

Summary
Immunity to respiratory virus infection is governed by complex biological networks that influence disease progression and pathogenesis. Systems biology provides an opportunity to explore and understand these multifaceted interactions based on integration and modeling of multiple biological parameters. In this review, we describe new and refined systems‐based approaches used to model, identify, and validate novel targets within complex networks following influenza and coronavirus infection. In addition, we propose avenues for extension and expansion that can revolutionize our understanding of infectious disease processes. Together, we hope to provide a window into the unique and expansive opportunity presented by systems biology to understand complex disease processes within the context of infectious diseases.
Keywords: systems biology, influenza, coronavirus, SARS‐CoV, H5N1, H1N1, MERS‐CoV
This article is part of a series of reviews covering Immunity to Viruses appearing in Volume 255 of Immunological Reviews.
Introduction
The systems biology paradigm argues that the study of complex biological systems requires predictions based on detailed integration of multiple biological parameters including genomic, proteomic, clinical, and pathologic experimental data 1, 2. It argues that reductionist approaches that focus on individual components within the system are inherently limited in both scope and potential 3. In contrast, systems‐based approaches provide the opportunity to identify novel insights, define systems wide interactions, and decipher the organization of higher order interaction networks that regulate the function of complex biological systems 4. For areas like infectious disease research and immunology, this approach holds significant promise in identifying common and unique features driving pathogenic outcomes in the host and between disparate pathogens 5, 6, 7.
For virulent respiratory viruses like sever acute respiratory syndrome (SARS), coronavirus (CoV), and influenza A virus (IAV), disease outcomes are governed by intricate interactions between multiple immune and viral processes. To understand these complex biological responses requires integration of multiple biological parameters through common experimental systems, uniform methodologies, and variable pathogenic inputs 8. The resulting datasets and subsequent modeling analysis predict key regulatory checkpoints, define protective/pathogenic outcomes, and require additional rounds of validation, data analysis, and model refinement, thus highlighting the iterative cycles intended by systems biology. In this way, the systems biology paradigm drives understanding of complex systems through identification of novel, important components.
The approach is not without criticism. Skeptics of systems biology criticize its high cost, slow pace, and descriptive results 2. Although these charges reflect the initial development of any new field, the complaints are not at the root of the disillusionment with systems biology. Rather, subscribers of systems biology also overemphasized the limitations of reductionist science and the corresponding lack of progress in complex diseases treatments for cancer, human immunodeficiency virus infection, and diabetes 9. Systems biology was heralded as the evolution and replacement of these approaches, providing novel insights through hypothesis‐neutral and discovery‐based approaches 4. However, it has also been fraught with setbacks and challenges, leading to criticisms on its reliance on reductionist‐based approaches for validation, lack of hypothesis‐driven direction, and declaration that the entire endeavor as a fishing expedition with a limited, non‐specific catch 9. This conflict highlights the great irony of systems biology; an approach designed to integrate biological data has successfully divided the scientific field.
Despite the conflict, systems biology and reductionist‐based approaches are not antithetical 10. While the individual merits can be argued, the combination of these approaches has the potential to revolutionize our understanding of biology. Reductionist‐based science can benefit from the expansion of understanding generated by the systems‐based approach to link pathways and targets to disease outcomes. Similarly, systems biology can utilize mechanistic insights and rigorous validation studies derived from reductionist approaches to refine and expand their models. In this way, these competing methods can form a symbiotic relationship and drive forward our understanding of complex biological systems. In this review, we offer a path to reconcile the systems biology paradigm with reductionist science employing global tenets, demonstrating specific examples, and outlining future directions. Our hope is that this blueprint can provide a bridge between the two approaches and result in expanded understanding of immunology and infectious disease going forward.
The foundation of systems virology
Although new technologies, high‐throughput datasets, and substantial resources play important roles, the key factor in the success of a systems biology group is the ability to communicate and interact across disciplines. The systems biology paradigm requires the integration of numerous components to successfully interrogate and evaluate the connectivity that exists between biological and genomic information (Fig. 1). Merging massive biological and clinical correlates with mathematical and computational modeling followed by validation and extension represents one of the most challenging aspects of the systems biology approach. Building a multidisciplinary team of independent researchers provides a means to rapidly integrate these different disciplines; however, the process is intrinsically difficult and requires dedicated effort and continuous interaction. For example, mathematical modeling often can exclude 99% of possible targets, leaving 1% for examination; yet for transcriptional datasets, this can result in >1000 possible gene targets which prove unmanageable for testing and validation. Similarly, the absence of mock treatments or sufficient sample numbers greatly reduces the power and utility of any modeling algorithm. In each case, consistent communication is required to harmonize key issues in study that maintain experimental and statistical rigor as well as model performance. In our experience, constant dialog through multimedia channels, and more importantly, via face‐to‐face meetings provides an essential dynamic for developing coordinated experimental and modeling platforms that maintain statistical rigor, inform across disciplines, and promote key relationships for program performance. Without this underlying foundation, the systems biology paradigm would ultimately fail to produce any useful insights into biological functions.
Figure 1.
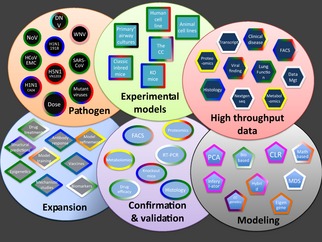
Systems biology requires integration across numerous components and disciplines. The experimental, modeling, and validation components utilized within our systems biology program divided into six functional categories. Each color within the border of the component signifies the various laboratory groups contributing to this component generation or analysis. Green – Baric RS (UNC); Blue – Heise MT (UNC); Red – Kawaoka Y(Wisconsin); Teal – Messer W(OHSU); Maroon – De Silva A(UNC); Pink – Gale (Univ. Washington); Purple – Pardo Manuel de Villena (UNC); Black – UNC Cystic Fibrosis Center; White – Katze (Univ. Washington); Yellow – Metz (PNNL); Fuchsia – Waters (PNNL); Orange – McWeeney (OHSU).
The tenets of our systems biology approach
The basic model of systems biology builds on an iterative cycle that includes experimental design, high‐throughput sample acquisition, data analysis and integration, predictive modeling, biological validation, and model refinement. From this approach, systems biology was expected to provide comprehensive knowledge, testable models, and new insights on improved, personalized care, diagnostics, and therapeutics for the treatment of human diseases. In some respects, this goal has been achieved in a variety of areas including vaccine efficacy, cancer predictions, viral diagnostics and prognostics, and most recently, infectious diseases 11, 12, 13, 14, 15, 16. However, many critics argue that most systems‐based reports are descriptive in nature, lack functional consequence, and omit necessary biological validation 2, 9. With this in mind, we applied the tenets described below to address these criticisms while maintaining the basic systems biology model structure (Fig. 2).
Figure 2.
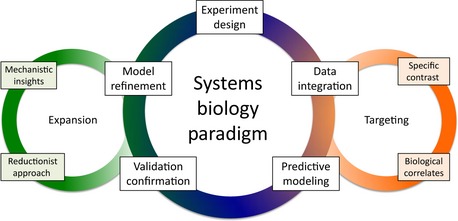
A refined systems biology paradigm layers in targeting and expansion. The standard systems biology paradigm includes an iterative cycle consisting of experimental design, data integration, predictive modeling, confirmation/validation, and model refinement (Blue circle). The new approach adds a targeting module that incorporates specific contrasts and biological knowledge to refine and improve the modeling outputs (Orange circle). Similarly, an expansion module adds the opportunity to explore the insights with further analysis using both systems‐ and reductionist‐based approaches to derive mechanistic insights (Green circle).
Examination of specific contrast
In its infancy, systems biology reports were primarily driven by the novelty of the technology and computing rather than the basic biological findings 2. These descriptive reports had minimal experimental contrast which limits biological insight providing silage for the critics of the approach. As the field developed, biologist added significant variation to their experimental design to generate contrast, but validated functional consequences remained the exception, rather than the rule. The main reason for this was the scope of the contrast. Studies modeling variation between mutant and wildtype (WT) virus infections could generate hundreds of differential targets for a specific time point; multiplied over the time course and infection conditions resulted in too many targets and not enough validation options.
To address this problem, our group applied specific contrast as a means to refine our target list and generate possible avenues of validation. The first step was to identify conditions in which specific differences are found in one or more biological output, but remain similar in other areas. Examples include difference in global transcription at early/intermediate/late time points 17, changes to overall lethality (18), and variation in functional gene subsets across different viral pathogens (19, Menachery VD, Eisfeld AJ, Josset L, Sims AC, Schaefer A, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Matzke MM, Webb‐Robertson BJ, Shukula AK, Burkett S, Metz TO, Pickles R, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS, in review). Using contrast as an initial parameter, modeling approaches can then focus on these specific conditions and time points, removing the majority superfluous data and extinguishing much of the noise within the system. The results are refined targets that relate to the specific contrasts within the systems that we seek to explore.
Incorporation of known modeling and biological data
In the effort to generate novel targets, systems biology approaches have often ignored known aspects of the experimental systems that they employ. The purpose of this is to generate data and targets that are not biased by the previous knowledge base. Although this premise is critically important for experimental design and data acquisition, target generation is inherently a biased approach. In the systems biology landscape, the bias is typically toward mathematical models built upon expansive biological databases; this methodology allows generation of novel targets independent of the previous knowledge bases. However, this approach discounts the prior experimental understanding and fails to utilize it in refining targets. Therefore, after the generation of unbiased gene sets that discriminate between different biological stimuli (e.g. different viruses, mutants, age, dose), we sought to incorporate known correlates into our modeling to refine and produce better targets for confirmation and validation studies. An example of this approach is integration of a known viral protein function or analysis of a process shown to be important to the infection. In addition, observation from broad systems‐based studies can also be utilized to refine the predictive model. In this way, we are able to improve the quality of target selection for downstream validation studies.
