

Abstract
Background:
P300 is a member of the mammalian histone acetyl transferase (HAT) family, an enzyme that acetylates histones and several non-histone proteins including P53 (the most important tumor suppressor gene) during stress, which plays an important role in the apoptosis of tumor cells. Hereby, this study describes the potency of CTB (Cholera Toxin B subunit) as a P300 activator to induce apoptosis in a breast cancer cell line (MCF-7) and a lung fibroblast cell line (MRC-5) as a non-tumorigenic control sample.
Materials and Methods:
MCF-7 and MRC-5 were cultured in RPMI-1640 and treated with or without CTB at a concentration of 85.43 μmol/L, based on half-maximal inhibitory concentration (IC50) index at different times (24, 48 and 72 h). The percentage of apoptotic cells were measured by flow cytometry. Real-time quantitative RT-PCR was performed to estimate the mRNA expression of P300 in MCF-7 and MRC-5 with CTB at different times. ELISA and Bradford protein techniques were used to detect levels of total and acetylated P53 protein generated in MCF-7 and MRC-5.
Results:
Our findings indicated that CTB could effectively induce apoptosis in MCF-7 significantly higher than MRC-5. We showed that expression of P300 was up-regulated by increasing time of CTB treatment in MCF-7 but not in MRC-5 and the acetylated and total P53 protein levels were increased more in MCF-7 cells than MRC-5.
Conclusion:
CTB could induce acetylation of P53 protein through increasing expression of P300 and consequently induce the significant cell death in MCF-7 but it could be well tolerated in MRC-5. Therefore, CTB could be used as an anti-cancer agent.
Keywords: Acetylation, apoptosis, Cholera toxin B subunit, P300, P53
INTRODUCTION
Histone acetyltransferases (HATs) are members of enzymes that play pivotal roles in regulation of gene expression (involved in the acetylation) and act as transcriptional co-activators.[1] The HATs induce transfer of acetyl group to lysine amino acid residues, which present the histone protein tails from acetyl CoA to form ε-N-acetyl lysine. Therefore, they associate with open chromatin sequences that facilitate the accessibility of transcription factors to DNA (transcriptional activation).[2] On the other hand, the histone deacetylases (HDACs), despite HATs, cause increase in lifespan but in order to regulate transcription, they cooperate by acetylation and deacetylation. They remove acetyl groups from histone tails thus preventing the attachment of transcription factors.[3] Importantly, alteration of gene expression in cancer based on interaction of these epigenetic modifications (post-translation) with each other, play a significant role in tumorigenesis.[4] In diseases such as cancer, often there occurs an imbalance between expression of transcriptional co-activator proteins that contain HAT and HDAC families.[5] P300 is a member of the mammalian HAT protein family with histone acetyl transferase activity, which suggests that this molecule is competent of acetylating all core histone proteins and it is an important transcriptional co-activator, which may play a distinct role in regulation of a wide range of biological processes such as cell growth, proliferation, survival and apoptosis through histone acetylation.[6] In several diseases, such as some human cancers, it has been shown that HAT activities were disrupted, which is often associated with malignant alterations.[7] The HATs would often down-regulate and get inactivated in several types of tumors.[8] However, histones are not the only proteins that can be acetylated, P300 presumably can also catalyze acetylation of several non-histone proteins such as P53 (the most important tumor suppressor gene)[9] that shows to be functionally regulated by acetylation and its stability changes.[10] While more than half of human tumors often have mutations in P53[11] alterations in tumor suppressor genes are not always due to mutations, they may also be due to this sort of epigenetic alteration.[12] The P53 tumor suppressor protein plays a major role in cellular response to DNA damage and other genomic aberrations.[13] Activation of P53 can lead to cell cycle arrest, DNA repair and apoptosis.[14] Following DNA damage, human P53 acetylates at Lys382.[15] Inactivation of P300 mediates deacetylation of P53 and negatively regulates activity of this protein.[16] In normal cells, P53 is a short-lived protein due to activity of Mdm2 (a negative regulator), as a ubiquitin ligase,[17] to inhibit and destabilize P53, so P53 levels are undetectable and inactive to induce apoptosis. In response to various types and levels of stress that causes DNA damage, HAT family mediates acetylation of the P53 in C terminus and some of the major P53 ubiquitination sites are blocked by Mdm2.[18] This function leads to P53 protein stabilization and activation of P53 protein in human cells.[19] Hyperacetylation of P53 can also cause the hyperactivity of this protein.[20] It seems that P300 is able to acetylate and activate P53 and induce apoptosis in response to DNA damage in some cancer cells.[21] The balance of P53 acetylation (positively regulates P53 activity) and deacetylation (negatively regulates P53 activity), respectively mediated by the HATs (particularly P300) and HDACs, is usually well-regulated, but the balance often gets upset in diseases such as cancer.[22] Inactivation of P300 is encountered in several types of tumors such as in certain types of human cancers including breast carcinomas.[23] Breast cancer is the most common cancer among females and is the second leading cause of death among women worldwide.[24] Each year about 1.15 million cases of female breast cancer is diagnosed worldwide while about 502,000 die from the disease.[25] Some reports suggest that inactivation of P300 probably mediates P53 deacetylation and inhibits P53, possibly mediates apoptosis in response to various types of stress in some of the malignancies.[26] The human breast carcinoma cell line MCF-7 has a wild-type P53 but this tumor suppressor gene is responsible for epigenetic event (hypoacetylation P53), and is not functional and cannot induce apoptosis.[27] These effects seem to be reversed in cancer cells by activation of P300.[28] The studies suggest that pharmacologic activation of P300 may promote apoptosis by direct hyperacetylation of P53 in cancer cells and could be used as an anti-cancer strategy. CTB is the only known small molecule activator of HATs [specific to P300 by the alteration of P300 structure[29] ], which is synthesized from salicylic acid and anacardic acid. The CTB goes into the hydrophobic compartments of the P300 enzyme and leads to modify the α-helix and ring structured amino acids within the histone tail of the HAT domain, which is presumably responsible for the activation of P300 activity.[30] However, only a small number of small molecules are known to have effects on the P300 gene and the CTB drug so far. As far as we know, there are no such reports on effects of CTB as an anti-tumor or of its effects on histone acetyltransferases P300 (activity of P300) to induce P53 acetylation in cancer cell lines though inhibitors of HDACs have been extensively studied as therapeutic targets.[31] We assumed that based on histone deacetylases inhibitors, pharmacologic activation of P300 may promote apoptosis by performing direct acetylation of P53 in some cancer cells and it has different effects on inducing apoptosis on normal and cancer cells, so it could be used as an anti-cancer strategy.[32] In this study, we investigated apoptotic effects of CTB, as the activator of P300 on P53 protein acetylation and consequent apoptosis in MCF-7 (breast adenocarcinoma) and MRC-5 (lung fibroblasts as non-tumorigenic control samples) cell lines.
