

Abstract

Distinction between closely related and morphologically similar cells is difficult by conventional methods especially without labeling. Using nuclear-targeted gold nanoparticles (AuNPs) as intracellular probes we demonstrate the ability to distinguish between progenitor and differentiated cell types in a human neuroblastoma cell line using surface-enhanced Raman spectroscopy (SERS). SERS spectra from the whole cell area as well as only the nucleus were analyzed using principal component analysis that allowed unambiguous distinction of the different cell types. SERS spectra from the nuclear region showed the developments during cellular differentiation by identifying an increase in DNA/RNA ratio and proteins transcribed. Our approach using nuclear-targeted AuNPs and SERS imaging provides label-free and noninvasive characterization that can play a vital role in identifying cell types in biomedical stem cell research.
Keywords: Gold nanoparticles, surface-enhanced Raman scattering, cellular imaging, nuclear localization signal, cellular distinction
Gold nanoparticles (AuNPs) have been shown to be of great use and advantage for delivering drugs into cells and tissues, cellular imaging such as transmission electron microscopy (TEM), as well as for diagnostic and therapeutic applications in biomedicine.1−3 In particular for biomedical applications involving the intracellular localization of AuNPs, their size,4 shape,4,5 concentration,6 charge,7 surface modification,8,9 and exposure time6 play an important role in their ability to enter and leave cells. Different mechanisms are known to be involved in the cellular uptake of AuNPs such as phagocytocis, pinocytosis, and macropinocytosis as well as clathrin- and caveolin-mediated endocytosis. Studies on various cell lines have shown that energy-dependent receptor-mediated endocytosis (RME) is the predominant mechanism.5,6,10−12 Generally, this involves taking the nanomaterial into phospholipid membrane-bound vesicles, called endosomes. These vesicles fuse and release their contents, which eventually end up in lysosomes rather than free in the cytosol.10−16 After being processed, removal of AuNPs is facilitated by exocytosis whereby this process shows a strong dependency on the extracellular NP concentration.17−19 In order to make AuNPs suitable for biomedical approaches, many studies have focused on their intracellular toxicity. It has been shown that the cytotoxicity correlates with the size of single particles and clusters. Cellular integrity largely remains unaffected for AuNPs bigger than 15 nm and cytotoxicity increases with decreasing particle size and increasing concentration with high toxicity observed for particles as small as 1.4 nm.1,12,16,19
Since the normal intracellular localization of AuNPs is determined by endocytosis, engineering the nanomaterial is necessary to avoid this. To achieve this objective, AuNPs have been conjugated with cell-penetrating peptides,11 protein transduction domains,20 and adenoviral RME peptides.21 This manipulation allows for the targeting of NPs to specific cell organelles such as the cell nucleus. Nuclear localization signal peptides (NLS) such as the SV40 large T antigen, HIV-1 Tat protein NLS and adenoviral NLS are well-known for their role as a “Trojan horse”, delivering cargo to the nucleus.7,11,13,21,22 To achieve nuclear translocation, engineered AuNPs must first get into and remain free in the cytoplasm. Tkachenko et al. showed that NLS bound to 20 nm AuNPs incubated in cell culture medium allowed the NPs to enter HeLa cells within an hour. Even though the NLS-AuNPs were mostly found in intracellular vesicles that appeared to be endosomes, they observed a few particles inside the nucleus after a total incubation time of two to three hours indicating that some of them had managed to escape the endosomal pathway.7 Other studies have also demonstrated the successful entry of NLS-AuNPs into the nucleus after their microinjection directly into the cytoplasm.7,22,23
AuNPs have been utilized extensively as transducers for surface-enhanced Raman spectroscopy (SERS), which is a molecular fingerprinting method. It utilizes vibrational signatures of bonds in a molecule for their highly sensitive, label-free, nondestructive and noninvasive detection.24,25 SERS is being used increasingly in biomedical systems for biosensing and detection of target molecules such as recreational drugs,26 therapeutic substances27 and metabolites like cancer markers.28 AuNPs have also been employed as intracellular probes facilitating cancer detection in saliva,29 blood cells,30 and tissues.31,32 It has also been demonstrated that SERS using AuNPs can be applied to living cells in order to monitor cellular functions33 and dynamics,15 cell response to stress,34 and cell death.35 However, only a few studies have focused on using SERS to interrogate the functional state of specific intracellular organelles such as the cell nucleus.36 The cell nucleus is one of the most important organelles that directs, as well as reflects, the complex intracellular processes underlying cellular differentiation.37,38
The discrimination of closely related cell phenotypes in a noninvasive and label-free manner is still a challenge using conventional methods of optical microscopy. In this study, we show the successful segregation of closely related cell phenotypes using the noninvasive method of SERS. We employed intracellular SERS active nanoprobes for targeting the cytoplasm as well as the cell nucleus of undifferentiated and fully differentiated SH-SY5Y cells, a human neuroblastoma cell line. Furthermore, nuclear-targeted AuNPs allowed us to investigate changes in nuclear content induced by cellular differentiation.
Our method employed the SV-40 large T NLS bound to 40 nm AuNPs (see Supporting Information for details on their characterization) as intracellular SERS probes. We use a customized NLS peptide sequence (CGTG-PKKKRKV-GGK(Flu)) comprising a fluorescein tag at the C-terminus and a cysteine for binding the AuNP to the NLS. Co-localization of fluorescence from the attached fluorescein probe with intracellular AuNPs was used to confirm the successful binding of the peptide to the NPs (Figure 1a–d). The successful attachment of NLS is further supported by a small (∼1 nm) shift in the SPR peak of AuNPs (Supporting Information Figure S3). Neither the NLS attachment nor the incubation in cell culture medium itself results in any significant aggregation (see Supporting Information Figure S3). To allow for cellular uptake, NLS-AuNPs were added to the standard culture medium at a concentration of 6.75 × 1010 NLS-AuNPs/mL. Following an incubation time of up to 72 h, cells were fixed with 4% formaldehyde and kept in phosphate buffered saline. Fixation with formaldehyde is widely used for biological sample preparation and has been shown to have no discernible effects on acquired spectra.39,40
Figure 1.