Confirmation and validation of functional consequences
In addition to their descriptive nature, systems biology–based reports have also been criticized for the lack of experimental verification. Often, the numerous targets make it nearly impossible to adequately demonstrate validation of the broad pathways identified. However, the first two tenets of our systems biology approach begin to address this scope issue. To fully address this criticism, our group began by making a distinction between confirmation and validation. Confirmation, for our purposes, is verification of the same biological output by a different method or in a different experimental condition. For example, array data generated from a cell line can be confirmed by either real‐time reverse transcriptase polymerase chain reaction of targeted genes or by repeating the experiment in an independent culture system. In contrast, validation occurs when initial findings are verified with a different biological output. For example, RNA expression data can be validated by proteomics and/or immunohistochemical staining. Together, confirmation and validation are paramount in our systems biology approach and are necessary to provide robust verification of systems‐based targets. Therefore, we have added increased emphasis to this area to allow pursuit of further studies through both iterative studies and the approach outlined in our final tenet.
Extension by reductionist‐based approaches
The iterative model of system biology builds upon validated targets to refine its approach and to design perturbations to generate new datasets for analysis. In this way, systems biology can produce novel insights, identify complex interactions, validate targets, and continuously add to our biological understanding of infectious disease. However, this process requires significant capital in terms of both time and resources. Importantly, continuation of the iterative cycle may not extend the observed finding, but rather identify new areas to explore. Therefore, our group examined ways to extend system biology findings beyond the iterative cycle by using standard reductionist techniques. Typically, these experiments query mechanistic aspects, seeking to identify required viral components or molecular processes involved in protective or pathogenic response and disease outcomes. Examples include studies of functional assays, identification of structural motifs, and/or utilization of knockout animals. Together, this approach effectively provides a second level of validation and provides similar outputs as reductionist‐based studies. In addition, these types of analysis can be programmed into the iterative cycle of systems biology and provide the perturbation necessary for the next round of data generation.
When considered collectively, the tenets outlined here do not represent a monumental shift in the systems biology model. In fact, many of the elements discussed above have been employed and advocated for by other systems biology groups 20. Instead, these tenets provide a means to meld systems biology and reductionist approaches to move away from broad, discovery‐based, and descriptive analysis and toward novel insights with validated functional consequence. Using this modified approach also provides an opportunity for extended understanding with either systems‐ or reductionist‐based approaches. The examples highlighted below demonstrate the efficacy of this methodology and provide a window into a possible future direction for systems‐based analysis.
Expanding understanding of viral proteins and host interaction
For decades, virology research has focused on understanding the function of viral proteins during infection. Utilizing overexpression systems, functional assays, and a variety of viral mutants, numerous RNA and DNA virus proteins have been characterized by reductionist approaches, leading to important biological findings and translational products like vaccines and therapeutics. These proteins have primarily been associated with either the viral life cycle (viral polymerases, structural proteins, etc.) or viral–host interaction like immune antagonism. However, due to their limited genetic capacity, RNA viruses typically encode multifunctional proteins that perform numerous processes beyond the scope of viral replication or immune evasion. Often, identifying these additional processes may be very difficult to reveal using traditional reductionist approaches. In these situations, systems biology provides an opportunity to utilize detailed experimental data to predict and identify novel roles for viral proteins. This fact is illustrated by our examination of SARS‐CoV open reading frame 6 (ORF6) protein 21.
Previous work from our laboratory and others 22, 24 had established and initially characterized a unique group of specific ORF proteins at the 3′ end of the SARS‐CoV genome including ORF6. Although non‐essential for in vitro replication, initial screens quickly identified ORF6 as an interferon antagonist 23. Subsequent experiments established that ORF6 interfered with signal transducer and activator of transcription 1 (STAT1) signaling via binding karyopherin α2, trapping both the import factor and its partner in complex karyopherin β125, 26. The resulting block prevented STAT1 nuclear localization and interfered with induction of the antiviral state within the host. However, despite its role in antagonizing the type I interferon (IFN) response, mutant SARS‐CoV lacking ORF6 (ΔORF6) grows to titers equivalent to WT in both IFN competent (CALU3) and incompetent cells (Vero) 21. Similarly, type I IFN pretreatment has only a modest impact on viral replication of ΔORF6, resulting in reduced titers similar to WT SARS‐CoV (data not shown). These results argued that the absence of ORF6 is complemented by the myriad of other SARS‐CoV IFN antagonist 27. However, in vivo infection revealed significant attenuation of ΔORF6 pathogenesis; notably, this attenuation did not extend to viral titers within the lung. Together, the data argued that ORF6 plays a role beyond just IFN antagonism.
To fully investigate the impact of ORF6 on viral infection, we employed a systems biology–based approach that blended host RNA expression data with the previously known aspects of ORF6 function 21. The resulting analysis revealed enhanced transcription of host genes following ΔORF6 infection and identified >6000 differentially regulated gene as compared with WT. Modeling the data with a focus on gene ontology confirmed augmented expression of antiviral genes. However, the results also revealed stark differences in terms of nuclear signaling, cell proliferation, cell cycle, as well as metabolic processes, and demonstrated a role for ORF6 beyond just IFN antagonism. Additional analysis filtered the dataset by the known ability of ORF6 to inhibit karyopherin‐based transport; the resulting modeling revealed important transcriptional hubs that play a critical role in differential regulation of cellular processes. These hubs, which include CREB1, VDR, and p53, were then confirmed in primary human airway epithelial cultures (HAE) and validated using both proteomic and chromatin immunoprecipitation approaches. Together, the results suggested that in addition to IFN antagonism, ORF6 regulates several karyopherin‐dependent transcriptional hubs and alters host cell processes that impact SARS‐CoV pathogenesis.
The identification of ORF6 activity beyond IFN antagonism provided novel targets to analyze during infection. By exploring and understanding the impact of these host processes, we can develop new insights on the viral life cycle and the subsequent host response. For example, CREB1, VDR, and p53 have been identified as key regulators mediating differences between WT and ΔORF6 virus; knock‐out animals exist for these genes allowing further study and identification of additional downstream effectors. In addition, these areas may provide ORF6‐based targets for therapeutic development of vaccine and drug treatments that may be effective against SARS and other CoVs. Similarly, Creb1 and p53 have been implicated in impacting influenza pathogenesis, and therapeutics may also have efficacy against IAV 28, 29, 30, 31. Current efforts in our laboratory seek to confirm and validate these findings in vivo; modeling mice infected with ΔORF6 and WT SARS‐CoV provides both confirmation and prioritization of in vitro generated targets. Combining these datasets will also provide an avenue for increased understanding of ORF6 function as well as a means to test possible therapeutics. These studies are currently ongoing.
The approach taken in these studies has also been used by other groups to query important aspects of pathogenesis using mutant viruses; the resulting analysis has provided novel insights following SARS‐CoV 32, HSV‐1 33, and influenza 34 infection. Together, these studies illustrate a trend that has implications on future systems‐based analysis of viral proteins. As mentioned previously, one tenet of our approach focuses on contrasts within the system. Using ΔORF6 studies as a template 21, mutants with ablated viral protein activity can be examined and compared with WT virus to quickly identify differential host gene expression. These host responses can then be modeled to determine broad differences based on gene ontology, pathway disruption, or a variety of other transcription‐based categorizations. This type of analysis provides a window into the possible functions of a specific viral protein and an avenue to begin further studies. Notably, known functions derived from reductionist approaches can also be integrated into the modeling which provides both refined targeting as well as incorporating the second tenet of our systems‐based approach. Next, identified targets can be confirmed and validated by a variety of approaches including proteomics analysis, drug treatments, or knock‐down studies to demonstrate functional significance in vitro. Finally, further model refinement and validation can be provided by in vivo systems‐based analysis. Together, these steps outline a blue print for systems‐based characterization of novel viral protein function and have been initiated for several viral proteins in both SARS‐CoV (ExoN, NSP16, ORF3) and IAV (NS1, PB1, PB2).
Identifying host factors that contribute to pathogenesis
Host responses to virus infection are usually regulated by oligogenic traits, resulting in disparate disease outcomes following infection. To understand these complex relationships between viral and host factors, in vivo experimentation represents an important area of viral pathogenesis research. While significant insights can be achieved by in vitro analysis, animal models provide a robust and complex biological system to evaluate the numerous parameters of pathogenesis including viral replication, immune cell infiltration, and other systemic host responses. This fact explains the existence and reliance on mouse models of infection for numerous viral pathogens including SARS‐CoV and IAV. However, traditional in vivo studies have focused on either known target genes that regulate immunity or have utilized viral mutants for comparative analysis with WT viruses. The result has been extensive examination and understanding of innate and adaptive immune components that regulate viral disease mostly in terms of viral replication. Meanwhile, elements that regulate other aspects of viral disease including systemic inflammation, clinical disease severity, and tissue repair remain undefined and understudied. Our systems‐based approaches provide an opportunity and a means to assay these less studied elements.