MATERIALS AND METHODS
Cell lines, drug, treatment and culture condition
Human breast cancer MCF-7 and human lung fibroblasts MRC-5 were purchased from the National Cell Bank of Iran-Pasteur Institute. CTB [N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-benzamide], an activator of P300, was purchased from Sigma (C6499, USA). All cell lines used in the present study were cultured in RPMI-1640 medium (Sigma) supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% penicillin-streptomycin (Sigma), and incubated at 37°C and in humidified atmosphere containing 5% CO2. CTB was dissolved in stock solutions and for treatments the compounds were diluted in DMSO to the appropriate concentrations according to reported procedures.[17] After the cells were >80% confluent and growing exponentially in 10 cm diameter culture dishes, 105 cells (MCF-7 or MRC-5) were counted and plated in 3 cm diameter culture dishes and kept in RPMI-1640 culture medium for 24 hours which were then incubated with certain concentrations of CTB, based on IC50 index, at different times (24, 48 and 72 h). Photographs of cultures were taken before and after treatment with CTB at different times under inverted microscope (Nikon, TE 2000-U, Japan).[33]
IC50 assay
The IC50 values for the CTB in MCF-7 groups were acquired after 24 hours of treatment. Briefly, 104 cells (MCF-7) were counted and placed into each well of a 24-well micro plate and were treated with various drug concentrations (0, 6.25, 12.5, 25, 50, 100, 150, 200 μM doses) for 24 hours, and the MTT survival assay was then carried out for evaluating the cell viability with different drug concentrations of MCF-7 groups. A graph of viability versus drug concentration was used to calculate IC50 values for MCF-7 cell line.[2,28]
Flow-Cytometric analysis
The percentage of apoptotic cells was measured by flow cytometry following AnnexinV (FL1-H) and PI (FL2-H) labeling. A minimum of 5 × 105 cells/ml were analyzed for each sample. Cells were treated with CTB (85.43 μmol/L) for 24, 48 and 72 hours and then washed in PBS and re-suspended in binding buffer (1×; 5 μl). AnnexinV-FITC was added to 195 μl cell suspension and then analysis was carried out according to the manufacturer's protocol (BMS500F1/100CE AnnexinV-FITC, eBioscience, USA). Finally the apoptotic cells were counted by FACScan flow cytometry (Becton Dickinson, Heidelberg, Germany). These experiments were carried out in triplicate and were, independently, repeated at least 3 times.[19,12]
Reverse transcription and real-time PCR analysis
Real-time quantitative RT-PCR was performed to quantitatively estimate the mRNA expression of P300 in MCF-7 and MRC-5 cells before and after treatment with CTB at different times. Total RNA was isolated by RNeasy mini kit (Qiagen), treated by RNase-free DNase set (Qiagen) to eliminate the genomic DNA. The RNA concentration was determined using a biophotometer (Eppendorf). Total RNA (100 ng) was reverse-transcribed to cDNA by using the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas) according to the manufacturer's instructions. The Maxima SYBR Green Rox qPCR master mix kit (Fermentas) was used for real-time RT-PCR. Primer sequences are shown in Table 1. Real-time PCR reactions were performed using Step one plus (Applied Biosystem). The PCR amplification conditions consisted of 10 min at 95°C followed by 40 cycles of denaturation step at 95°C for 15 sec and annealing and extension for 1 min at 60°C. Data were analyzed using the Comparative Ct (ΔΔct) method. The relative expression level of P300 was calculated by determining a ratio between the amount of P300 and that of endogenous control. Melting curve analysis (60°C→95°C increment of 0.3°C) was used to determine the melting temperature of specific amplification products and primer dimers. These experiments were carried out in triplicate and were independently repeated at least 3 times.[34]
Table 1.