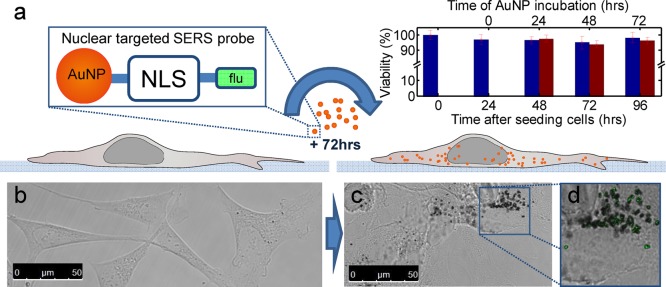
Preparation and intracellular interaction of the nuclear-targeted SERS probe. (a) Schematic showing AuNPs linked via cysteine to a SV-40 large T nuclear localization signal peptide having a fluorescein (flu) tag at the C-terminus. NLS-AuNPs were added to the culture medium without compromising cellular viability. The viability of differentiated cells incubated with NPs for up to 3 days (red bars) was tested and compared with cells left untreated (blue bars). (b) Bright-field image of differentiated SH-SY5Y cells without AuNPs. (c) After 72 h of incubation, cells show intracellular AuNPs. The fluorescein tag is colocalized with AuNPs in bright field confirming the successful binding of NLS to NPs.
Undifferentiated, dividing cells (n = 20) formed the first cell group (UDCs) while fully differentiated cells (n = 20) formed the second cell group (DCs) of study (see Figure 1b). Both cell groups adopt a neuronal phenotype and show closely related morphologies.41 For the two respective cell groups NLS-AuNPs were incubated with them either before or after differentiation was complete. SERS spectra from cells could be acquired at shorter incubation times as well (see Supporting Information Figure S9 for data after 24 and 48 h incubation), however they were strongest and most numerous at 72 h especially from the nucleus; hence, this incubation time was used. This was because after an incubation time of 72 h cells showed many aggregates of cytoplasmic NLS-AuNPs as well as some within the nucleus due to higher uptake over time (Figure 1c,d, see also Figure S1 for dark-field images in Supporting Information). Furthermore there was no evidence of compromised cell viability as assessed using Trypan blue (inset Figure 1a).
Spectral images (Figure 2) of cells were acquired using a 633 nm laser in streamline mode with a Renishaw inVia Raman microscope, where an area of 200 nm × 600 nm corresponds to a single pixel on the image. Intense SERS signals were obtained from areas where aggregates of NLS-AuNPs were localized; exemplar spectra from such areas are shown in Figure 2a. Spectra from different cellular regions show different peak positions and intensities; cytoplasmic spectra (blue) reveal peaks assigned to proteins and fatty acids, whereas spectra from the nuclear region (red) mostly reveal peaks characteristic for nucleic acids and proteins. For example, this can be seen in the spectral range of 500–900 cm–1, where significant peaks can be observed within the nuclear region of the cell (red lines, Figure 2a) compared to less peaks in the cytoplasmic spectra (blue lines, Figure 2a). We found, as expected, that spectra corresponding to nucleic acids were acquired from the nuclear region of the cell (white ellipse) indicating the successful translocation of our NLS-modified SERS nanoprobes to the nucleus. To further verify the nuclear localization of NLS-AuNP probes, we tested our experimental approach using Hoechst 33342, a common cell stain known to bind to DNA, as a marker for selective staining of the nucleus. Characteristic SERS peaks for Hoechst dye, bound to DNA,42 were exclusively found in the spectra from the nuclear region which also contained peaks corresponding to nucleic acids while spectra from the cytoplasmic area of the cells did not show these (see Figure S5 in Supporting Information). The appearance of characteristic SERS peaks of bound-Hoechst and those corresponding to nucleic acids confirmed that some NLS-AuNPs were translocated into the nucleus. Subsequent experiments were carried out without Hoechst staining. The intracellular location of nucleic acids and proteins are highlighted in Figure 2c,d, respectively, and DNA, RNA as well as the DNA/RNA backbone vibrational modes are shown in red, green and yellow, in Figure 2c.
Figure 2.
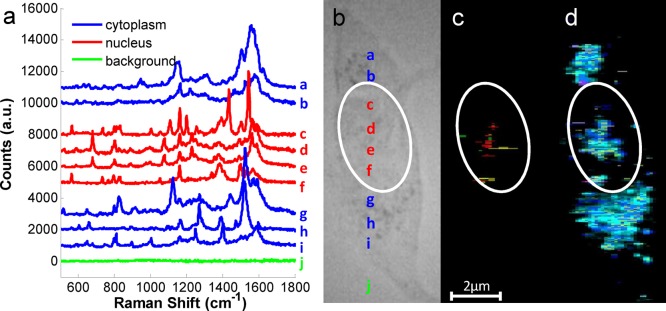
SERS imaging. (a) SERS spectra obtained from different positions (a–j) within the culture such as cytoplasm (blue), cell nucleus (red) and the surrounding environment (green) show significant differences in terms of their intensity and peak positions. (b–d) Bright-field image (b) and SERS map from the same differentiated SH-SY5Y cell highlighting the intracellular distribution of the sugar phosphate backbone (895 and 1050–1100 cm–1, yellow) as well as nucleic acids of DNA (typical bands such as 670, 830, 1375, and 1580 cm–1, red) and RNA (815 cm–1, green) (c) as well as proteins (yellow: symmetric ring CC stretch; blue: NH3+ deformation; magenta: CH2 deformation; cyan: Amide II) (d). The cell nucleus is indicated by a white ellipse (200 nm × 600 nm pixel size).