The systems virology animal model employed by our group infects 20‐week‐old C57BL6/J (B6) mice with a dose range of either SARS‐CoV or IAV over a 7‐day time course. Both mouse age and dose range were used to provide a robust spectrum of disease measurements that included viral replication kinetics, clinical disease severity, pathology changes, and variation in host transcript and proteomics. Using the transcriptomics dataset from both SARS‐CoV and IAV challenge, the modelers analyzed the data with a context likelihood of relatedness method (CLR); this approach uses correlation of expression profiles to score interactions between regulator and target genes to generate networks that encompassed significant elements of in vivo disease 35, 36. For SARS‐CoV and IAV, CLR‐based methodologies resulted in identification of differential networks associated with transcription regulation, chemotaxis, lymphocyte activation, lipid metabolism, and others (Fig. 3). However, the results required additional narrowing to identify target pathways that could be validated. Therefore, we applied the first two tenets of our systems biology approach: contrast and incorporation of biological knowledge.
Figure 3.
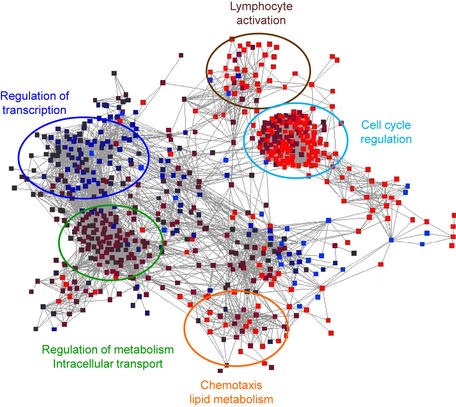
Context likelihood of relatedness method reveals functional modules that impact SARS ‐CoV or influenza infection in vivo . RNA expression data from in vivo infection of SARS‐CoV and H5N1‐VN1203 were utilized to infer coexpression networks between genes and help identify functional modules important during respiratory virus infection. Topological analysis permits identification of bottlenecks of network communication and provides potential target areas for knockdown studies. Data representative of RNA expression 4‐day post‐SARS‐CoV infection (100 pfu dose) with red representing upregulation and blue representing downregulation relative to mock.
We focused on the SARS‐CoV dataset and used the measured outputs to identify and explore areas of contrast. In our examination, we noted similarities between the two highest SARS‐CoV doses in terms of viral replication kinetics, global gene expression changes, and pathology elements (18). However, these doses sharply diverge in terms of weight loss at late time points and overall lethality. This contrast provided an opportunity to explore pathways and networks that were different between lethal and sublethal disease. To incorporate these biological data, we utilized a weighted gene coexpression network analysis (WGCNA); this approach uses underlying correlation structures to partition gene expression into modules not biased by existing knowledge bases 37. Connectivity inferred by WGCNA is a strong predictor of biological function, and the most highly connected hub genes provide candidate mediators of disease that have been used in a variety of fields including cancer, plant, and genetics 37, 38, 39, 40, 41, 42, 43. Gene ontology analysis of this module revealed a strong categorization for extracellular matrix (ECM) remodeling and wound healing pathways. While previously shown to be important in lung diseases 44, 45, these pathways had been largely unstudied within the context of viral respiratory disease. To further narrow candidate pathways for confirmation and validation purposes, we evaluated the connectivity and module membership within the known biological pathways. The resulting analysis found urokinase pathway enriched by both criteria making it an ideal network for validation studies.
Returning to the plethora of systems generated data, we sought to address our third systems biology tenet: confirmation and validation. As a part of the broad ECM remodeling network, the urokinase pathway is primarily associated with regulation of plasminogen resulting in fibrin degradation/deposition 46, 47, 48, 49, 50. Therefore, we utilized paired in vivo proteomics data to analyze differences in urokinase pathway–related components following the lethal and sublethal doses. Our results showed a significant increase in several urokinase pathway–related proteins during the lethal dose as compared with the sublethal dose (18). These proteins, which include fibrin chains, fibronectin, and plasminogen, confirmed enhanced urokinase activity following the lethal challenge. Similarly, histology staining for fibrin deposition confirmed augmented staining during lethal SARS‐CoV infection. Finally, we chose to employ a standard reductionist approach, gene knock‐out, to further validate the role of urokinase pathway in vivo. Mice lacking Serpine1, an important negative regulator within the urokinase pathway, were challenged with SARS‐CoV and had increased weight loss, lung hemorrhage, and increased lethality as compared with control mice. Together, these data confirm a role for the urokinase pathway in regulating lethal SARS‐CoV disease and validated a target generated by our systems‐based modeling.
The approach taken to characterize the urokinase pathway's contribution in protecting from lethal SARS‐CoV disease was built upon the tenets of our systems biology approach. Collecting unbiased measurements from in vivo infection, we identified areas of contrast within the SARS‐CoV dataset, modified the models to incorporate this biological data, and generated refined targets for confirmation and validation. This approach clearly demonstrated an important role for the urokinase pathway in protection against lethal SARS‐CoV infection and may possibly lead to the development of therapeutics based on targets within this pathway. A similar approach has also been used to identify and validate other networks that regulate SARS‐CoV and IAV pathogenesis in vivo; these include complement components (18) as well as TNFRSF1b 14 and Kepi1 (McDermott JE, Gralinski LE , Eisfeld AJ, Mitchell HD, Bankhead A, Josset L, Tchitchek N, Chang J, Neumann G, Tilton SC, Li C, Fan S, Schäfer A, McWeeney S, Kawaoka Y, Baric RS, Waters KM , Katze MG, in review). Importantly, while analysis of the Serpine1 −/− data provides an avenue for reiterative systems biology studies and continued model refinement, we also plan to explore mechanistic and translational aspects of the urokinase pathway modification using traditional reductionist‐based approaches. Together, the combination of these two methods provides a means to expand our knowledge base in two different, but important directions.
The study also illustrates the utility of this systems biology–based approach in broadening the scope of viral pathogenesis research. While the wound healing and ECM remodeling pathways had been clearly established as important to lung disease, traditional viral pathogenesis research was unlikely to have explored these areas with sufficient depth to generate strong conclusions. As a result of this study, the urokinase pathway will likely be the subject of future experimentation by both systems‐based and reductionist approaches with a variety of pathogen, effectively expanding the scope of the research. The importance of this fact should not be diminished as this is a primary goal of systems biology that is beginning to be achieved.
Revisiting known biological correlates with expanded depth and breadth
The systems biology paradigm seeks to drive understating of complex biological systems by incorporating detailed integration of experimental data to construct models that predict and identify novel findings 2. However, although identification of novel insights is paramount, it would be short sighted to not apply these vast datasets to other areas of interest. The effort to collect comparable, unbiased data across pathogens and platforms provides a unique opportunity to both identify new insights through systems‐based modeling, and reexamine previously charted areas with increased depth and coverage. Driven by biological intuition rather than mathematical modeling alone, analysis based on systems biology data may provide novel insights into well‐studied areas of biology and enhance our understanding of complex biological systems. The efficacy of this approach is illustrated in our examination of cytokine responses, most notably interferon‐stimulated gene (ISGs) induction.
Cytokine responses following infection have been extensively studied over the past few decades and have resulted in well‐defined pathways for important processes including innate and adaptive immunity, inflammation, antiviral defense, etc. However, insights from these types of studies are hampered by the lack of uniform platforms, infection conditions, and data collection methods. Systems virology datasets alleviated these problems and permit analysis beyond a single treatment or infection condition. Therefore, in lieu of the typical unbiased modeling approach, we incorporated a network with known biological importance to virus infection: type I IFN and ISGs. Type I IFN induces a signaling cascade that arms immune defenses and serve to provide the first line of defense against viral pathogens 51, 52. Upon binding through the type I IFN receptor, IFN signaling initiates transcription of hundreds of ISGs that have antiviral, immune modulatory, and cell regulatory functions. Expression of these genes within host cells renders an inhospitable antiviral state critical to limiting viral infection in vitro and in vivo. While several prominent ISGs such as Mx, PKR, and OAS have been studied in depth, the vast majority have never been fully evaluated. However, the ISGs impact on viral infection is clear; in the absence of the type I IFN cascade, the host fails to mount effective immune responses and becomes very susceptible to viral challenge 53. Successful viral pathogens, including SARS‐CoV and IAV, have developed means to overcome specific IFN and ISG effector functions 27, 54, 55. Globally, ISGs as a class have broad functions beyond just direct viral antagonism including increased MHC class I and II expression, augmented immune infiltration and trafficking, and host protein shutoff 51. These processes can impede viral replication and pathogenesis; therefore, viruses likely attempt to short circuit global ISG induction, potentially through different means.
With the biological filter set, we next sought to evaluate the ISG response within the parameters of systems‐based datasets. Because ISGs vary based on cell and tissue type 56, Calu3 cells were treated with type I IFN to establish a consensus ISG list specific for respiratory cells. With this list, we then examined RNA expression of ISGs following SARS‐CoV, IAV H1N1‐CA04, and H5N1‐VN1203 infection; the resulting analysis revealed stark differences in terms of ISG induction between all three respiratory pathogens. Whereas IAV H1N1‐CA04 robustly induced the majority of ISGs following infection, H5N1‐VN1203 actively manipulated the ISG response with both up‐ and downregulation of ISG subsets. Finally, SARS‐CoV successfully delayed ISG until after peak viral titers had been achieved. Together, the data demonstrated the utility of incorporating known biological factors as a means to filter and identify novel patterns across pathogens.