Primers used in real-time PCR

Bradford protein assay
Total (intracellular) protein concentration was determined by Bradford method. Bradford protein quantification assay is an accurate procedure for determining the concentration of protein in solution based on binding of Coomassie Blue dye to proteins. This method was carried out before enzyme linked immunosorbent assay (ELISA) assay. Total proteins extracted from MCF-7 and MRC-5 cells before and after CTB treatment will be described later in ELISA assay. Bovine serum albumin (BSA) was used at 9 different concentrations (0.25, 0.5, 1, 1.5, 2, 3, 4, 5 and 6 mg/ml) to prepare a protein standard. After diluting the protein standards, the stock dye reagent was prepared (500 mg Coomassie Blue was dissolved in 500 ml methanol and was added to 100 ml phosphoric acid and 50 ml ddH2O) that was diluted in 8 ml ddH2O. Two milliliters of dye reagent was added to each tube of protein standard and was incubated at room temperature for at least 5 min. Absorbance of the protein standards and experimental samples were carried out by the spectrophotometry (Bausch and Lomb, Germany) at 595 nm and, finally, a standard curve was plotted.[35]
Acetylated and total P53 sandwich ELISA assay
ELISA was used to specifically detect endogenous levels of total and acetylated p53 protein generation in MCF-7 and MRC-5 cells in the presence or absence of CTB at different times (24, 48 and 72 h). Acetylated and total P53 ELISA Kit were prepared by Cell Signaling Technology and cell lyses were prepared at first step. Briefly, cells were harvested under treated conditions by CTB at different times, media was removed and cells were washed with cold PBS. PBS was removed and 0.5 ml of ice-cold cell lyses buffer with 1 mM phenyl methyl sulfonyl fluoride (PMSF) was added to each plate and incubated on ice for 5 min. Cells were scraped off the plate and were transferred to an appropriate tube, and a freeze-and-thaw test was performed 3 times. The tubes were micro-centrifuged at 4°C, for 10 min, and the supernatant was transferred to a new tube. This supernatant was the cell lysates. For doing ELISA assay at first, the concentration of total protein extract in both cell lysates was determined using the Bradford assay. Sandwich ELISA was performed according to the manufacturer's protocol. Finally, the absorbance of samples were read in ELISA reader (Hyperion, Germany) at 450 nm wavelength and ELISA analysis was calculated based on control index. All experiments were carried out in triplicate.[36]
Statistical analysis
All the quantitative data were presented as the mean ± standard deviation. One-way analysis of variance (ANOVA) with LSD post-hoc test was performed to determine statistical significance among different groups by using SPSS software package 16.0. Significance was accepted at a level of P < 0.05.
RESULTS
IC50
After the treatment of MCF-7 cells with MTT solution in this assay, the dark blue formazan crystals were seen in cells, which indicated their metabolic activity. The reduction in the number of cells was dependent on the cell type as shown by the half-maximal inhibitory concentration (IC50) index. The IC50 values for the CTB were established [Figure 1]. The results showed that the essential CTB concentration to achieve the IC50 in MCF-7 cells at 24 hours was 85.43 μmol/L [Figure 1].
Figure 1.
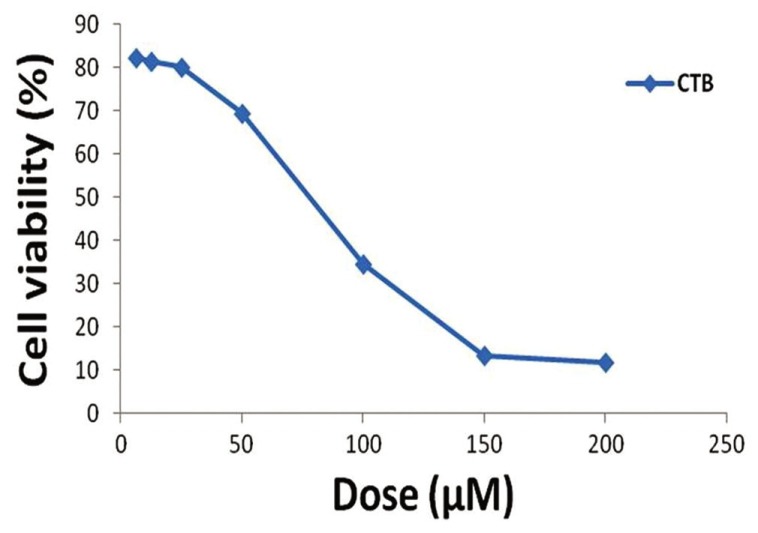
IC50 assay for half-maximal inhibitory concentration analysis of CTB in MCF-7 cancer cell lines after 24 hours of treatment. Cells were incubated with or without the CTB using 0, 6.25, 12.5, 25, 50, 100, 150 and 200 μM doses, and the relative amount of viable cells were estimated by measuring the absorbance of the cell suspension after incubation with MTT assay was carried out and a graph of viability versus drug concentration was used to calculate IC50 values based on the trend line equation (Y = −0.421 × +85.97) for MCF-7 cell line (85.43 μM)
Flow cytometry
To establish the anti-apoptosis potential of the CTB, we first investigated the effects of this P300 activator on the proliferation of the breast carcinoma cell line (MCF-7). The flow cytometry results showed that the 85.43 μmol/L concentration of CTB based on IC50 index at different times (24, 48 and 72 h) could significantly induce apoptosis in MCF-7 cells and it was increased with ascending time (P < 0.001) [Figure 2a and 2m]. CTB treatment arrested MCF-7 cell proliferation (≥95% of inhibition) in 72 hours, whereas its inhibition on MRC-5 cells proliferation in all different times were negligible, although different times (24, 48 and 72 h) could significantly induce apoptosis in MRC-5 cells and it was increased with ascending time (P > 0.05) [Figures 2a-c]. MCF-7 apoptotic cells showed a sharp increase at all times in comparison with MRC-5 cells (P < 0.001). DMSO was used in the control sample (vehicle CTB) and a small amount of cell death in both cell lines at different times was observed (P < 0.05) [Figure 2c and 2b]. These results were supported by morphologic examination using an inverted microscope [Figure 3].