Following spectral map acquisition, data analysis was carried out with a multivariate, unsupervised data reduction technique of principal component analysis (PCA) using MATLAB R2010b employing a graphical user interface toolkit (see Supporting Information). SERS mapping generates large, complex data sets creating the need for adequate data reduction and analysis. It has been shown that PCA is a suitable tool for this as well as it facilitates sample group discrimination in SERS imaging.35,43,44 PCA generates loadings and score plots from the derived principal components (PCs) of the initial or preprocessed data. Gained PC loadings are correlation coefficients between the original data and PC scores. PC loadings identify the importance of each variable (i.e., peak in a SERS spectrum). The correlation of a variable to a PC reflects its contribution to the variation in the data set. Therefore, PC loadings plots reveal vibrational modes (wavenumbers) corresponding to the variation which allows for distinction between groups. Thus, we chose PCA on our hyperspectral data in order to fulfill two aims: data reduction to less dimensions and accomplishment of an objective distinction between the two cell groups (UDCs and DCs). PCA was applied on the data acquired from the whole cell (see Supporting Information). Figure 3a compares PC loadings for both cells groups. There is a clear segregation of both cell groups using PC1 and PC2 loadings spectra in which UDCs (red) and DCs (blue) show distinct differences in peak positions and variance. UDCs always show less variance and narrower peaks in their loadings compared to DCs that may be due to their more homogeneous, undifferentiated state. In DCs, protein synthesis and cellular protein content are increased as a result of their differentiation.45 Peak positions in PC loadings are directly related to the SERS peaks of single spectra. Peaks of both cell groups are assigned exclusively to proteins, showing different positions between the cell groups. PC loadings of UDCs show peaks at 604 (tyrosine (tyr)), 840 (tyr), 1015 (phenylalanine (phe)), 1177 (tyr), 1240 (Amide III), and 1606 cm–1 (phe, tyr), whereas DCs show peaks at 1004 (phe), 1162 (methionine), and 1550 cm–1 (Amide II).35,46−51 Differences in variance and peak positions allow for the clear segregation of the two cell groups. The NLS peptide itself does not spectrally interfere in the analysis (see Supporting Information Figure S4).
Figure 3.
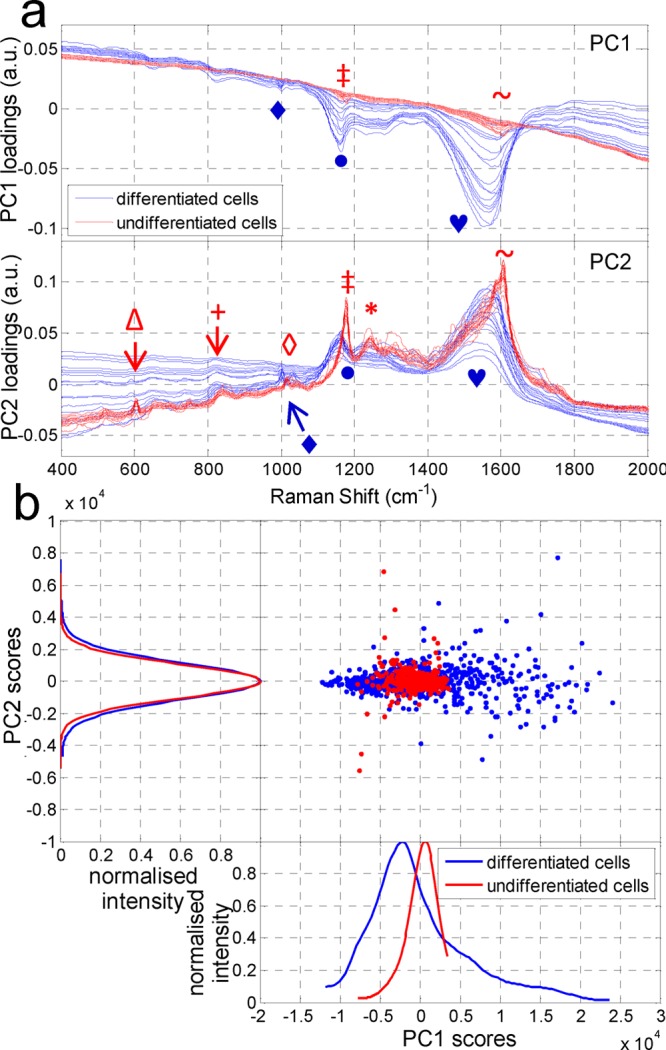
Principal component analysis of whole cell data. (a) Spectral plot of loadings of differentiated (DCs) (blue, n = 20) and undifferentiated cells (UDCs) (red, n = 20) for PC1 (upper) and PC2 (lower) showing clear distinctions between the cell groups especially in terms of intensity and peak positions. Peaks are exclusively assigned to proteins but reflect the change of intracellular protein composition due to differentiation. Peaks were found at the following (in cm–1): Δ, 604; +, 840; ⧫, 1004; ◊, 1015; •, 1162; ‡, 1177; *,: 1240; heart, 1550; and ∼, 1606. (Shapes above the curve refer to UDCs (red), below to DCs (blue).) (b) Scatter plot of PC1 vs PC2 scores with attached 1D intensity line. PCA of SERS scans of the whole cell, UDC (red) and DC (blue) groups were each formed from five different single cell data sets. The explained variance of the PC1 scores accounts for 76.4 and 93.9% of the variation for UDCs and DCs, respectively (see Supporting Information for explained variance of higher PCs). The PC1 intensity plot demonstrates that cell groups can be segregated using the mean and standard deviation of the PC1 scores. Higher PC scores show intensity distributions similar to PC2 while only the standard deviation permits for cellular distinction.