Having established significant differences across IAV and SARS‐CoV strains, we next sought to confirm and validate these results. Examination of primary HAE demonstrated that ISG downregulation was maintained for SARS‐CoV and that robust induction was observed for H1N1‐CA04. Proteomics analysis from Calu3 cells also found robust production of ISG proteins for both SARS‐CoV and H1N1‐CA04; however, ISG protein production was delayed with similar kinetics as RNA expression following SARS‐CoV infection. In contrast, H5N1‐VN1203 infection failed to increase any detectable ISG production and resulted in reduced protein levels of ISGs expressed at a basal level. Together, these results validated the RNA expression analysis with paired protein measures.
Fulfilling the major tenets of our systems biology approach, we next sought to expand our studies to explore the mechanism of action for both SARS‐CoV and IAV. To answer these questions, we utilized reductionist‐based approaches but applied systems generated data where possible. Using RNA expression data, we asked and confirmed that ISG delay following SARS‐CoV was primarily due to delayed type I IFN production. Modeling downstream gene expression patterns, we also determined that H5N1‐VN1203 did not target transcriptional factors to mediate ISG antagonism. Next, we utilized RNA expression patterns to identify target genes for differential histone modification. The resulting experiment confirmed that H5N1‐VN1203 mediated incorporation of a repressive histone modification in the 5′ untranslated region of downregulated ISGs; this modification was absent in upregulated ISGs. Having established a role for epigenetic regulation in the ISG antagonism, we next sought to identify the involved viral components. Multidimensional scaling (MDS) analysis, a statistical technique to visualize similarities and differences between datasets, revealed informative insights for both SARS‐CoV and IAV infection (Fig. 4). MDS analysis comparing ISG expression following WT and ΔORF6 SARS‐CoV infection demonstrated little contrast between the two, suggesting a limited role for ORF6 in global ISG antagonism. In contrast, MDS analysis of H5N1 mutants revealed NS1 truncation resulted in closer alignment with H1N1‐CA04 ISG expression than WT; PB1 and PB2 mutants had some change, but aligned closer to the WT. Subsequent experiments revealed that NS1 truncation ablated ISG downregulation and no incorporation of the repressive histone marker. These findings suggest that epigenetic regulation via NS1, in addition to interference with sensing/signaling, contributes to IAV control of the type I IFN response. Together, these results established novel mechanistic insights into ISG antagonism by both SARS‐CoV and IAV.
Figure 4.
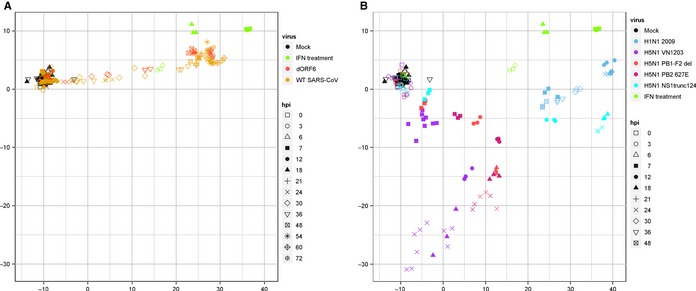
Multidimensional scaling analysis illuminates variation between wildtype and mutant viruses. Multidimensional scaling analysis provides a statistical method to evaluate ISG expression of a virus relative to the expression from other mutants. Analysis completed on Calu3 cells following mock (Black), type I interferon treatment (Green), or infection with (A) wildtype (WT) SARS‐CoV (Orange), ΔORF6 (Red) and (B) WT H5N1‐VN1203 (Purple), NS1‐trunc124 (Teal), PB1‐F2del (Red), PB2‐627E (Maroon), and WT H1N1‐CA04 (Blue).
The approach outlined in this section marks a significant departure from the traditional systems biology paradigm that focuses primarily on novel insights derived from unbiased mathematical modeling. Instead, this methodology requires well‐defined sets of biological phenomena and effectors (e.g. ISG effector genes, MHC class I/II expression networks, stress response, apoptotic markers) that are differentially regulated after virus infection to drive the analysis forward. In addition, cross‐virus comparisons provide contrast, validation, and variant response networks which combine to inform hypotheses for future reductionist and mechanistic studies. In the case of ISGs, it had been well established that these genes play an important role during viral infection; however, limitations in terms of cell type, pathogen, and experimental conditions had made analysis difficult. By applying this question to the systems virology dataset, we were able to filter our scope and generate a robust analysis comparing three highly pathogenic respiratory viruses with great depth and coverage. The results produced novel insights into how these virulent viruses manipulate an important arm of the immune response and have major implications for future studies and the development of therapeutics.
The study demonstrates the utility of mining the depth and breadth of systems biology data independent of traditional systems‐based analysis. With this in mind, our group has initiated similar studies to examine areas known to be important for viral pathogenesis including inflammation, cell‐cycle regulation, and autophagy. However, the most important implication may be the ability to duplicate this approach in examination of other areas of scientific research. One of the stated goals of systems biology is to provide datasets and research tools to the scientific community. However, although vast datasets are readily available, their utility has been limited by the complexity and multidisciplinary nature of traditional systems‐based analysis. The approach outlined here provides a means for traditional scientists to integrate known biological parameters to filter these massive datasets into useful, targeted analysis for their specific studies. The scope of this approach extends beyond just infectious disease, as other scientific disciplines including lung physiology, allergy research, and aging might benefit from examining the systems virology datasets. In this way, the approach effectively provides accessibility to these datasets and facilitates utilization by the scientific community.
Expanding systems‐based studies to translational applications
A major complaint of the systems biology approach is the glacial pace required to generate novel findings 2. Working through the systems biology model requires extensive high‐throughput data generation, significant modeling development, and substantial validation experiments to identify and verify discovery‐based hypotheses. Incorporation of all these components takes significant time and effort slowing the process for data discovery. However, once established, the systems‐based approach provides an ideal framework to quickly and accurately examine new experimental conditions utilizing the template from earlier experiments. The result can be rapid evaluation and insights into novel area of viral infection as illustrated by our examination of novel Middle East Respiratory Syndrome Coronavirus (MERS‐CoV).
In September 2012, a novel coronavirus, MERS‐CoV, was identified as a causative agent in two cases presenting with acute respiratory distress and renal failure. Subsequent surveillance revealed 15 additional cases derived from individuals either from or having recently traveled to the Middle East resulting in 11 deaths and a mortality rate or 64.7% (11/17). The severe respiratory infection coupled with the high mortality rate quickly drew comparisons with SARS‐CoV outbreak in 2002–2003; however, sequence lineage and host range suggested significant differences between these viruses 57. In addition, it was quickly established that MERS‐CoV did not use the SARS‐CoV entry receptor, human ACE2 58. Instead, dipeptidyl‐peptidase 4 has been identified as the functional MERS‐CoV entry receptor 59. Other differences observed between the viruses include cytopathic effect, tissue tropism, and sensitivity to type I IFN 58, 60. However, although these initial studies provided a small window into the pathogenesis of HCoV‐EMC, very little data existed beyond viral characterization. With this in mind, we sought to examine host responses to MERS‐CoV within the context of our systems virology paradigm 19.
Utilizing the same experimental design that was used for both SARS‐CoV and IAV, human airway cells were infected with MERS‐CoV at a high multiplicity of infection (MOI). Preliminary screening had confirmed rapid cytopathic effect in these cells; therefore, samples for viral replication and RNA analysis were harvested over the first 24 h, mimicking the time course used for the highly pathogenic H5N1‐VN1203 61. The results indicated that both SARS‐CoV and MERS‐CoV maintain similar levels of viral replication 19. Although MERS‐CoV induced significant cytopathic effects, both viruses grew to robust, equivalent titers over the time course. Examination of the host response also revealed broad similarities between SARS‐CoV and MERS‐CoV including biosynthesis/degradation pathways, IL17 activation, and innate immune recognition.
The host expression profile also indicated major differences between the viruses in terms of induction kinetics, pathway activation, and possible viral antagonism. MERS‐CoV induced a much more rapid host response as compared with SARS‐CoV with massive dysregulation by 18‐ and 24‐h postinfection. Although these gene changes shared some similarities with SARS‐CoV infection at later time point, the data suggested that the global host response is quite distinct between the two CoVs, especially in areas that included ubiquitination, lymphocyte signaling, and antigen presentation. In addition, we compared MERS‐CoV with IAV strains H5N1‐VN1203; the results demonstrated similarities beyond just high cytopathic effect including similarities in ISG manipulation between the two highly virulent viruses (Menachery VD, Eisfeld AJ, Josset L, Sims AC, Schaefer A, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Matzke MM, Webb‐Robertson BJ, Shukula AK, Burkett S, Metz TO, Pickles R, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS, in review). Together, the rapid assessment of the host response illustrates the utility of systems‐based approaches to newly emergent respiratory virus. By using specific contrast with systems‐based pathogen data and considering biological correlates like enhanced CPE, the analysis was able to provide novel insights into the pathogenesis of this newly emergent MERS‐CoV.
