

Abstract
Peutz–Jeghers syndrome is a rare autosomal dominantly inherited condition, characterized by the presence of hamartomatous gastrointestinal polyps and mucocutaneous pigmentation. Patients with this syndrome can be associated with other neoplasms such as ovarian neoplasms known as sex-cord tumor with annular tubules that are associated in one third of the cases with this syndrome and other types of malignancies. We report a 42-year-old woman with a history of Peutz–Jeghers Syndrome and bilateral breast cancer that presented with abnormal uterine bleeding. Total abdominal hysterectomy with bilateral salpino-oophorectomy was done and an ovarian sex cord tumor with annular tubules was incidentally diagnosed. By reviewing literatures and in agreement with previous studies we suggest routine screening for malignancies in patients with Peutz–Jeghers syndrome.
Keywords: Breast cancer, ovarian cancer, Peutz–Jeghers syndrome, sex cord tumor
INTRODUCTION
Peutz–Jegher's syndrome is a rare autosomal dominant disorder characterized by mucocutaneous pigmentation, hamartomatous polyposis, and predisposition to benign and malignant tumors of the gastrointestinal tract, breast, ovary, uterine cervix, and testis.[1,2,3,4,5,6,7] Germline-inactivating mutations in one allele of the STK11/LKB1gene at chromosome 19p13.3 have been found in most Peutz–Jegher's syndrome.[8,9,10,11,12] Genital tract neoplasms in the female patients with PJS include ovarian neoplasms from the epithelium and stromal cells, adenoma malignum of the cervix, and adenocarcinomas of the endometrium. The most common ovarian neoplasm found in patients with Peutz–Jegher's syndrome is the sex cord tumor with annular tubules, and this is followed by Sertoli cell tumor of the ovary, mucinous epithelial ovarian tumor, serous tumor, and ovarian mature teratoma.[13]
CASE REPORT
In 2003, a 40-year-old woman consulted a doctor because of feeling a palpable mass in the right breast and bleeding discharge from both breasts. Mammography was taken and revealed bilateral mass with infiltrating margins. Excisional biopsy was performed on both breasts. In histopatholgical examination, grossly; the right side showed a nonencapsulated mass with 5.0 × 4.0 × 1.0 cm size as well as relatively dense consistency and the left one showed the same mass with 6.0 × 3.0 × 2.0 cm size. In microscopic examination it was showed carcinoma in atypical sclerosing papilloma of both breasts [Figure 1].
Figure 1.
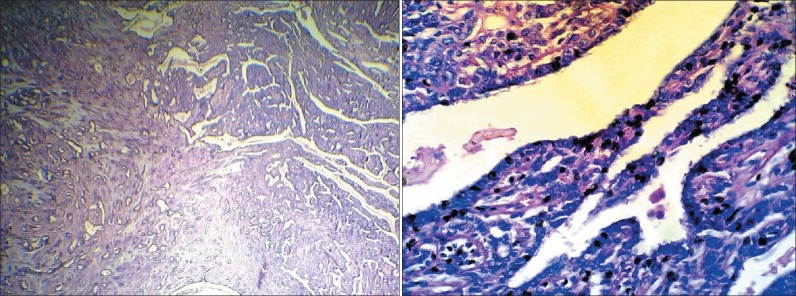
Carcinoma in atypical sclerosing papilloma
One month later bilateral modified radical mastectomy and axillary lymph nodes dissection were performed. In microscopic examination there was no residual of primary tumor and the only abnormality is atypical ductal hyperplasia. Lymph-nodes, skin, and nipple were free. Three months later a colonoscopy was performed and multiple polyps were found in the large bowel. Biopsy was taken and the result was hamartomatous polyp [Figure 2].
Figure 2.