Figure 2.

CTB induces apoptosis in cancer cells lines (MCF-7) but not fibroblasts (MRC-5). Relative levels of apoptotic cells in MCF-7 cancer cell lines and cultured fibroblasts (MRC-5) treated with 85.43 μM CTB for different times. Cells incubated with the vehicle (DMSO) were used as a control. (a, b). The percentage of apoptotic cells was measured using the AnnexinV FITC and PI assay as described in Materials and Methods. ****P <0.001 vs. all other groups MCF-7 cells treated with CTB. ***P<0.05 vs. all other groups MRC-5 cells. **P< 0.05 vs. all other groups MCF-7 cells incubated with the vehicle (DMSO) were used as a control. *P< 0.05 vs. 48 and 72 hours groups MRC-5 cells incubated with the vehicle (DMSO) were used as control. (c-m) MCF-7 and MRC-5 cells were treated with 85.43 μM of CTB, and apoptosis was measured by flow cytometry following AnnexinV (FL1-H) and PI (FL2-H) staining. Cells that are AnnexinV-positive and propidium iodide negative are in early apoptosis, as phosphatidyl serine (PS) translocation has occurred, although the plasma membrane remains intact. Cells that are positive for both AnnexinV and PI either are in the late stages of apoptosis or are already dead, as PS translocation has occurred, and the loss of plasma membrane integrity is visible
Figure 3.
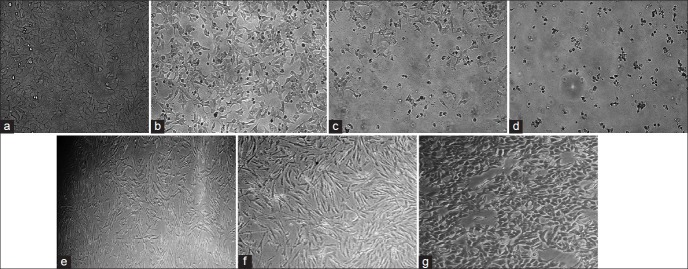
Representative invert microscopy images of MCF-7and MRC-5 cells before and after treatment with 85.43 μmol/L CTB at different times and the sample by using DMSO without CTB (vehicle DMSO), magnification, ×20
Real-Time PCR
It was suggested earlier that apoptotic induction in cancer cell lines by CTB requires the activation of P300 gene expression. To examine this hypothesis, we used 2 cell lines, MCF-7 as cancerous cell line and MRC-5 as non-cancerous cell line. We examined the inhibitory effects of 85.43 μmol/L CTB (based on IC50 index) at different times on the mRNA expression of P300 on MCF-7 and MRC-5 cells using RT-PCR. The P300 gene expression was dramatically up-regulated by CTB treatment with an ascending time in MCF-7 cells, in particular, at 72-hour treatment its increased expression was significantly raised [Figure 4, P < 0.01]. In MRC-5 cells the expression of P300 was also increased 72 hours after the CTB treatment (P > 0.05) but it was not statistically significant in different times (P > 0.05) [Figure 4]. However, the effect of CTB treatment on up-regulation of P300 expression was significantly higher in MCF-7 cells in comparison with MRC-5 cells [Figure 4, P < 0.01].
Figure 4.
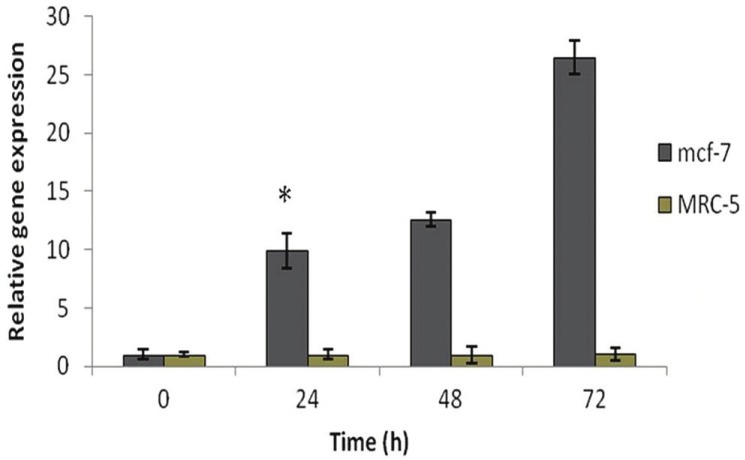
Results of real-time quantitative PCR before and after CTB at different times on the P300 mRNA expression in MCF-7 and MRC-5 cells. Relative expression levels of each gene were obtained by using the Comparative Ct (ΔΔct) method. HAT activator-caused epigenetically activated P300 values were the means of triplicate experiments **P <0.001 vs. control (non-treated CTB) and other MCF-7 groups. No significant difference was seen in other groups
Acetylated and total P53 sandwich ELISA
To investigate further distinct effects of CTB (P300 activator) on cell apoptosis, the ELISA analysis was conducted in MCF-7 (wild-type p53) and MRC-5 cells. The cells were treated at different times (0, 24, 48 and 72 h) with 85.43 μmol/L CTB, to study its effects on the acetylation status of the P53 as targets of P300. The results of ELISA analysis was calculated based on control index. The results showed that CTB could induce P53 acetylation in MCF-7 and MRC-5 cells and significantly increase in the total protein levels with ascending time until 48-hour treatment in MCF-7 cells but not in MRC-5 cells (P < 0.05). Interestingly, between 48 to 72 hours, decrease of protein levels was observed in MCF-7 cells [Figure 5a]. Notably, after treatment by CTB at all different times, the acetylated P53 protein levels in MCF-7 cells was significantly higher than in MRC-5 cells (P < 0.05) [Figure 5a]. Consistently, we also performed the mentioned method to examine the total P53 protein levels in both the cell lines. These results were similar to results of acetylated P53 except for an increase in the total P53 protein levels in MRC-5 cells until 48 hours after treatment (P < 0.05) [Figure 5b]. In the control samples (using DMSO without CTB), there was negligible effect on inducing total and acetylated P53 in both cell lines at the different times of study (P > 0.05) [Figure 5a and b].