SERS data sets (150 data points each) for each cell group were analyzed individually using PCA and the results are shown as 2D scatter plots for PC scores of UDCs (red) and DCs (blue) (see Figure 3b). The 1D intensity plots are smoothed histograms of the PC data and allow for easy characterization of the data distribution. They were generated using Kernel smoothing computed probability distributions (see Supporting Information) and are shown alongside. PC score intensities are normally (Gaussian) distributed (see Supporting Information) and characterized by their mean μ and standard deviation σ. For PC1 scores, the mean value μPC1 is always negative for DCs and positive for UDCs. Additionally, the standard deviation σPC1 is smaller for UDCs (<700) than for DCs (>700) revealing a broader distribution of data points within the DC group. The standard deviation of PC2 also gives values similar to σPC1. In contrast, the mean μPC2 is in the range of ±60, independent of cell group affiliation and hence is not useful in separating the two groups of cells. Higher PC scores feature 1D intensity distributions analogous to that of PC2 (Figure 3b) and thus were not considered further for analysis. Since both cell groups are closely related, an overlap in distribution would be expected. Even though cellular differentiation does change the intracellular composition, most subcellular structures remain the same or similar. A greater variety of intracellular proteins are reflected in a wider distribution of PC intensity plots characterized by a higher standard deviation in DCs. Nevertheless, the above results show that using data sets from the whole cell, PC1 loadings and scores distribution including 1D intensities allow for unambiguous and successful segregation of the cell types into two classes. Furthermore, PC2 (and higher PCs) loadings as well as 1D intensity of the scores were successfully tested and allowed segregation of the two cell groups. Summarizing, PCA (PC loadings as well as PC score intensities) on an unknown sample would allow for cell group classification.
As stated earlier, only a smaller fraction of AuNPs aggregates were found in the cell nucleus as identified by the SERS peaks assigned to nucleic acids. Since analysis of the whole cell data masks the developments in the nucleus, we analyzed nuclear SERS spectra separately. Figure 4a shows nuclear 2D scatter plots of PC1 and PC2 scores and their 1D intensity line plots. PC1 scores show an explained variance of 61.5 and 93.4% for UDCs and DCs, respectively. Nuclear PC1 scores show clear similarities to those of the whole cell data. Likewise, the nuclear mean value μPC1_nuc is always positive for UDCs and negative for DCs. The standard deviation σPC1_nuc is larger than for whole cell data but is still smaller for UDCs (<2400) compared to DCs (>2400). The 1D intensity plots of PC1 (with clearly different μPC1_nuc and σPC1_nuc) allow for accurate cellular distinction, while those for higher PCs do not. Cellular differentiation in the cytoplasm is characterized by a compositional change in proteins transcribed. In contrast, the cell nucleus undergoes a change in the ratio of the molecular content as well as morphological and structural development.52 In particular, the reorganization of chromatin plays an important role during the transition from a proliferating cell to a nondividing cell. While in dividing cells (UDCs) the nucleus is subject to continuous changes in chromatin formation during cell division, nondividing cells (DCs) do have a steady nuclear formation. This homogeneity within the population of DCs is also consistent with the higher explained variance observed for this cell group.
Figure 4.

Analysis of nuclear SERS spectra. (a) PCA analysis of nuclear spectra shown as a scatter plot and 1D intensity plots for PC1 against PC2 scores. The explained variance of the PC1 scores account for 61.5 and 93.5% for UDCs and DCs, respectively. PC1 scores allow for the correct cellular segregation of UDCs (red) and DCs (blue) as the results show distinct differences in mean and standard deviation. (b) Combined bar and line plots showing the normalized difference in the spectrum of nuclear peak occurrences (bars) and the PC1 loadings. DCs and UDCs are characterized by bars/peaks pointing to the upper and lower side of the plot, respectively. Marked peak positions (1–24) are assigned in (c).