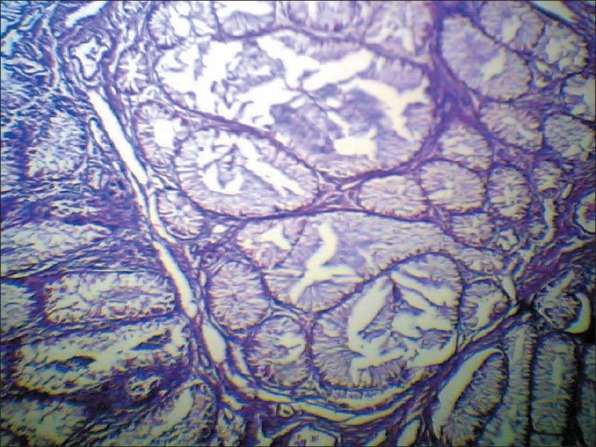
Hamartomatous polyp (×100)
Two years later in 2005, she was referred to the hospital again because of abnormal uterine bleeding (menometrorrhagia). Pelvic ultrasound showed increased endometrial thickness with normal adnexal regions. Bilateral salpingo-oophorectomy was performed. In histopathological examination, grossly, the uterus was measured 10.0 × 8.0 × 3.0 cm. The right ovary was measured 4.0 × 2.0 × 1.0 cm and the left one 4.0 × 3.0 × 1.0 cm. The cut surface of both ovaries showed nonhomogenous appearance with no obvious lesion. In microscopic examination, both ovaries showed sharply circumscribed rounded epithelial nests composed of ring-shaped tubules encircling the hyalinized basement membrane-like material. The nests have two basic patterns: The simple pattern is that of a single tubule encircling a central rounded hyaline mass, and the complex pattern is characterized by communicating tubules encircling multiple hyaline masses. These features are compatible with sex cord tumor with annular tubules that are usually associated with Peutz–Jeghers syndrome [Figure 3]. Except simple hyperplasia of endometrium; no any pathologic feature was found in the endometrium.
Figure 3.
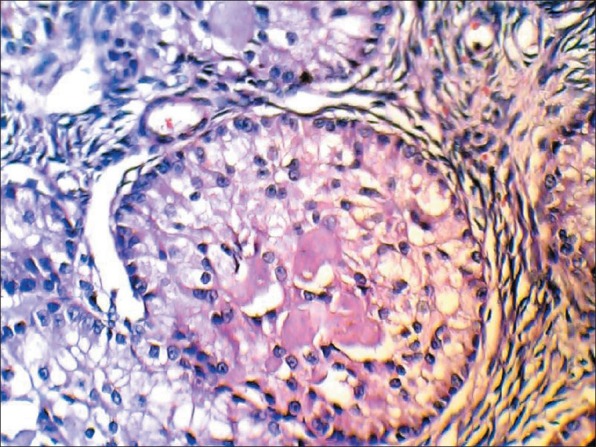
Sex cord tumor with annular tubules (×400)
DISCUSSION
Peutz–Jegher's syndrome is a rare disease. The disease was first recognized in 1921 by Peutz in a Dutch family; the pedigree of this family continues to be followed.[14,15] It is an autosomal dominant disorder with a high degree of penetrance for both polyposis and skin pigmentation. The incidence is 1 in 30000 to 120000 live births[16] and males and females are equally affected. The PJ gene in most families has been mapped to chromosomal 19p13.3 close to marker D19S886.[11,12]
The characteristic mucocutaneous pigmentations (melanin spots) are present in more than 95% of patients and are caused by pigment laden macrophages in the dermis. They are typically flat, bluish-gray to brown spots, 1 to 5 mm in size that look like freckles [Figure 4].
Figure 4.