With its ability to cause severe respiratory disease and its high mortality rate, novel drug and vaccines against HCoV‐EMC, as well as future emerging CoVs, are also high priorities. Therefore, we utilized systems‐based approaches to generate and validate therapeutic options for MERS‐CoV infection. Our approach focused on early changes following infection, identifying upstream regulators and other compounds that may reverse host expression networks. Disruption of these networks potentially interferes with important aspects of the virus life cycle or replication and could effectively limit pathogenesis. The analysis resulted in identification of possible drug targets with significant potential based on Z‐score. We validated the most robust of these targets, SB203580, demonstrating efficacy against both HCoV‐EMC and SARS‐CoV in vitro 19. While in vivo studies in young and highly vulnerable aged animal models are needed, the results demonstrated the utility of systems‐based analysis in evaluating novel, emergent viruses and delivered on some of the promise of this approach in terms of therapeutic translation.
Globally, this approach highlights a powerful means to rapidly evaluate and characterize novel, emergent viruses. The analysis builds upon the infrastructure of systems biology using previously evaluated pathogens to provide a useful comparison. With these data in hand, MERS‐CoV was rapidly evaluated against the related SARS‐CoV in a common culture system. The result was a plethora of new data that greatly aided our understanding of the virus/host interactions. In addition, the systems biology data provided the opportunity to compare the novel CoV with other respiratory viruses such as H5N1‐VN1203. These comparisons allow identification of similarities and differences in terms of host response that might inform treatment and therapeutic options. Similarly, the analysis of multiple pathogens permits the development of expression signatures associated with virulence. For example, ISG manipulation by both H5N1‐VN1203 and MERS‐CoV could be used as a signature to rapidly evaluate the pathogenic potential of emergent or zoonotic viral strains. Similar approaches with other host expression pathways and the addition of other virulent and avirulent pathogens across several viral families might provide a mosaic of signatures to quickly and rapidly assess the danger posed by a particular emergent pathogen. Together, the data illustrate that the infrastructure of systems biology provides a unique and novel platform to evaluate emergent viruses and demonstrates the rapid analysis not typically associated with systems‐based analysis 62.
Moving systems biology forward
Systems‐based approaches have made significant and lasting contributions to a wide array of scientific fields over the past decade 11, 12, 13, 14, 15, 16. However, with its increased interest, expanded resources, and refined techniques, systems biology also has an opportunity to improve its targeting, results, and global impact. In the past, these advances have relied on application of new technologies for data generation 2; however, as systems biology matures, improvement will require novel approaches in terms of integration, modeling, and validation in addition to the newest data generation methods. Below, we highlight elements in each of these areas and how our group plans to incorporate them to improve our systems‐based approach.
Data expansion
The foundation of systems biology is built upon acquisition of high‐throughput, unbiased datasets from varying experimental conditions to model and predict important aspects of complex biological systems. In our initial approach, these studies utilized a blend of uniformity with variability achieved through common culture and animals systems, uniform experimental protocols, and variation in viral pathogens. The approach resulted in diverse, high‐throughput datasets that served as the basis for the development and application of our mathematical models. Our targets were further refined by other measurements including weight loss, lethality, and cell infiltration via lung histology. Together, this approach yielded novel insights that were subsequently confirmed, validated, and expanded. However, these conditions and readouts represent only a fraction of the systems‐based datasets that can be generated and analyzed by this platform.
As we move forward, our group seeks to use expanded experimental conditions to increase our input database. As demonstrated by our previous works, comparison across pathogen has yielded significant insight into respiratory infection. Whereas our current program has studied a variety of CoV and IAV precursors as well as mutants, future work endeavors to explore and compare these families to other respiratory pathogens including respiratory syncytial virus (RSV), pathogenic hantaviruses, and other microbial pathogens. Similarly, we plan to expand our experiments beyond the Calu3 cell line and HAE cultures used in our initial studies; primary type II pneumocytes, alveolar macrophage cultures, endothelial cells, and infiltrating immune cell populations are important targets for viral infection, and their response to infection is likely governed by cell‐type and tissue‐specific factors. Exploring this tissue specificity of respiratory pathogens provides insights into disease progression, keys to severity, and possible cell‐specific targets for therapeutic development. With regards to in vivo experiments, we plan to use mouse resources like the collaborative cross (discussed below) and age‐dependent models to provide additional scope and contrast for systems‐based analysis. In addition to altered and expanded experimental conditions, we also plan to increase the measured outputs available for analysis. In the past, high‐throughput data had been limited to RNA expression and proteomics data; moving forward, the newest technologies provide the opportunity to assess complex readouts like host genetic variation or phenotypic readouts like respiratory function, cell infiltration, phosphoproteomics, and metabolism. Together, these expanded experimental conditions and readouts represent just a few areas that systems biology can use moving forward to reveal complex insights into disease pathogenesis and severity. Below, we briefly describe the application of the data expansion tools to our systems‐based approaches.
The collaborative cross
Genetic variation plays a critical role in human disease as demonstrated by numerous genome‐wide association studies (GWAS) for a variety of ailments. However, exploring genetic diversity had been limited by the lack of experimental systems that model human outbred populations. The collaborative cross (CC), a novel panel of approximately 300 genetically related recombinant inbred lines, solves these problems as it was designed to model genetic diversity in outbred populations like humans 63, 64, 65, 66, 67, 68. Derived from eight founder lines, the CC provides extensive variation [47 million single nucleotide polymorphisms (SNP) and 4 million insertions/deletions, on the same order as those observed in the human genome], high reproducibility of genetically identical mouse lines, and customization via inbred intercrosses. Screening infectious disease outcomes in this resource provides the opportunity to understand how genetic variation within a population affects the host response to viral infection. Importantly, the CC is powerful enough to untangle the role of monogenic and oligogenetic traits in complex disease phenotypes by providing a diverse range of independent readouts ideal for modeling areas like inflammation, innate immunity, and lymphocyte responses 67. For example, SARS‐CoV infection in the founder lines results in dramatically divergent disease outcomes ranging from mild respiratory and clinical disease symptoms (<5% weight loss) to fulminant lethal disease in 2 or 3 days and a wide range in lethal doses ranging from <102 to >106, depending on the line (data not shown). Another initial study infected the precursor to the CC (the pre‐CC) with IAV and was limited to a single animal each from 155 lines 69. However, these data from these genetically unique animals at a single time point were sufficient to define quantitative trait loci associated with host responses including virus‐induced weight loss, titer, pulmonary edema, neutrophil recruitment, and transcriptional expression. Similar results with SARS‐CoV (data not shown) demonstrated the success of this approach even on such a small‐scale experiment. Subsequently, our group is seeking to identify natural polymorphisms and oligogenetic traits that regulate the immune response following SARS‐CoV, IAV, and West Nile Virus (WNV) infection in the full CC over the next 5 years. Measuring a wide array of immune parameters at multiple time points in >150 distinct lines, this project will effectively produce the largest and most comprehensive database for in vivo immune responses, host expression patterns, and disease outcomes in a reproducible animal model of outbred human populations following viral infection. Applying systems‐based techniques to these datasets provides the opportunity to understand the virus–host interaction networks that regulate disease severity and protective immunity, leading to new therapeutic intervention strategies. Together, the approach has the potential to revolutionize our understanding of the immune system and make significant contributions to the understanding and treatment of human disease.
Whole‐body plethysmography
For respiratory pathogens such as SARS‐CoV and IAV, small animal models recapitulate many elements of human disease including viral replication, cell infiltration, and adaptive immune responses. However, measurement of clinical disease has been primarily limited to indirect measures like weight loss and lethality which have little direct correlation with human disease. Although other measurements, including histology and flow cytometry analysis, can provide a window into respiratory function in vivo, previous studies suggest that they do not necessarily correlate well with pathogenesis and survival 69. Therefore, we sought to add an additional dimension to systems‐based analysis with measurement of lung function following viral infection. Using the Buxco whole‐body unrestrained plethysmography system, respiratory function can be tracked in the same animal over a time course and has already been utilized in studies for RSV, WNV, and IAV 69, 70, 71. In our system, SARS‐CoV, IAV, or mock infected animals are placed into the plethysmography chamber, and respiratory outputs are measured every 2 s for a 5 min duration following a set acclimation period. Measured over a time course, the outputs include a variety of direct and derived values that indicate aspects of respiratory function including calculated airway resistance (penH), breath frequency, lung volumes, and flow rates. Together, these readouts provide a direct measure of respiratory function following viral infection and when combined with resources like the CC, generate high‐throughput datasets ideal for systems‐based analysis.
Measuring immune infiltration
While far from a novel procedure, analysis of lung infiltration by flow cytometry and histopathology has primarily been used for confirmation and validation purposes rather than as inputs for systems‐based modeling. However, the combination of pathogens, dose scaling, and varying time points found in our systems biology infrastructure yields an enormous amount of high‐throughput data ideal for target generation. Flow cytometry analysis of infected lungs evaluates the composition, kinetics, and magnitude of the immune infiltrate for a wide array of immune cells. Similarly, histopathology provides information on the structural aspects of lung infiltration including airway denuding, perivascular cuffing, and hyaline membrane formation. Differences in both flow and histopathology have been previously shown to influence pathogenesis following viral infection, yet the genetic factors and host response patterns that regulate pathogenic or protective responses remain poorly understood, especially in outbred populations. Therefore, application of systems‐based analysis to these data will likely yield fruitful targets for further examination by transcriptional or epigenetic profiling of specific cell types following infection.