In order to characterize nuclear spectra two methods of analysis were implemented: nuclear peak occurrence (blue bars) and differential PC1 loadings (red line) with results shown in Figure 4b. The nuclear peak occurrence (NPO) method is based on a manual frequency count (with 5 cm–1 bin size) of all peak positions found within nuclear spectra of single cells (n = 15) for each cell group. Nuclear peak occurrence reflects only peak positions irrespective of their intensities. In contrast, PC1 loadings (obtained as described above using PCA) involve both peak intensities as well as position. Hence, the cell groups were analyzed separately and the loadings were normalized. This was followed by subtraction of the average loadings of each cell group from each other to create a differential loading spectrum (see Figure S8 in Supporting Information). As SERS spectra in general show higher intensities for peaks of proteins than for DNA/RNA, and since PC loadings also consider peak intensities the resulting difference spectrum shifts toward DCs in protein rich regions such as 1130–1200 or 1450–1550 cm–1. Apart from slight differences in these regions, analysis from both methods shown in Figure 4b are in good agreement. The differences highlighted by these analyses allow for the description of molecular changes in the nucleus since PC loadings as well as the more straightforward method of NPO are directly linked to vibrational modes. Peaks (1–24) pointing up or down therefore characterize DCs and UDCs respectively. All peaks are either assigned to nucleic acids, the DNA/RNA backbone (bkb) or proteins as presented in Figure 4c.34,37,45−51,53−56 Significantly, the number of protein peaks increases during cellular differentiation from only a few (peaks as in Figure 4b,c, 1, 16, 20) in UDCs to many peaks (peaks as in Figure 4b,c, 3, 7, 9–11, 14, 15) in DCs suggesting higher protein content and variety. Peak positions corresponding to nucleic acids and their backbone also change. UDCs show distinct peaks at 780 cm–1 (uracil, cytosine) and 805–819 cm–1 (symmetric O–P–O phosphodiester stretch in RNA), while for DCs, the SERS peaks assigned to DNA are observed at 793 and 835 cm–1 (symmetric and asymmetric O–P–O phosphodiester stretch). Furthermore, both cell groups show SERS bands at 1060–1092 cm–1 corresponding to the PO2– stretch of the DNA backbone. In addition, peak positions and assignments of peak (7) and (9–11) are in agreement with those of histones,57 which are proteins found in the nucleus associated with DNA. These characteristic peaks suggest a higher nuclear packaging in DCs than in UDCs. Furthermore, SERS peaks of nucleic acids in DCs mostly correspond to DNA and proteins (histones) indicating DNA packaged with chromosomal proteins in heterochromatin while, UDCs feature SERS peaks of DNA/RNA reflecting a looser packaging similar to euchromatin.
In order to prove our hypothesis, we used Hoechst 33342 to visualize nuclear chromatin density in UDCs and DCs as shown in Figure 5a-b, respectively, where chromatin density correlates to fluorescence intensity in the image. Dividing cells such as UDCs pass through different phases of the cell cycle featuring changes in chromatin density such as decondensed chromatin during interphase or chromosomal condensation during mitosis and cytokinesis. Nucleus 1 in Figure 5a of a representative interphase cell shows an evenly distributed chromatin content with a large, oval, or rounded nucleus while nuclei 2 and 3 show very dense, frayed chromatin (condensed chromosomes) as expected during the late state of mitosis (late telophase) or the beginning of cytokinesis. Figure 5b shows denser chromatin in nondividing DCs with the bright fluorescence on Hoechst staining confirming tighter packaging. Furthermore, staurosporine was used as a differentiating agent in our experiments (Materials and Methods in Supporting Information) and in fact is a commonly used potent maturation agent for the differentiation of the SH-SY5Y cell line at low concentrations such as 10nM (≈5 ng/mL).58 At this concentration, which was used in our experiments, cells are arrested at the G2 phase (late interphase) of the cell cycle which is accompanied by changes in nuclear morphology such as partly condensed chromosomes and increased DNA ploidy (up to octaploid DNA) depending on the cell line.58,59 Thus our results are consistent with the known effects of staurosporine confirming the utility of our approach to measure nuclear status and cellular differentiation in this cell line.
Figure 5.
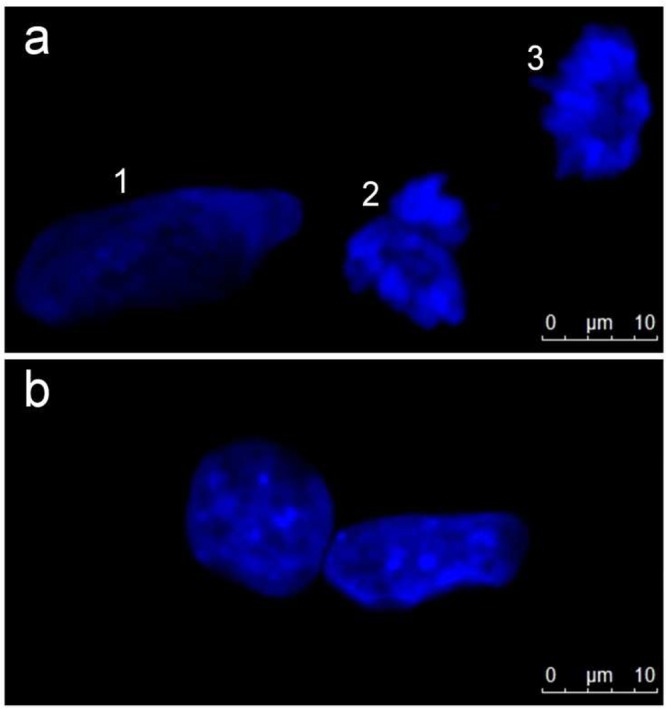
Fluorescence staining of cell nuclei of SH-SY5Y cells with Hoechst 33342. (a) UDCs show different chromatin states during the cell cycle; in interphase they show evenly distributed, decondensed chromatin (1) whereas in the late stage of mitosis or beginning of cytokinesis the chromatin appears highly condensed (2, 3). (b) DCs show areas of high fluorescence indicating condensed chromatin.
In summary, employing NLS-AuNPs as intracellular probes for cellular SERS imaging, we have been able to segregate undifferentiated from differentiated cells in a human neuronal cell line using PCA analysis for whole cell scans as well as nuclear spectra. Furthermore, our results using a novel method of NPO and the more commonly used PC1 loadings has also allowed us to characterize nuclear development during this process of cellular differentiation. This work therefore suggests that this approach using NLS-AuNPs and SERS imaging could be of great importance in the future characterization of different cells types in biomedical stem cell research.