Figure 5.
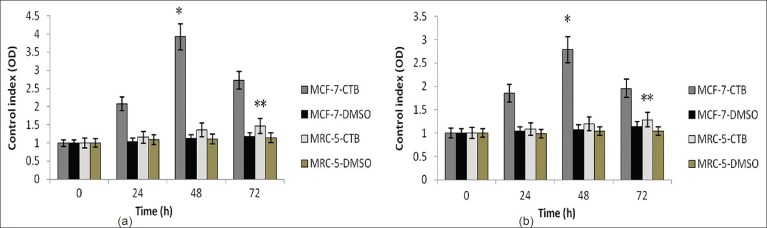
The results of ELISA analysis based on the control index for total and acetylated P53 protein generated in MCF-7 and MRC-5 cells were treated with and without CTB at different times. Cells were treated with 85.43 μmol/L of CTB for 0, 24, 48 and 72 hours. Values are mean + SE of triplicate experiments. (a) *P<0.001 vs. all other groups in different time. **P< 0.05 vs. total P53 in MRC-5 with Salermide treatment at 72 hours. (b) *P< 0.001 vs. all other groups in different time. **P< 0.05 vs. total P53 in MRC-5 without CTB treatment and with CTB treatment at time 48 and 72 hours. No significant difference was shown in total and acetylated P53 content of other groups
DISCUSSION
The potencies and functional mechanisms of the recently developed P300 activator (CTB) were studied at the concentration that was confirmed by IC50 on the MCF-7 as breast cancer cell line and MRC-5 as non-tumorigenic control cells. CTB IC50 result was generally matched with the previously published data of Mantelingu et al.[28] about the efficacy of CTBP on P300 activation. Recent reports have presented that this compound in mammalian cells can specifically enhance the P300 activity.[29] In recent years, a few researchers have described the therapeutic effect of P300 activation but there are no reports of its effects on the diverse types of cancerous and non-cancerous cells.[29,28] The role of P300 during stress is complex and its activator effect is probably cell context-specific.[37,38] Till date, no clear explanation exists about molecular mechanisms of the P300 activators (particularly CTB) in different cells or about comparing their effects on cancerous and normal cells at different times. It is notable that in this study, the apoptotic potency of CTB was examined on the MCF-7 because expression level of P300 is down-regulated, P53 is wild-type, and this kind of cancer is the most prevalent malignancy in woman. In this study, treatment by 85.43 μmol/L of CTB at various time showed a time-dependent increase in apoptotic cell count of the cancerous cells but not in non-tumorigenic MRC-5 cells as measured by flow-cytometric assay. On the other hand, although CTB could effectively induce activation of P300 and subsequent cancer cell death it did not have such an effect on fibroblastic cells (poor cell apoptosis). These results revealed that probable apoptotic sensitivity of MRC-5 cells to CTB stimulation was negligible, which agreed with the previous findings of Fermento et al.[23] stating that increasing activity of p300 by TGF-β in normal skin and lung fibroblasts cells induces stimulation of collagen synthesis in fibrotic lesions. Also Iyer et al.[39] showed that activities of P300 among mammary tumors and normal mammary glands are different, so the inactivation of P300 in breast carcinoma and other solid tumors is confirmed. The severe apoptotic effect in the MCF-7 was observed after 72 hours of incubation with CTB, so as approximately 4% of MCF-7 cells, were viable while at this time MRC-5 cells showed only a slight increase in level of apoptosis (only about 30%). It seems that this result is because of the presence of the wild-type P53. Our results were supported by the findings of Janknecht et al.,[38] which shows modulation of P300 function may consider novel therapies directed against tumors with wild-type P53. Therefore, it was assumed that in MCF-7 cells, degradation of P300 expression promoted cell survival and CTB alone could induce apoptosis in these cancer cells with wild-type P53 in a time-dependent manner. Our results were matched with the findings of Isharwal et al.[5] They state that the increase of P300 expression correlates with the fate of cancerous cells, and it has a prognostic value in predicting biochemical recurrence-free survival or apoptosis in tumor cells. In addition, our results were consistent with the findings of Vempati et al.[6] and Kristin et al.[37] who demonstrated that P300 may be an important regulator of wild-type P53 function and P300 fails to acetylate mutant P53. It was observed that CTB was ineffective to promote acetylation of P53 and it seemed that it cannot activate P53 to induce apoptosis in fibroblastic cells. This finding was supported by morphological examination using invert microscope. Once it was determined that CTB anti-tumor activity was primarily because of the promotion of apoptosis, we decided to study the molecular mechanisms involved in this process. We first studied the expression of P300 in CTB-mediated apoptosis by using real-time PCR. The results of real-time PCR assay indicated that CTB was responsible for the over-expression of P300 in a time-dependent manner in MCF-7 cells (progressive increase in P300 mRNA levels after 24, 48 and 72 h) by exposure to CTB. This was in accordance with the findings of Mantelingu et al.,[28] which showed that incubation of P300 with increasing concentration of CTBP results in a dose-dependent enhancement of P300 HAT activity by HAT assay. It was found that P300 expression level in MRC-5 cells by the treatment of CTB slightly increases in a time-dependent manner only in 72 hours. No alteration of P300 expression levels between the other groups of (non-treatment, 24 and 48 h) MRC-5 samples (normal P300 expression) was found. More importantly it was discovered that in the non-cancerous cells, P300 expression may increase because CTB was at least equivalent to MCF-7 breast cancer cells. Subsequently, we observed that strong P300 transcription occurred particularly after 48 hours of CTB treatment in MCF-7 cells. However, relative stability of P300 expression until 48 hours of treatment and only little enhancement after 72-hour incubation was not effective on the cell apoptosis and viability of the non-cancerous cells. Importantly, it seemed that low levels of apoptosis, which was observed in all MRC-5 samples after treatment with CTB, was more significantly relevant to apoptotic effects of DMSO as carrier and solvent of CTB on fibroblastic cells. This was in agreement with the findings of Ikushima et al.[40] and Polley et al.