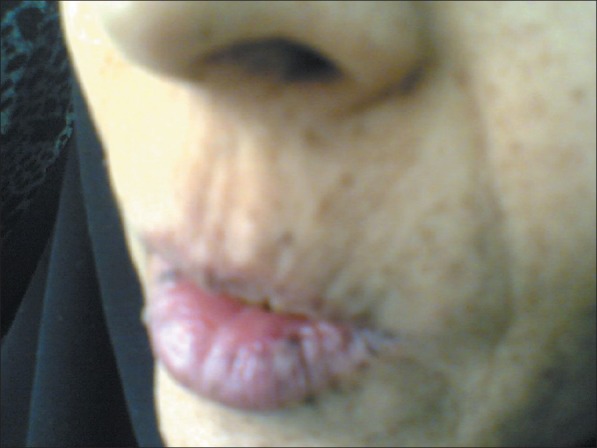
Melanin pigmentation of the lip
In one series of 202 patients, the diagnosis was based upon the skin lesions in 52 patients and related to gastrointestinal polyps in 150.[17] This study found the following frequency and location of polyps in patients with Peutz–Jegher's syndrome:
Small intestine — 64%
Colon — 64%
Stomach — 49%
Rectum — 32%
The Peutz–Jegher's syndrome is associated with an increased risk of gastrointestinal and nongastrointestinal malignancies.[17,18,19] It is estimated that the relative risk of cancer at any site is up to 15-fold higher than the general population. In a meta-analysis that included 210 individuals who had been described in six previous publications, the cumulative risk for all cancers was a remarkable (93%) from the age of 15 to 64.[20] Markedly increased lifetime risks of cancer were observed for cancers of the stomach (29%), small intestine (13%), colon (39%), pancreas (36%), and breast (54%). There were also significantly increased estimates of lifetime risk of cancer of the esophagus (0.5%), lung (15%), uterus (9%), and ovary (21%).[20]
Females have an increased incidence of cervical, uterine, ovarian, and breast cancers (often bilateral).[19] Cervical tumors include cervical adenoma malignum, a highly differentiated mucinous adenocarcinoma with a highly aggressive behavior.[21] In addition, small, asymptomatic, benign ovarian tumors known as sex-cord tumors with annular tubules occur commonly in women with this syndrome.[22]
We discovered a woman with this syndrome while evaluating a patient with bilateral breast cancer. Then ovarian sex cord tumor with annular tubules was incidentally diagnosed.
Sex-cord tumors with annular tubules is the most common ovarian neoplasm found in the patients with PJS.[13] Among the patients diagnosed with sex-cord tumors with annular tubules, 36% of them are affected by Peutz–Jegher's syndrome.[22] This neoplasm is a distinctive ovarian neoplasm, and the predominant component of this tumor has morphologic features that are intermediate between those features of the granulosa cell tumor and those features of the sertoli cell tumor; focal differentiation into either granulosa cell or sertoli cell tumor may occur. It is capable of producing both estrogen and progesterone. Symptoms suggestive of hyperestrogenism have been described in approximately 50% of cases including irregular menstrual bleeding, postmenopausal bleeding, and isosexual precocity have been described.[22,23] In our case, the hyperstrogenism was presented as abnormal uterine bleeding. When this condition occurs in association with Peutz–Jegher's syndrome, the ovarian sex-cord tumors with annular tubules are almost always benign and they are typically multifocal, calcified, bilateral, very small or even microscopical in size.[22] In contrast, those unassociated with Peutz–Jegher's syndrome, are unilateral and large, and they sometimes have malignant behavior. Microscopically, the predominant appearance of the tumor is similar to that encountered in patients with the syndrome, but variations from the characteristic pattern are seen in minor portions of the tumor.
Breast cancer that is usually ductal, but occasionally it is lobular carcinoma, seems to be found in patients with Peutz–Jegher's syndrome with an increased frequency.[24]
An atypical papilloma of breast displays focal areas within the papillary processes with proliferation of a monotonous cell population identical to the cribriform or other variants of low-grade non-necrotic intraductal carcinoma.[25,26] The myoepithelial cell layer may or may not be retained beneath these areas.[26] Such areas may be interpreted as carcinoma arising in a papilloma when they occupy at least a third but less than 90% of the lesion. If less than a third of the lesion show these changes, the designation of atypical papilloma would be appropriate.