Acknowledgments
We gratefully acknowledge EPSRC (Engineering and Physical Sciences Research Council), United Kingdom, funding support (EP/H028757/1; EP/G060649/1) for this work. We thank Professor Jeremy Baumberg for his kind support and use of facilities in the Nanophotonics Centre. We also thank Ian Davies for his introduction to MATLAB, Dr. Torsten Krude for helpful discussions and Daniel Sigle for help with SEM imaging.
Glossary
Abbreviations
- SERS
surface-enhanced Raman scattering
- AuNPs
gold nanoparticles
- NLS
nuclear localization signal peptide
- PCA
principal component analysis
- RME
receptor-mediated endocytosis
- UDCs
undifferentiated cells
- DCs
differentiated cells
- PC
principal component
- tyr
tyrosine
- phe
phenylalanine
- A
adenine
- C
cytosine
- G
guanine
- U
uracil
- T
thymine
- str
stretch
- def
deformation
- br
breathing
- NPO
nuclear peak occurrence
- bkb
backbone
Supporting Information Available
Additional information, figures, tables, and references. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.H. primarily did all the work. W.L.K. and R.B. providing the cell lines and supported A.H. in her cell biology related work. R.B. and S.M. conceptualized the project. S.M. supervised the work. The first draft of the manuscript was written by A.H. and S.M. All authors contributed and have given approval to the final version of the manuscript.
The authors declare no competing financial interests.
Supplementary Material
References
- Murphy C. J.; Gole A. M.; Stone J. W.; Sisco P. N.; Alkilany A. M.; Goldsmith E. C.; Baxter S. C. Gold Nanoparticles in Biology: Beyond Toxicity to Cellular Imaging. Acc. Chem. Res. 2008, 41121721–1730. [DOI] [PubMed] [Google Scholar]
- Lévy R.; Shaheen U.; Cesbron Y.; Sée V. Gold Nanoparticle Delivery in Mammalian Live Cells: A Critical Review. Nano Rev. 2010, 1, 4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisselier E.; Astruc D. Gold Nanoparticles in Nanomedicine: Preparations, Imaging, Diagnostics, Therapies and Toxicity. Chem. Soc. Rev. 2009, 3861759–1782. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Li J.; Lykotrafitis G.; Bao G.; Suresh S. Size-Dependent Endocytosis of Nanoparticles. Adv. Mater. 2009, 214419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E. C.; Zhang Q.; Xia Y. The Effect of Sedimentation and Diffusion on Cellular Uptake of Gold Nanoparticles. Nat. Nanotechnol. 2011, 66385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironava T.; Hadjiargyrou M.; Simon M.; Jurukovski V.; Rafailovich M. H. Gold Nanoparticles Cellular Toxicity and Recovery: Effect of Size, Concentration and Exposure Time. Nanotoxicology 2010, 41120–137. [DOI] [PubMed] [Google Scholar]
- Tkachenko A. G.; Xie H.; Liu Y.; Coleman D.; Ryan J.; Glomm W. R.; Shipton M. K.; Franzen S.; Feldheim D. L. Cellular Trajectories of Peptide-Modified Gold Particle Complexes: Comparison of Nuclear Localization Signals and Peptide Transduction Domains. Bioconjugate Chem. 2004, 153482–490. [DOI] [PubMed] [Google Scholar]
- Lynch I.; Cedervall T.; Lundqvist M.; Cabaleiro-Lago C.; Linse S.; Dawson K. A. The Nanoparticle–Protein Complex as a Biological Entity; a Complex Fluids and Surface Science Challenge for the 21st Century. Adv. Colloid Interface Sci. 2007, 134–1350167–174. [DOI] [PubMed] [Google Scholar]
- Alkilany A. M.; Nagaria P. K.; Hexel C. R.; Shaw T. J.; Murphy C. J.; Wyatt M. D. Cellular Uptake and Cytotoxicity of Gold Nanorods: Molecular Origin of Cytotoxicity and Surface Effects. Small 2009, 56701–708. [DOI] [PubMed] [Google Scholar]
- Thurn K.; Brown E.; Wu A.; Vogt S.; Lai B.; Maser J.; Paunesku T.; Woloschak G. Nanoparticles for Applications in Cellular Imaging. Nanoscale Res. Lett. 2007, 29430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Stellacci F. Effect of Surface Properties on Nanoparticle–Cell Interactions. Small 2010, 6112–21. [DOI] [PubMed] [Google Scholar]
- Albanese A.; Chan W. C. W. Effect of Gold Nanoparticle Aggregation on Cell Uptake and Toxicity. ACS Nano 2011, 575478–5489. [DOI] [PubMed] [Google Scholar]
- Oyelere A. K.; Chen P. C.; Huang X.; El-Sayed I. H.; El-Sayed M. A. Peptide-Conjugated Gold Nanorods for Nuclear Targeting. Bioconjugate Chem. 2007, 1851490–1497. [DOI] [PubMed] [Google Scholar]
- Conner S. D.; Schmid S. L. Regulated Portals of Entry into the Cell. Nature 2003, 422692737–44. [DOI] [PubMed] [Google Scholar]
- Ando J.; Fujita K.; Smith N. I.; Kawata S. Dynamic Sers Imaging of Cellular Transport Pathways with Endocytosed Gold Nanoparticles. Nano Lett. 2011, 11125344–5348. [DOI] [PubMed] [Google Scholar]