Exploring adaptive immunity
While reductionist‐based approaches have provided a foundation for our understanding, they have fallen short in terms of modeling the adaptive immune response for human diseases. This is because end‐stage effector functions like antibody production and T‐cell activation are influenced by numerous host pathways and complex interactions in a time‐dependent fashion. To understand these pathways requires probing the adaptive response to virus infection with systems‐based analysis. However, although virus, dose, and time point are critical elements in initiating the response, a diverse genetic background is required to generate sufficient contrast to model and identify key elements of the adaptive immune response. This diverse system will be achieved with incorporation of the CC. Building on the diversity of the CC, viral‐specific T‐cell responses from different lines will be identified by intracellular cytokine staining, surrogate activation marker identification, as well as peptide tetramer staining of antigen‐specific T cells; acquiring these data will identify key regulators through differential expression in the high‐throughput datasets. Similarly, antibody neutralization titers and ELISA titers will be employed to distinguish genes and pathways that contribute to the end‐stage B‐cell function. Together, this technique adds adaptive immune responses as a biological correlate to refine modeling and identify novel targets related to induction of adaptive immune responses.
Revolutionizing and rethinking biological modeling
With the development of new outputs and experimental conditions, the task of modeling and integrating multiple variables gains increased importance. In its infancy, systems biology relied primarily on RNA expression data as the main input for its mathematical modeling and target generation. As the field has matured, new high‐throughput data sources were established as raw data for these modeling approaches. Similarly, the expansion of systems biology also led to the development and application of new modeling paradigms based on statistical tests (Fischer's summary statistic), module mapping (WGCNA), and fold‐change z‐score (CLR). However, for the most part, these modeling systems existed in isolation interacting primarily with simple datasets and experimental conditions. Advancement of systems‐based modeling requires integration of these high‐throughput datasets, varying experimental systems, and novel mathematical approaches.
Several groups, including ours, have sought to meld these diverse aspects to develop models that refine and enhance targets for validation and confirmation within systems biology. One approach utilizes different high‐throughput data types to generate common, refined targets. For example, our SARS‐ΔORF6 studies utilized CLR‐based modeling of both transcriptomics and proteomic data to identify transcriptional hubs regulated by karyopherins; this approach narrowed the targets and provided manageable validation avenues. Another approach seeks to examine modeling approaches across experimental systems with common biological stimuli. Studies with IAV H5N1 exemplify this approach as conservation of network modeling was observed between varying in vitro and in vivo models for interferon responses, inflammasome, and hypercytokinemia 73. A third approach attempts to bridge varying mathematical and computational approaches to yield a more complete picture of complex biological systems. Using this method, our group combined CLR‐based analysis with WGCNA module mapping to create a blended model system that helped to identify important regulators influencing pathogenesis including Tnfrsf1b and Kepi1 (JE McDermott, LE Gralinski, AJ Eisfeld, HD Mitchell, A. Bankhead, L Josset, N Tchitchek, J Chang, G Neumann, SC Tilton, C Li, S Fan, A Schäfer, S McWeeney, Y Kawaoka, RS Baric, KM Waters, MG Katz, in review). Another example generated meta‐analysis from SARS‐CoV, IAVs H5N1‐VN1203, and H1N1‐1918 infection data at multiple doses using statistical tests, module mapping, and z‐score–based analysis 74. The study suggested that increased magnitude, rather than differential gene expression, drove differences in host damage between low and high pathogenic infection conditions. Together, these studies illustrate simple steps taken toward integrating aspects of the modeling approaches; however, significant advancement is still necessary as systems biology research moves forward.
Among the areas of primary need is the ability to multiplex high‐throughput data in the computation and mathematical models. With the development and incorporation of numerous new high‐throughput data sources including the CC, the next leap in model design must move beyond single and dual input analysis. Incorporating multiple biological parameters is required to understand complex biological systems involved during infectious disease pathogenesis. Along with developing new models for areas like whole‐body plethysmography, immune infiltration, and metabolomics, systems biology teams must devote significant resources toward integrating these models with other output measures. In addition, incorporation of multiple modeling approaches will help refine and advance the responses; new algorithms like the inferelator and cross‐validation approaches provide a blueprint to these advances 61, 75. Similarly, modeling approaches including discriminant analysis via mixed integer programming or principal component analysis have been utilized by other systems biology groups with great success and could be combined with our approaches to produce more robust models of disease 14, 15. In addition, efforts to ‘train’ the modeling approaches are underway, utilizing known biological signatures and datasets to help refine and improve targeting in similar, yet unknown conditions. For example, studies with SARS‐CoV mutants lacking NSP16 have defined important pathways independent of any modeling approach (Menachery VD, Eisfeld AJ, Josset L, Sims AC, Schaefer A, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Matzke MM, Webb‐Robertson BJ, Shukula AK, Burkett S, Metz TO, Pickles R, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS, in review); these data were then used to screen the efficacy of newly developed models to examine other mutant viruses with similar infection kinetics. A similar approach was utilized in examination of influenza vaccine responses 15. Together, these efforts are just a few of the ways that systems‐based modeling can be refined to provide efficient, effective targeting. While significant commitment is needed for this possibility to come to fruition, the resulting models would provide an opportunity to revolutionize our understanding of complex biological systems.
Validation, translation, and extension
Lack of validation has been a major source of criticism of systems‐based analysis 2, and a concerted effort by the field is essential for maximizing the long‐term sustainability of the approach. However, the use of reductionist‐based approaches for validation and extension is inherently limited to our areas of scientific understanding. Often, these studies utilize knock‐out mice, drug treatment, or other reagents previously generated in the course of reductionist‐based science. While some of these areas are novel for infectious disease research (Kepi1, Serpine1, etc.), limiting analysis and validation to these areas is a problem. Therefore, as systems biology matures, novel methods and experimental systems to validate and test targets must be developed beyond the current scope of reductionist‐based approaches. In addition, these types of experiments should seek to make direct parallels to human disease and foster development of drug and therapeutic treatments. Many of the novel techniques and methods outlined in the data expansion section above also fit into these parameters. Below we describe several additional areas of expansion that are applicable to our respiratory virus infection model.
Examination of allelic variation
Reductionist‐based validation approaches often rely on knock‐out and overexpression studies to provide understanding, using the absence or overabundance of a molecule as a means to define its function. However, these types of null genetic mutation are rarely observed in nature, making it difficult to directly translate these finding into human disease models. For validation to have a translational impact on human disease, our studies must use genetic diversity to alter target gene/pathways functions rather than knock‐out or knock‐down studies. The CC allows this type of analysis through generation of customizable recombinant inbred intercrosses 76, 77. For example, studies with SARS‐CoV have identified Serpine1, a key component of the urokinase pathway and an important player in tissue damage/repair following infection. Serpine1 deficiency results in enhanced disease and pathogenesis following SARS‐CoV infection, likely due to increased lung pathology (18). However, the impact of these data remains unclear with regards to human disease: the vast majority of people maintain functional Serpine1, but genetic diversity in this gene or region of the genome may have a significant impact on pathogenic outcomes and disease severity. To assay this possibility, we can utilize the CC to generate mouse lines with allelic diversity in this region of the genome, while maintaining genetic identity throughout the rest of the genome (Fig. 5 A). Similarly, the genome region could remain static and the background shifted to assess the impact of a specific allele. For Serpine1 in the CC, this means the possibility of examining the impact of six haplotypes during in vivo infection (Fig. 5 B). Importantly, other analysis can be layered onto the CC RIX experimental model including predictions based on SNP consequences (Fig. 5 C), structural alterations 78 (Fig. 5 D), and/or epigenetic regulatory factors. In addition, multiple genes could be analyzed in parallel to assay their allelic impact on each other; while either holding constant or randomizing the majority of the genome, allelic variants can be shuffled between the two gene regions, creating combinations to assay their interaction with each other and the global response (Fig. 5 A). For example, systems vaccinology has identified important genes impacting the antibody response (TNFRS17 & CaMKIV) and T‐cell response (EIF2AK4 & C1QB) following yellow fever and influenza vaccination 15. Using the CC, the varying allele combinations could be examined in the same background and provide a window into the interactions of different gene combinations and pathways.
Figure 5.
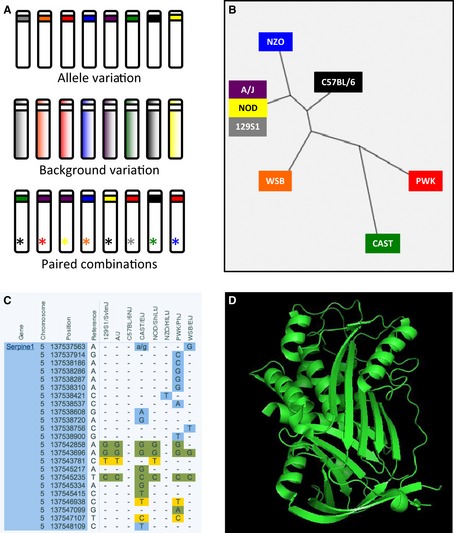
The collaborative cross offers the opportunity to examine allelic variation in systems‐based gene targets. (A) A schematic of three approaches used to evaluate how allelic diversity in a target gene may alter viral pathogenesis. Allele variation holds the background relatively constant and shifts the possible gene alleles. Background variation assesses a single allele in multiple backgrounds. Finally, paired combinations examine the relationship between allele combinations in multiple systems or genome‐wide association studies‐based targets. (B) Phylogenetic tree of mouse Serpine1 within the collaborative cross. (C) Consequences of single nucleotide polymorphisms within various CC strains. A. single nucleotide polymorphisms consequences include untranslated region (Blue); synonymous coding (Green); and non‐synonymous coding (Yellow). (D) Structure of human Serpine1 78.