[11] that Smad complex and TGF-β recruit co-activators such as P300 to induce growth arrest and/or apoptosis through P53 protein interaction in cancerous cells, and loss of P300 genes could lead to tumor progression, which in contrast with normal human epithelial cells seemed to be refractory to P300 activation. Inversely, these observations disagree with the findings of Chen et al.,[41] Bedford et al.[42] and Goodman[43] that knockdown and lack of P300 gene suppress cell growth and increase apoptosis effect in the cancerous cells. However, these data could not completely explain the failure of CTB to induce cell death in MRC-5 cells. The results indicated that function of P300 was different in MCF-7 and MRC-5 cells and that P300 activation might enable MCF-7 cancer cell apoptosis, but it seems non-essential for the apoptosis of lung fibroblast cells. These results were also similar to the study of Karamouzis et al.[44] and Chan et al.[26] that the down-regulation of P300 expression was observed in breast cancer cells, and P300 may provide a tumor suppressor-like function although the tumor-suppressor function of P300 is still unclear. These results were dissimilar with the findings of Fermento et al.,[23] which showed up-regulation of P300 expression in murine mammary adenocarcinoma LM3 cells. Our results showed that the effect of 24 and 48 hours of treatment of MRC-5 cells by CTB on the expression of P300 mRNA was similar to the non-treatment condition, and we observed that after 48 hours of treatment, P300 mRNA levels were elevated. Subsequently, we used real-time PCR to evaluate P300 expression in both the cell lines before treatment. We observed lower expression of P300 in MCF-7 cells in comparison to MRC-5 cells, which might be explained by the fact that decrease of P300 expression in MCF-7 cells leads to inhibited apoptosis and mediated survival in response to stress. Therefore, these results suggest that P300 and HDACs keep a balance of specific acetylation levels for proper cellular function and mediate survival in normal cells. This finding was in accordance with the previous findings of Peck et al.[36] that indicate that the degree of acetylation is largely mediated by a balance between histone acetyltransferase and histone deacteylase in normal cells. Our findings indicate that stimulation of cell death by CTB requires the activation of P300 gene, showing its potential anti-tumor effect. This observation, similar to a recent study by Chen et al.,[45] has showed that small molecule activators of P300 may act as anti-cancer agents. To investigate further and determine the total and acetylated status of P53 in response to CTB in the cells, ELISA analysis was performed after ensuring the level of total protein concentration using the Bradford method. We found a remarkable increase in P53 acetylation level in a time-dependent manner until 48 hours in MCF-7 but not in MRC-5 cells. Consistently, CTB induced a similar increase of total P53 in both the cell lines, as proposed earlier. In MRC-5 cells, we saw a little increase of total and acetylated P53 protein levels in a time-dependent manner of CTB treatment compared to MCF-7 cells, which showed CTB over-expression of P300 resulted in up-regulation of acetylated P53 and subsequently P53 activation in MCF-7 cells (based on previous results by flow-cytometry and real-time PCR assay). Different researchers reported various data about how P300 activation could induce [Doran et al.[7] or not induce [Vempati et al.[6] P53 acetylation in cancer cell lines. A direct correlation between total and acetylated P53 protein levels and CTB toxicity in the MCF-7 cell line was discovered. These results suggested that incubation of MCF-7 with CTB might induce hyperacetylation of P53 protein and apoptosis in MCF-7 cells. Our results indicated that slight decrease of total and acetylated P53 was evident at 72-hour incubation in MCF-7 cells. We suggested that although increase in total and acetylated P53 levels in response to P300 activation at this time was accorded, P53 protein was undetectable by ELISA assay due to the release of proteases and degradation process inside the cancer cells after 48 hours of cell death. A decrease in total P53 level in response to CTB incubation after 48 hours in the control MRC-5 cells was not observed, which indicated that CTB could not induce significant apoptosis even after 48 hours in MRC-5 cells. These observations showed that although in MCF-7 cells P53 was wild-type it was a target for deacetylation and dysfunction of P300 and so couldn’t induce apoptosis due to this aberrant epigenetic event. These data were in accordance with the findings of Iyer et al.[39] that P300 contributes to maintain P53 stability by regulating its ubiquitination and P53 acetylation (activate P53 function) may promote the P53-P300 complex (epigenetic changes). Our findings suggested that acetylation of wild-type P53 as a tumor suppressor might lead to activation of apoptotic program and was integral to cytotoxic activity of the CTB to induce massive apoptosis in less than 24 hours of treatment in MCF-7 cells. This finding further highlights our theoretical assumptions, indicating that activation of P300 is required for the induction of cell death and P53 acetylation in cancer cells only for wild-type P53, and CTB triggers neither cell death nor P53 acetylation in normal cells. Our results are similar to the previous study of Gu et al.[46] that indicates a novel pathway for wild-type P53 acetylate by co-activator, P300 to induce apoptosis in some cell types. Cui et al.[47] showed that Lys-CoA as inhibitor of P300 has anti- proliferative activity against tumor cell lines, which was in contrast to our findings. Treatment with chemotherapeutic drugs also induced cell death indistinguishable from triggers by activation of P300 that also was accompanied by P53 acetylation. We suggested that P300 activators, such as CTB, may function through common pathways and mediate their cytotoxic effects through targeting P53 and its acetylation, which was similar to the study by Gauthier et al.[48] that treatment of HT-29 cells (HIV-1) with forskolin and adenylate cyclase activates CBP (homologous gene of P300) leading to new strategies for reducing virus production in HT-29 cells through P300 (CBP) activation.