- Connor E. E.; Mwamuka J.; Gole A.; Murphy C. J.; Wyatt M. D. Gold Nanoparticles Are Taken up by Human Cells but Do Not Cause Acute Cytotoxicity. Small 2005, 13325–327. [DOI] [PubMed] [Google Scholar]
- Chithrani D. B. Intracellular Uptake, Transport, and Processing of Gold Nanostructures. Mol. Membr. Biol. 2010, 277299–311. [DOI] [PubMed] [Google Scholar]
- Chithrani B. D.; Chan W. C. W. Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007, 761542–1550. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Neuss S.; Leifert A.; Fischler M.; Wen F.; Simon U.; Schmid G.; Brandau W.; Jahnen-Dechent W. Size-Dependent Cytotoxicity of Gold Nanoparticles. Small 2007, 3111941–1949. [DOI] [PubMed] [Google Scholar]
- de la Fuente J. M.; Berry C. C. Tat Peptide as an Efficient Molecule to Translocate Gold Nanoparticles into the Cell Nucleus. Bioconjugate Chem. 2005, 1651176–1180. [DOI] [PubMed] [Google Scholar]
- Tkachenko A. G.; Xie H.; Coleman D.; Glomm W.; Ryan J.; Anderson M. F.; Franzen S.; Feldheim D. L. Multifunctional Gold Nanoparticle–Peptide Complexes for Nuclear Targeting. J. Am. Chem. Soc. 2003, 125164700–4701. [DOI] [PubMed] [Google Scholar]
- Feldherr C. M.; Akin D. Regulation of Nuclear Transport in Proliferating and Quiescent Cells. Exp. Cell Res. 1993, 2051179–186. [DOI] [PubMed] [Google Scholar]
- Feldherr C. M.; Lanford R. E.; Akin D. Signal-Mediated Nuclear Transport in Simian Virus 40-Transformed Cells Is Regulated by Large Tumor Antigen. Proc. Natl. Acad. Sci. U.S.A. 1992, 892211002–11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie S.; Emory S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 27553031102–1106. [DOI] [PubMed] [Google Scholar]
- Schlücker S. Sers Microscopy: Nanoparticle Probes and Biomedical Applications. ChemPhysChem 2009, 109–101344–1354. [DOI] [PubMed] [Google Scholar]
- Faulds K.; Smith W. E.; Graham D.; Lacey R. J. Assessment of Silver and Gold Substrates for the Detection of Amphetamine Sulfate by Surface Enhanced Raman Scattering (SERS). Analyst 2002, 1272282–286. [DOI] [PubMed] [Google Scholar]
- Farquharson S.; Gift A.; Shende C.; Inscore F.; Ordway B.; Farquharson C.; Murren J. Surface-Enhanced Raman Spectral Measurements of 5-Fluorouracil in Saliva. Molecules 2008, 13102608–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seballos L.; Zhang J. Z.; Sutphen R. Surface-Enhanced Raman Scattering Detection of Lysophosphatidic Acid. Anal. Bioanal. Chem. 2005, 3835763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Yang T.; Lin J. Spectral Analysis of Human Saliva for Detection of Lung Cancer Using Surface-Enhanced Raman Spectroscopy. J. Biomed. Opt. 2012, 173037003–037005. [DOI] [PubMed] [Google Scholar]
- Feng S.; Chen R.; Lin J.; Pan J.; Wu Y.; Li Y.; Chen J.; Zeng H. Gastric Cancer Detection Based on Blood Plasma Surface-Enhanced Raman Spectroscopy Excited by Polarized Laser Light. Biosens. Bioelectron. 2011, 2673167–3174. [DOI] [PubMed] [Google Scholar]
- Kim B.; Han G.; Toley B. J.; Kim C.-k.; Rotello V. M.; Forbes N. S. Tuning Payload Delivery in Tumour Cylindroids Using Gold Nanoparticles. Nat. Nanotechnol. 2010, 56465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailor M. J.; Park J.-H. Hybrid Nanoparticles for Detection and Treatment of Cancer. Adv. Mater. 2012, 24283779–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y.; Chung A.; Erickson D. Surface Enhanced Raman Spectroscopy and Its Application to Molecular and Cellular Analysis. Microfluid. Nanofluid. 2009, 63285–297. [Google Scholar]
- Escoriza M. F.; VanBriesen J. M.; Stewart S.; Maier J. Raman Spectroscopic Discrimination of Cell Response to Chemical and Physical Inactivation. Appl. Spectrosc. 2007, 618812–823. [DOI] [PubMed] [Google Scholar]
- Ong Y. H.; Lim M.; Liu Q. Comparison of Principal Component Analysis and Biochemical Component Analysis in Raman Spectroscopy for the Discrimination of Apoptosis and Necrosis in K562 Leukemia Cells: Errata. Opt. Express 2012, 202225041–25043. [DOI] [PubMed] [Google Scholar]
- Xie W.; Wang L.; Zhang Y.; Su L.; Shen A.; Tan J.; Hu J. Nuclear Targeted Nanoprobe for Single Living Cell Detection by Surface-Enhanced Raman Scattering. Bioconjugate Chem. 2009, 204768–773. [DOI] [PubMed] [Google Scholar]
- Pajerowski J. D.; Dahl K. N.; Zhong F. L.; Sammak P. J.; Discher D. E. Physical Plasticity of the Nucleus in Stem Cell Differentiation. Proc. Natl. Acad. Sci. U.S.A. 2007, 1044015619–15624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch A. R. Higher Levels of Organization in the Interphase Nucleus of Cycling and Differentiated Cells. Microbiol. Mol. Biol. Rev. 2000, 641138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J. W.; Taylor D. S.; Thompson D. L. The Effect of Cell Fixation on the Discrimination of Normal and Leukemia Cells with Laser Tweezers Raman Spectroscopy. Biopolymers 2009, 912132–139. [DOI] [PubMed] [Google Scholar]