The allele‐based examination using the CC provides a means to directly measure the impact of specific gene alterations identified in human disease models. This contrast the traditional knock‐out/knock‐in approaches and has implication for targets generated by both reductionist‐based approaches and GWAS. Notably, for systems biology targets, the CC allows validation of genes and pathways without the requirement of previously generated reagents alleviating one of the major issues with validation. Because the CC is based on natural genetic variation rather than genetic deletions, this resource also allows for investigations into the role of genes that cannot be knocked out or knocked down in traditional validation approaches. This overcomes a major challenge of systems‐based approaches, where many candidate genes cannot be targeted due to their essential function. Together, these approaches represent a major leap in the utilization and validation of systems‐based analysis.
Drug targeting
The initial purpose of systems biology was not only greater understanding of complex biological systems but also as a platform approach to rapidly identify candidate drugs and therapies to treat human diseases. While there have been some successes in this area 79, development of these reagents for treatment of infectious disease has been lacking. Moving forward, focus on clinical treatments provides both validation to systems‐based targets as well as develop useful therapeutics for treating patients. To achieve this purpose, our group used functional genomics and computation biology to diagnose virus etiology, forecast disease severity in the lung, and develops a highly portable screening platform that analytically pinpoints and then tests the ability of lead compounds to attenuate virus disease 19. Built upon a database of characterized small molecules and FDA‐approved drugs [e.g. connectivity map database, Canadian DrugBank, the Pharmacogenomics Knowledge base, etc.], the approach uses pattern recognition and multiparametric, multiobjective optimization techniques to match gene expression changes with drug targets that possibly reverse those trends and could be repurposed for use as antiviral therapies.
Using these drug targets, validation studies would analyze the efficacy of the drugs both in vitro and in vivo for toxicity, viral replication, and disease pathogenesis. Beginning in common cell lines with known effective doses (Veros, Hela, Calu3), we can evaluate the ability of the drug to impact viral replication using low viral doses to maximize effect. Subsequent in vitro studies would use tissue‐specific cell lines and primary cultures to define cell toxicity, viral replication, and host expression changes as measured by microarray. Drug targets effective in these areas would be further tested in inbred models of infection for in vivo efficacy. Finally, a diverse panel of CC mice would also be tested to ascertain possibility of drug failure due to genetic diversity. This approach provides a means to quickly develop drug treatments for human disease and also validate targets generated by systems biology.
The utility of this approach is illustrated by identification of drug targets for both SARS‐CoV and EMC‐CoV. Detailed transcriptomic analysis of SARS‐CoV and EMC2012 host response networks had demonstrated early upregulated genes (approximately 200) that are downstream of p38 map kinase signaling pathways. A systematic review of the FDA database indicated that SB203580, a pyridinyl imidazole inhibitor of p38 mitogen‐activated protein kinase, significantly downregulated overlapping sets of genes that are induced by high path coronaviruses 80. To test compound effectiveness, we infected Vero cells with SARS‐CoV at a MOI of 0.01 and then treated cells with type I IFN or SB203580. Importantly, SB203580 significantly reduced both EMC‐CoV and SARS‐CoV growth (and CPE) in culture, and in some cases, was equivalent to IFN treatment, supporting our hypothesis that lead compounds for therapeutic testing and evaluation can be identified by modeling approaches that compare contrasting expression patterns during infection and drug treatment. Further testing in tissue‐specific cells and in vivo continues for SB203580, but the data illustrate conceptual support for this approach. A similar method is planned for other systems biology targets suggesting that identified drugs may enhance protective responses mitigated by the Urokinase pathway (e.g. protein kinase C) or stimulate the TLR3/TLR4/myd88 signaling pathways (e.g. TLR3‐ PolyIC, Poly AU, TLR4‐Monophosphoryl lipid A) 80, protecting against lethal disease.
Predictive, diagnostic biomarkers
Infectious disease susceptibility is governed by numerous factors including underlying genetics, the age of the infected, environmental factors, and the dose of the pathogen exposure. These factors make treatment more difficult to prescribe as it is nearly impossible to determine the appropriate use of aggressive or conservative treatment options at early time points. Whereas the systems biology approaches have provided identification of key pathways that play protective or pathogenic roles after infection, the next stage of development requires linking these pathway outcomes with early events to provide prognostic biomarkers to aid in treatment of disease 82, 83, 84, 85. These studies will take advantage of the massive functional genomics datasets to identify early etiologic and prognostic signature that track with disease severity across pathogens and in vivo systems including human (PBMC), mouse, and non‐human primate models. Next, natural genetic variation on biomarker sensitivity and specificity can be evaluated using resources such as the CC, a reproducible model of human genetic diversity. Finally, key validated biomarker and prognostic indicators will be integrated into assays for differential diagnosis and prognosis, linked to computational algorithms that forecast FDA drugs and other treatments that may attenuate pathogenesis. In this way, development of biomarkers via systems‐based approaches has the potential to revolutionize the way human diseases are treated with an eye toward likely outcome dictating the intervention strategies.
Conclusion
Systems biology has documented potential for enhancing our understanding of complex biological systems. Based primarily on genomic high‐throughput data, modern modeling approaches, and reductionist‐based validation methods, systems‐based studies have provided novel insights for infectious disease aspects including innate immunology, clinical disease, viral–host interactions, drug targeting, adaptive immunity, vaccine efficacy, etc. 11, 12, 13, 14, 15, 16, 21, 86.
In many ways, systems biology is still in its infancy, and new developments in data generation, modeling, and validation approaches will continue to expand, providing enormous opportunities for furthering our understanding of complex virus–host interaction paradigms and disease (Fig. 6). For example, high‐throughput data has expanded to include data areas like respiratory function, phosphoproteomics, next‐generation sequencing, and metabolomics that provide a different type of output to model with the possibility for unique targets. Similarly, the constant evolution, application, and refinement of modeling approaches provide improved targets for downstream analyses and increased understanding. Finally, both reductionist‐ and systems‐based groups provide novel techniques and new areas for validation and incorporation. Together, these current and future elements will exponentially extend the power of systems‐based analysis and permit examination of even more complex questions including the role of genetic variation in infectious disease processes, the creation of novel therapeutics for respiratory virus infection, and integration of human GWAS findings into model systems for validation.
Figure 6.

Novel experimental conditions, modeling approaches, and validation methods will continue to expand the power of systems biology and provide further insights into complex biological interactions. Depictions of current and future data types are divided into the three faces on the systems biology cube. The right face includes experimental approaches that have and will be utilized for high‐throughput data generation. The left face illustrates modeling approaches and outputs from current and future studies. Finally, the top face shows results for validation and expansion of systems‐based targets.
As potential becomes reality with regards to data, techniques, and resources, a significant change must also accompany our overall research mindset. The interaction between disciplines like virology and computational modeling must be strengthened and expanded to leverage the utility of the new outputs and models. In addition, a renewed effort is required to develop hybrid approaches that employ the best elements of both reductionist‐ and systems‐based methods. Finally, training a new generation of scientist with these tenets is paramount in the acceptance and maximum utilization of this approach. While reductionist‐based approaches have provided the foundation of our understanding of many critically important biological phenomena, a new era of big datasets is emerging which will not only provide a more holistic view of complex diseases but also reveal new virus–host paradigms that regulate disease severity and provide for new opportunities for the treatment and amelioration of human disease.
Acknowledgements
We thank J. McDermott and L. Josset for generating Figs 2 and 3, respectively. In addition, we thank L. Gralinski for critical reading of the text. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under contract no. HHSN272200800060C and award numbers U19AI100625 and F32AI102561. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors have no conflicts of interest to declare.