CONCLUSION
We formulated the hypothesis that dysfunction of P300 could play crucial roles in inhibiting pro-apoptotic protein expression in cancer cells so it seems that P53 aberrantly repressed in MCF-7 cells. Thus, the treatment with CTB could predominantly induce apoptosis through enhancing activity of P300 and consequent hyperacetylation and reactivation of tumor suppressor P53 to induce cell death in MCF-7 cancer cells. We described that CTB as an activator of P300 could make a promising novel class of agents to the future anti-tumorigenic drugs that target acetylation of proteins, which are epigenetically activated by P300 and thus might be a target for cancer therapy.
ACKNOWLEDGEMENTS
We would like to appreciate Dr. Mohammad Kazemi, Dr. Nafiseh Esmaeili, Dr. Marjan Gharagozloo and Dr. Zahra Babazadeh for their sincere help without which this study could not be performed.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Zou XM, Li YL, Wang H, Cui W, Li XL, Fu SB, et al. Gastric cancer cell lines induced by Trichostatin A. World J Gastroenterol. 2008;14:4810–5. doi: 10.3748/wjg.14.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giahuai T, Shundong C, Yuehua M, Richard LP, Delong L. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. Hematol Oncol. 2010;3:15–28. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lara E, Mai A, Calvanese V, Altucci L, Lopez P, Chantar ML, et al. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptic effect. Oncogene. 2009;28:781–91. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 4.Dworkin AM, Hung TH, Toland AE. Epigenetic alteration in breast: Implication for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009;19:165–71. doi: 10.1016/j.semcancer.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isharwal S, Miller MC, Marlow C, Makarov M, Partin AW, Veltri RW. P300 (histone acetyl transferase) biomarker predicts prostate cancer biochemical recurrence and correlates with changes in epithelia nuclear size and shape. Prostate. 2008;68:1097–104. doi: 10.1002/pros.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D. P300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem. 2010;285:28553–64. doi: 10.1074/jbc.M110.149393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doran D, Shimuizo H, Perkins N, Hupp T. DNA-dependent acetylation of P53 by the transcription coactivators P300. J Biol Chem. 2003;278:13431–41. doi: 10.1074/jbc.M211460200. [DOI] [PubMed] [Google Scholar]
- 8.Emanuele S, Lauricella M, Tesoriere GN. Histone deacetylase inhibitors: Apoptotic effects and clinical implications. Int J Oncol. 2008;33:637–64. [PubMed] [Google Scholar]
- 9.Cazzalini O, Perucca P, Savio M, Necchi D, Bianchi L, Stivala LA, et al. Interaction of P21 with PCANA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008;36:1713–22. doi: 10.1093/nar/gkn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huanh W, Chen C. Akt phosphorylation of P300 at Ser-1834 is essential for its histone acetyltransferases and transcriptional activity. Mol Cell Biol. 2005;25:6592–602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polley S, Guha S, Roy NS, Kar S, Sakaguchi K, Chuman Y, et al. Differential recognition of phosphorylated transactivation domains of P53 by different p300 domains. J Mol Biol. 2008;376:8–12. doi: 10.1016/j.jmb.2007.11.082. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, Bae SC. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–79. [PMC free article] [PubMed] [Google Scholar]
- 13.Cazzalini O, Perucca P, Savio M, Necchi D, Stivala LA, Ducommun B, et al. Interaction of P21CDKN1A with PCNA regulates the histone acetyltransferases activity of P300 in nucleotide excision repair. Nucleic Acids Res. 2008;36:1713–22. doi: 10.1093/nar/gkn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozaki T, Nakagawara A. P53: The Attractive Tumor Suppressor in the cancer research field. J Biomed Biotechnol. 2011;10:11–23. doi: 10.1155/2011/603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leeuwen I, Lain S. Sirtuins and P53. Cancer Res. 2009;9:230–55. doi: 10.1016/S0065-230X(09)02005-3. [DOI] [PubMed] [Google Scholar]
- 16.Cang S, Ma Y, Liu D. New clinical developments in histone deacetylase inhibitors for epigenetic therapy of cancer. J Hematol Oncol. 2009;2:22. doi: 10.1186/1756-8722-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu G, Chen X. Regulation of the P53 transcriptional activity. J Cell Biochem. 2006;97:448–58. doi: 10.1002/jcb.20700. [DOI] [PubMed] [Google Scholar]
- 18.Robbins SL, Kumar V. 7th ed. Vol. 11. Elserier: Saunders; 2010. Robbins and cotran pathologic basis of disease; pp. 19–54. [Google Scholar]
