storb B. Stem cell-stromal cell interactions. Curr Top Microbiol Immunol. 1992;177:123–36. doi: 10.1007/978-3-642-76912-2_10. [DOI] [PubMed] [Google Scholar]
- 2.Trentin JJ. Determination of bone marrow stem cell differentiation by stromal hemopoietic inductive microenvironments (HIM) Am J Pathol. 1971;65:621–8. [PMC free article] [PubMed] [Google Scholar]
- 3.McCulloch EA, Siminovitch L, Till JE, Russell ES, Bernstein SE. The cellular basis of the genetically determined hemopoietic defect in anemic mice of genotype sl-sld. Blood. 1965;26:399–410. [PubMed] [Google Scholar]
- 4.Huang E, Nocka K, Beier DR, Chu TY, Buck J, Lahm HW, et al. The hematopoietic growth factor KL is encoded by the sl locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell. 1990;63:225–33. doi: 10.1016/0092-8674(90)90303-v. [DOI] [PubMed] [Google Scholar]
- 5.Williams DE, Eisenman J, Baird A, Rauch C, Van Ness K, March CJ, et al. Identification of a ligand for the c-kit proto-oncogene. Cell. 1990;63:167–74. doi: 10.1016/0092-8674(90)90297-r. [DOI] [PubMed] [Google Scholar]
- 6.Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- 7.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19:257–67. doi: 10.1016/s1074-7613(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 8.Barker JE. Sl/Sld hematopoietic progenitors are deficient in situ. Exp Hematol. 1994;22:174–7. [PubMed] [Google Scholar]
- 9.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 11.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Nagasawa T. A chemokine, SDF-1/PBSF, and its receptor, CXC chemokine receptor 4, as mediators of hematopoiesis. Int J Hematol. 2000;72:408–11. [PubMed] [Google Scholar]
- 13.Mancini SJ, Mantei N, Dumortier A, Suter U, MacDonald HR, Radtke F. Jagged1-dependent notch signaling is dispensable for hematopoietic stem cell self-renewal and differentiation. Blood. 2005;105:2340–2. doi: 10.1182/blood-2004-08-3207. [DOI] [PubMed] [Google Scholar]
- 14.Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–9. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramakrishnan A, Torok-Storb BJ. The role of the marrow microenvironment in hematopoietic stem cell transplantation. Cell Ther Transplant. 2010;2:7–12. doi: 10.3205/ctt-2009-en-000072.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–36. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 18.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259–72. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. J Cell Physiol. 1977;91:335–44. doi: 10.1002/jcp.1040910303. [DOI] [PubMed] [Google Scholar]
- 21.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–50. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 22.Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet. 2008;371:1579–86. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz RE, Reyes M, Koodie L, Jiang Y, Blackstad M, Lund T, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109:1291–302. doi: 10.1172/JCI15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation. 2002;105:93–8. doi: 10.1161/hc0102.101442. [DOI] [PubMed] [Google Scholar]
- 25.English K, French A, Wood KJ. Mesenchymal stromal cells: Facilitators of successful transplantation? Cell Stem Cell. 2010;7:431–42. doi: 10.1016/j.stem.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Allison M. Genzyme backs osiris, despite prochymal flop. Nat Biotechnol. 2009;27:966–7. doi: 10.1038/nbt1109-966. [DOI] [PubMed] [Google Scholar]
- 27.Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell. 2012;10:244–58. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Balakrishnan I, Torok-Storb B, Pillai MM. Marrow stromal cell infusion rescues hematopoiesis in lethally irradiated mice despite rapid clearance after infusion. Adv Hematol. 2012:142530. doi: 10.1155/2012/142530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reyes M, Verfaillie CM. Characterization of multipotent adult progenitor cells, a subpopulation of mesenchymal stem cells. Ann N Y Acad Sci. 2001;938:231, 3. doi: 10.1111/j.1749-6632.2001.tb03593.x. discussion 233–5. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp Hematol. 2002;30:896–904. doi: 10.1016/s0301-472x(02)00869-x. [DOI] [PubMed] [Google Scholar]
- 31.Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–9. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 32.Check E. Stem cells: The hard copy. Nature. 2007;446:485–6. doi: 10.1038/446485a. [DOI] [PubMed] [Google Scholar]
- 33.Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416:545–8. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- 34.Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–5. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 35.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graf T. Historical origins of transdifferentiation and reprogramming. Cell Stem Cell. 2011;9:504–16. doi: 10.1016/j.stem.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 38.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–9. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. cloning in vitro and retransplantation in vivo. Transplantation. 1974;17:331–40. doi: 10.1097/00007890-197404000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Friedenstein AJ, Deriglasova UF, Kulagina NN, Panasuk AF, Rudakowa SF, Luria EA, et al. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol. 1974;2:83–92. [PubMed] [Google Scholar]
- 41.Simmons PJ, Torok-Storb B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood. 1991;78:55–62. [PubMed] [Google Scholar]
- 42.Deschaseaux F, Charbord P. Human marrow stromal precursors are alpha 1 integrin subunit-positive. J Cell Physiol. 2000;184:319–25. doi: 10.1002/1097-4652(200009)184:3<319::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 43.Stewart K, Monk P, Walsh S, Jefferiss CM, Letchford J, Beresford JN. STRO-1, HOP-26 (CD63), CD49a and SB-10 (CD166) as markers of primitive human marrow stromal cells and their more differentiated progeny: A comparative investigation in vitro. Cell Tissue Res. 2003;313:281–90. doi: 10.1007/s00441-003-0762-9. [DOI] [PubMed] [Google Scholar]
- 44.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: Nature, biology, and potential applications. Stem Cells. 2001;19:180–92. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 45.Majumdar MK, Banks V, Peluso DP, Morris EA. Isolation, characterization, and chondrogenic potential of human bone marrow-derived multipotential stromal cells. J Cell Physiol. 2000;185:98–106. doi: 10.1002/1097-4652(200010)185:1<98::AID-JCP9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 46.Gronthos S, Zannettino AC, Hay SJ, Shi S, Graves SE, Kortesidis A, et al. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J Cell Sci. 2003;116:1827–35. doi: 10.1242/jcs.00369. [DOI] [PubMed] [Google Scholar]
- 47.Buhring HJ, Battula VL, Treml S, Schewe B, Kanz L, Vogel W. Novel markers for the prospective isolation of human MSC. Ann N Y Acad Sci. 2007;1106:262–71. doi: 10.1196/annals.1392.000. [DOI] [PubMed] [Google Scholar]
- 48.Pillai MM, Yang X, Balakrishnan I, Bemis L, Torok-Storb B. MiR-886-3p down regulates CXCL12 (SDF1) expression in human marrow stromal cells. PLoS One. 2010;5:e14304. doi: 10.1371/journal.pone.0014304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Randolph-Habecker J, Iwata M, Torok-Storb B. Cytomegalovirus mediated myelosuppression. J Clin Virol. 2002;25(Suppl 2):S51–6. doi: 10.1016/s1386-6532(02)00092-6. [DOI] [PubMed] [Google Scholar]
- 50.Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85:997–1005. [PubMed] [Google Scholar]