- Kniggendorf A.-K.; Gaul T. W.; Meinhardt-Wollweber M. Effects of Ethanol, Formaldehyde, and Gentle Heat Fixation in Confocal Resonance Raman Microscopy of Purple Nonsulfur Bacteria. Microsc. Res. Tech. 2011, 742177–183. [DOI] [PubMed] [Google Scholar]
- Xie H.; Hu L.; Li G. Sh-Sy5y Human Neuroblastoma Cell Line: In Vitro Cell Model of Dopaminergic Neurons in Parkinson’s Disease. Chin. Med. J. 2010, 12381086–1092. [PubMed] [Google Scholar]
- Fore S.; et al. Raman Spectroscopy of Individual Monocytes Reveals That Single-Beam Optical Trapping of Mononuclear Cells Occurs by Their Nucleus. J. Opt. 2011, 134044021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S.; Lin J.; Huang Z.; Chen G.; Chen W.; Wang Y.; Chen R.; Zeng H. Esophageal Cancer Detection Based on Tissue Surface-Enhanced Raman Spectroscopy and Multivariate Analysis. Appl. Phys. Lett. 2013, 1024043702–043702–043704. [Google Scholar]
- de Groot P. J.; Postma G. J.; Melssen W. J.; Buydens L. M. C.; Deckert V.; Zenobi R. Application of Principal Component Analysis to Detect Outliers and Spectral Deviations in near-Field Surface-Enhanced Raman Spectra. Anal. Chim. Acta 2001, 4461–271–83. [Google Scholar]
- Sampath P.; Pritchard D. K.; Pabon L.; Reinecke H.; Schwartz S. M.; Morris D. R.; Murry C. E. A Hierarchical Network Controls Protein Translation During Murine Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell 2008, 25448–460. [DOI] [PubMed] [Google Scholar]
- Notingher I.; Bisson I.; Bishop A. E.; Randle W. L.; Polak J. M. P.; Hench L. L. In Situ Spectral Monitoring of Mrna Translation in Embryonic Stem Cells During Differentiation in Vitro. Anal. Chem. 2004, 76113185–3193. [DOI] [PubMed] [Google Scholar]
- Notingher I. Raman Spectroscopy Cell-Based Biosensors. Sensors 2007, 781343–1358. [Google Scholar]
- Boyd A.; McManus L.; Burke G.; Meenan B. Raman Spectroscopy of Primary Bovine Aortic Endothelial Cells: A Comparison of Single Cell and Cell Cluster Analysis. J. Mater. Sci.: Mater. Med. 2011, 2281923–1930. [DOI] [PubMed] [Google Scholar]
- Kneipp J.; Kneipp H.; Wittig B.; Kneipp K. Novel Optical Nanosensors for Probing and Imaging Live Cells. J. Nanomed. Nanotechnol. 2010, 62214–226. [DOI] [PubMed] [Google Scholar]
- Stone N.; Kendall C.; Smith J.; Crow P.; Barr H. Raman Spectroscopy for Identification of Epithelial Cancers. Faraday Discuss 2004, 126, 141–157discussion 169–183.. [DOI] [PubMed] [Google Scholar]
- Stewart S.; Fredericks P. M. Surface-Enhanced Raman Spectroscopy of Amino Acids Adsorbed on an Electrochemically Prepared Silver Surface. Spectrochim Acta A 1999, 557–81641–1660. [Google Scholar]
- Grigoryev S. A.; Bulynko Y. A.; Popova E. Y. The End Adjusts the Means: Heterochromatin Remodelling During Terminal Cell Differentiation. Chromosome Res. 2006, 14153–69. [DOI] [PubMed] [Google Scholar]
- Ghita A.; Pascut F. C.; Mather M.; Sottile V.; Notingher I. Cytoplasmic Rna in Undifferentiated Neural Stem Cells: A Potential Label-Free Raman Spectral Marker for Assessing the Undifferentiated Status. Anal. Chem. 2012, 8473155–3162. [DOI] [PubMed] [Google Scholar]
- Kneipp J.; Kneipp H.; McLaughlin M.; Brown D.; Kneipp K. In Vivo Molecular Probing of Cellular Compartments with Gold Nanoparticles and Nanoaggregates. Nano Lett. 2006, 6102225–2231. [DOI] [PubMed] [Google Scholar]
- Chan J. W.; Lieu D. K.; Huser T.; Li R. A. Label-Free Separation of Human Embryonic Stem Cells and Their Cardiac Derivatives Using Raman Spectroscopy. Anal. Chem. 2009, 8141324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.; Gu H. Probing of Cancer Cell Apoptosis by SERS and LSCM. Proc. SPIE 2009, 10.1117/12.835118. [DOI] [Google Scholar]
- Guillot J.-G.; Pézolet M.; Pallotta D. Laser Raman Spectra of Calf Thymus Histones H1, H2a, and H2b. Biochim. Biophys. Acta 1977, 4912423–433. [DOI] [PubMed] [Google Scholar]
- Bruno S.; Ardelt B.; Skierski J. S.; Traganos F.; Darzynkiewicz Z. Different Effects of Staurosporine, an Inhibitor of Protein Kinases, on the Cell Cycle and Chromatin Structure of Normal and Leukemic Lymphocytes. Cancer Res. 1992, 522470–473. [PubMed] [Google Scholar]
- Th’ng J. P.; Guo X. W.; Swank R. A.; Crissman H. A.; Bradbury E. M. Inhibition of Histone Phosphorylation by Staurosporine Leads to Chromosome Decondensation. J. Biol. Chem. 1994, 269139568–9573. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.