- 19.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacteylase family: Functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Olmosa Y, Brosensb JJ, Lama EW. Interplay between SIRT proteins and tumor suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat. 2011;14:35–44. doi: 10.1016/j.drup.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Chim CS, Wong AS, Kwong YL. Absence of P300 gene promoter methylation in acute leukemia. Cancer Genet Cytogenet. 2004;150:164–7. doi: 10.1016/j.cancergencyto.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Escande C, Chini CS, Nin V, Dykhouse KM. Deleted in breast cancer–1 regulates SIRT1 activity and contributes to high-fat diet–induced liver steatosis in mice. J Clin Invest. 2010;120:545–58. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fermento ME, Gandini NA, Lang CA, Perez JE. Intracellular distribution of P300 and its differential recruitment to aggresomes in breast cancer. Exp Mol Pathol. 2010;88:256–64. doi: 10.1016/j.yexmp.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Boniol M, Gavin A, Vatten LJ. Breast cancer mortality in neighboring European countries with different levels of screening but similar access to treatment: Trend analysis of who mortality database. BMJ. 2011;343:d441. doi: 10.1136/bmj.d4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montazeri A, Vahdaninia M, Harirchi I, Harirchi AM, Sajadian A, Khaleghi F, et al. Breast cancer in Iran: Need for greater women awareness of warning signs and effective screening methods. Asia Pac Fam Med. 2008;7:6. doi: 10.1186/1447-056X-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan HM, Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 27.Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis, and treatment. Semin Cancer Biol. 2009;19:165–71. doi: 10.1016/j.semcancer.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantelingu K, Kishore AH, Balasubramanyam K, Pavan Kumar GV, Altaf M, Swamy SN, et al. Activation of P300 histone acetyltransferases by small molecules altering enzyme structure: Probed by surface-enhanced raman spectroscopy. J Phys Chem B. 2007;111:4527–34. doi: 10.1021/jp067655s. [DOI] [PubMed] [Google Scholar]
- 29.Devipryia B, Paraneswari AR, Rajalakshmi G, Palvannan T, Kumaradhas P. Exploring the binding affinities of P300 enzyme activators CTBP and CTB using docking method. Indian J Biochem Biophys. 2010;47:364–9. [PubMed] [Google Scholar]
- 30.Wong P, Pickard A, Mccance DJ. P300 alters keratinocyte cell growth and differentiation through regulation of p21(Waf1/CIP1) PLoS One. 2010;5:e8369. doi: 10.1371/journal.pone.0008369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cen Y. Sirtuins inhibitors: The approach to affinity and selectivity. Biochim Biophys Acta. 2010;1804:1635–44. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Calvanese V, Fraga MF. Sirt1 brings sternness closer to cancer and aging. Aging (Albany NY) 2011;3:162–7. doi: 10.18632/aging.100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallinari P, Di MS, Jones P, Pallaoro M, Steinkuhler C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 34.Yang T, Fu M, Pestell R, Sauve AA. SIRT1 and endocrine signaling. Trends Endocrinol Metab. 2006;17:186–91. doi: 10.1016/j.tem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Jones AP, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 36.Helweg B, Gatbonton T, Schuler AD. Antitumor activity of a small – molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–77. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 37.Molversmyr AK, Saether T, Gilfillan S, Lorenzo PI, Kvaløy H, Matre V, et al. A SUMO-regulated activation function controls synergy of c-Myb through a repressor–activator switch leading to differential P300 recruitment. Nucleic Acids Res. 2010;38:4970–84. doi: 10.1093/nar/gkq245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janknecht R. The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol Histopathol. 2002;17:657–68. doi: 10.14670/HH-17.657. [DOI] [PubMed] [Google Scholar]
- 39.Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–31. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 40.Ikushima H, Miyazono K. TGFβ signalling: A complex web in cancer progression. Nat Rev Cancer. 2010;10:415–24. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 41.Chen LF, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med (Berl) 2003;81:549–57. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 42.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and P300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodman RH, Smolik S. CBP/P300 in cell growth, transformation and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
- 44.Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Roles of CREB-binding protein (CBP)/P300 in respiratory epithelium tumorigenesis. Cell Res. 2007;17:324–32. doi: 10.1038/cr.2007.10. [DOI] [PubMed] [Google Scholar]
- 45.Chen S, Feng B, George B, Megan CR. Transcriptional co-activator P300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab. 2010;298:E127–37. doi: 10.1152/ajpendo.00432.2009. [DOI] [PubMed] [Google Scholar]
- 46.Gu W, Roeder RG. Activation of P53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 47.Cui L, Miao J, Furuya T, Fan QI, Li X, Rathod PK, et al. Histone acetyltransferases inhibitor Anacardic acid causes changes in global gene expression during In vitro Plasmodium Falciparum development. Eukaryot Cell. 2008;7:1200–10. doi: 10.1128/EC.00063-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gauthier S, Tremblay MJ. Cholera toxin inhibits HIV-1 replication in human colorectal epithelial HT-29 cells through Adenylate cyclase activation. Antiviral Res. 2010;88:207–16. doi: 10.1016/j.antiviral.2010.08.015. [DOI] [PubMed] [Google Scholar]