References
- 1. Kitano H. Computational systems biology. Nature 2002;420:206–210. [DOI] [PubMed] [Google Scholar]
- 2. Aderem A, et al. A systems biology approach to infectious disease research: innovating the pathogen‐host research paradigm. MBio 2011;2:e00325–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev 2009;227:264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet 2001;2:343–372. [DOI] [PubMed] [Google Scholar]
- 5. Bumann D. System‐level analysis of Salmonella metabolism during infection. Curr Opin Microbiol 2009;12:559–567. [DOI] [PubMed] [Google Scholar]
- 6. Beiting DP, Roos DS. A systems biological view of intracellular pathogens. Immunol Rev 2011;240:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakaya HI, Pulendran B. Systems vaccinology: its promise and challenge for HIV vaccine development. Curr Opin HIV AIDS 2012;7:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng X, Chan EY, Li Y, Diamond DL, Korth MJ, Katze MG. Virus‐host interactions: from systems biology to translational research. Curr Opin Microbiol 2009;12:432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joyner MJ, Pedersen BK. Ten questions about systems biology. J Physiol 2011;589:1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Esparza J, Yamada T. The discovery value of “Big Science”. J Exp Med 2007;204:701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woods CW, et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE 2013;8:e52198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mani KM, et al. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B‐cell lymphomas. Mol Syst Biol 2008;4:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol 2011;7:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Querec TD, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 2009;10:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakaya HI, et al. Systems biology of vaccination for seasonal influenza in humans. Nat Immunol 2011;12:786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rasmussen AL, et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J Virol 2011;85:11646–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graham RL, Sims AC, Brockway SM, Baric RS, Denison MR. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J Virol 2005;79:13399–13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gralinski LE, et al. Mechanisms of SARS‐CoV Induced Acute Lung Injury. MBio 2013; doi: 10.1128/mBio. 00271–13. [DOI] [Google Scholar]
- 19. Merle H, et al. Alkali ocular burns in Martinique (French West Indies) Evaluation of the use of an amphoteric solution as the rinsing product. Burns 2005;31:205–211. [DOI] [PubMed] [Google Scholar]
- 20. Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity 2010;33:516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sims AC, et al. Release of SARS‐CoV nuclear import block enhances host transcription in human lung cells. J Virol 2013;87:3885–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yount B, et al. Severe acute respiratory syndrome coronavirus group‐specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol 2005;79:14909–14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol 2007;81:11620–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frieman MB, et al. SARS coronavirus accessory ORFs encode luxury functions. Adv Exp Med Biol 2006;581:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kopecky‐Bromberg SA, Martinez‐Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 2007;81:548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frieman M, Yount B, Heise M, Kopecky‐Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol 2007;81:9812–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2012;2:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Munoz‐Fontela C, et al. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J Immunol 2011;187:6428–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terrier O, et al. Cellular transcriptional profiling in human lung epithelial cells infected by different subtypes of influenza A viruses reveals an overall down‐regulation of the host p53 pathway. Virol J 2011;8:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Turpin E, Luke K, Jones J, Tumpey T, Konan K, Schultz‐Cherry S. Influenza virus infection increases p53 activity: role of p53 in cell death and viral replication. J Virol 2005;79:8802–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang J, et al. Epigenetic changes mediated by microRNA miR29 activate cyclooxygenase 2 and lambda‐1 interferon production during viral infection. J Virol 2012;86:1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeDiego ML, et al. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog 2011;7:e1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pasieka TJ, Baas T, Carter VS, Proll SC, Katze MG, Leib DA. Functional genomic analysis of herpes simplex virus type 1 counteraction of the host innate response. J Virol 2006;80:7600–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, Katze MG. A single N66S mutation in the PB1‐F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol 2011;85:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol 2010;8:717–729. [DOI] [PubMed] [Google Scholar]
- 36. Michoel T, De Smet R, Joshi A, Van de Peer Y, Marchal K. Comparative analysis of module‐based versus direct methods for reverse‐engineering transcriptional regulatory networks. BMC Syst Biol 2009;3:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langfelder P, Horvath S. Eigengene networks for studying the relationships between co‐expression modules. BMC Syst Biol 2007;1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol 2005;4. Article 17. [DOI] [PubMed] [Google Scholar]
- 39. Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 2008;24:719–720. [DOI] [PubMed] [Google Scholar]
- 40. Mason MJ, Fan G, Plath K, Zhou Q, Horvath S. Signed weighted gene co‐expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genomics 2009;10:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei H, et al. Transcriptional coordination of the metabolic network in Arabidopsis. Plant Physiol 2006;142:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Horvath S, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci USA 2006;103:17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marino‐Ramirez L, Tharakaraman K, Bodenreider O, Spouge J, Landsman D. Identification of cis‐regulatory elements in gene co‐expression networks using A‐GLAM. Methods Mol Biol 2009;541:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bertozzi P, et al. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med 1990;322:890–897. [DOI] [PubMed] [Google Scholar]
- 45. Levi M, Moons L, Bouche A, Shapiro SD, Collen D, Carmeliet P. Deficiency of urokinase‐type plasminogen activator‐mediated plasmin generation impairs vascular remodeling during hypoxia‐induced pulmonary hypertension in mice. Circulation 2001;103:2014–2020. [DOI] [PubMed] [Google Scholar]
- 46. Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 2003;31:S213–S220. [DOI] [PubMed] [Google Scholar]
- 47. Sisson TH, Hanson KE, Subbotina N, Patwardhan A, Hattori N, Simon RH. Inducible lung‐specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2002;283:L1023–L1032. [DOI] [PubMed] [Google Scholar]
- 48. Sisson TH, Hattori N, Xu Y, Simon RH. Treatment of bleomycin‐induced pulmonary fibrosis by transfer of urokinase‐type plasminogen activator genes. Hum Gene Ther 1999;10:2315–2323. [DOI] [PubMed] [Google Scholar]
- 49. Chapman HA, Allen CL, Stone OL. Abnormalities in pathways of alveolar fibrin turnover among patients with interstitial lung disease. Am Rev Respir Dis 1986;133:437–443. [DOI] [PubMed] [Google Scholar]
- 50. Hattori N, Sisson TH, Xu Y, Desai TJ, Simon RH. Participation of urokinase‐type plasminogen activator receptor in the clearance of fibrin from the lung. Am J Physiol 1999;277:L573–L579. [DOI] [PubMed] [Google Scholar]
- 51. Katze MG, He Y, Gale M Jr. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2002;2:675–687. [DOI] [PubMed] [Google Scholar]
- 52. Sarkar SN, Sen GC. Novel functions of proteins encoded by viral stress‐inducible genes. Pharmacol Ther 2004;103:245–259. [DOI] [PubMed] [Google Scholar]
- 53. Muller U, et al. Functional role of type I and type II interferons in antiviral defense. Science 1994;264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 54. Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 2008;89:2359–2376. [DOI] [PubMed] [Google Scholar]
- 55. Garcia‐Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res 2011;162:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res 2009;37:D852–D857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Boheemen S, et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012;3:e00473–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Muller MA, et al. Human coronavirus EMC does not require the SARS‐coronavirus receptor and maintains broad replicative capability in mammalian cell lines. MBio 2012;3:e00515–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Raj VS, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus‐EMC. Nature 2013;495:251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zielecki F, et al. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol 2013;87:5300–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li C, et al. Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. J Virol 2011;85:10955–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tisoncik JR, Belisle SE, Diamond DL, Korth MJ, Katze MG. Is systems biology the key to preventing the next pandemic? Future Virol 2009;4:553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aylor DL, et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res 2011;21:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Collaborative Cross C . The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 2012;190:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roberts A, Pardo‐Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome‐wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome 2007;18:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Szatkiewicz JP, Beane GL, Ding Y, Hutchins L, Pardo‐Manuel de Villena F, Churchill GA. An imputed genotype resource for the laboratory mouse. Mamm Genome 2008;19:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J 2011;52:24–31. [DOI] [PubMed] [Google Scholar]
- 68. Yang H, Bell TA, Churchill GA, Pardo‐Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nat Genet 2007;39:1100–1107. [DOI] [PubMed] [Google Scholar]
- 69. Ferris MT, et al. Modeling host genetic regulation of influenza pathogenesis in the collaborative cross. PLoS Pathog 2013;9:e1003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Julander JG, et al. Use of plethysmography in assessing the efficacy of antivirals in a mouse model of pandemic influenza A virus. Antiviral Res 2011;92:228–236. [DOI] [PubMed] [Google Scholar]
- 71. Morrey JD, Siddharthan V, Wang H, Hall JO. Respiratory insufficiency correlated strongly with mortality of rodents infected with West Nile virus. PLoS ONE 2012;7:e38672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stark JM, Khan AM, Chiappetta CL, Xue H, Alcorn JL, Colasurdo GN. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J Infect Dis 2005;191:387–395. [DOI] [PubMed] [Google Scholar]
- 73. Korth MJ, Tchitchek N, Benecke AG, Katze MG. Systems approaches to influenza‐virus host interactions and the pathogenesis of highly virulent and pandemic viruses. Semin Immunol 2012. doi: 10.1016/j.smim.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chang ST, Tchitchek N, Ghosh D, Benecke A, Katze MG. A chemokine gene expression signature derived from meta‐analysis predicts the pathogenicity of viral respiratory infections. BMC Syst Biol 2011;5:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bonneau R, et al. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems‐biology data sets de novo. Genome Biol 2006;7:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hua J, et al. Single‐locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 2003;100:2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zou F, et al. Quantitative trait locus analysis using recombinant inbred intercrosses: theoretical and empirical considerations. Genetics 2005;170:1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sharp AM, et al. The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Structure 1999;7:111–118. [DOI] [PubMed] [Google Scholar]
- 79. Hurle MR, Yang L, Xie Q, Rajpal DK, Sanseau P, Agarwal P. Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther 2013;93:335–341. [DOI] [PubMed] [Google Scholar]
- 80. Davis T, Bachler MA, Wyllie FS, Bagley MC, Kipling D. Evaluating the role of p38 MAP kinase in growth of Werner syndrome fibroblasts. Ann NY Acad Sci 2010;1197:45–48. [DOI] [PubMed] [Google Scholar]
- 81. Gnjatic S, Sawhney NB, Bhardwaj N. Toll‐like receptor agonists: are they good adjuvants? Cancer J 2010;16:382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cameron MJ, et al. Interferon‐mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol 2007;81:8692–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. de Lang A, et al. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS‐CoV‐infected macaques. PLoS Pathog 2007;3:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rockx B, et al. Early upregulation of acute respiratory distress syndrome‐associated cytokines promotes lethal disease in an aged‐mouse model of severe acute respiratory syndrome coronavirus infection. J Virol 2009;83:7062–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rockx B, et al. Comparative pathogenesis of three human and zoonotic SARS‐CoV strains in cynomolgus macaques. PLoS ONE 2011;6:e18558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Josset L, et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio 2013;4:e00165–13. [DOI] [PMC free article] [PubMed] [Google Scholar]