Once necrosis became evident in the cribriform areas, the lesion qualifies as carcinoma arising in a papilloma even if occupies less than 33% of the lesion.[26,27,28] Focal proliferation of a truly anaplastic cell population qualifies as carcinoma arising in a papilloma even if it occupies less than a third of a lesion. In our case more than 90% of the papilloma shows carcinomatous changes without any necrosis.[26,27] The clinical significance of carcinoma arising in a papilloma has not been established. It is possible that when only a third of a papillary lesion is replaced by intraductal carcinoma, its behavior would be different from one in which close to 90% of the lesion displays features of a carcinoma.[25]
CONCLUSION
Regarding to the pervious studies we suggest patients with Peutz–Jegher's syndrome should be regularly and closely monitored, because of the increased risk of cancer. Early diagnosis of this syndrome and routine screening for malignancy in these patients can detect some malignancies in early stages.
ACKNOWLEDGEMENTS
Authors wish to express their gratitude to woman who participated in this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Jeghers H, Mc Kusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:1031. doi: 10.1056/NEJM194912292412601. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharya S, Mahapatra SR, Ramlal Nangalia R, Amitabh Palit A, Morrissey JR, Ruban E, et al. Melaena with Peutz-Jeghers syndrome: A case report. J Med Case Rep. 2010;4:44. doi: 10.1186/1752-1947-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latchford AR, Neale K, Phillips RK, Clark SK. Peutz-Jeghers syndrome: Intriguing suggestion of gastrointestinal cancer prevention from surveillance. Dis Colon Rectum. 2011;54:1547–51. doi: 10.1097/DCR.0b013e318233a11f. [DOI] [PubMed] [Google Scholar]
- 4.Christiansen E, Nielsen R. Gastrointestinal bleeding and intussusception caused by Peutz-Jeghers syndrome. Ugeskr Laeger. 2011;173:2800–1. [PubMed] [Google Scholar]
- 5.Kondi-Pafiti A, Bakalianou K, Iavazzo C, Dastamani C, Hasiakos D, Liapis A. Endometrial carcinoma and ovarian sex cord tumor with annular tubules in a patient with history of Peutz-Jeghers syndrome and multiple malignancies. Eur J Gynaecol Oncol. 2011;32:452–4. [PubMed] [Google Scholar]
- 6.Sweetser S, Sugumar A, Boardman LA. A rare cause of obscure gastrointestinal bleeding. Diagnosis: Sporadic Peutz-Jeghers type jejunal polyp causing obsucre GI bleeding. Gastroenterology. 2011;141:1159. doi: 10.1053/j.gastro.2009.10.062. 1533. [DOI] [PubMed] [Google Scholar]
- 7.Takakura K, Kato T, Arihiro S, Miyazaki Y, Arai Y, Nakao N, et al. Selective ligation using a detachable snare for small-intestinal polyps in patients with Peutz-Jeghers syndrome. Endoscopy. 2011;43(Suppl 2 UCTN):E264–5. doi: 10.1055/s-0030-1256528. [DOI] [PubMed] [Google Scholar]
- 8.Sugars S. Peutz-Jeghers syndrome: A patient's view. Fam Cancer. 2011;10:473–9. doi: 10.1007/s10689-011-9453-y. [DOI] [PubMed] [Google Scholar]
- 9.Riegert-Johnson D, Roberts M, Gleeson FC, Krishna M, Boardman L. Case studies in the diagnosis and management of Peutz-Jeghers syndrome. Fam Cancer. 2011;10:463–8. doi: 10.1007/s10689-011-9438-x. [DOI] [PubMed] [Google Scholar]
- 10.Latchford AR, Phillips RK. Gastrointestinal polyps and cancer in Peutz-Jeghers syndrome: Clinical aspects. Cancer. 2011;10:455–61. doi: 10.1007/s10689-011-9442-1. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Churchman M, Avizienyte E, McKeown C, Davies S, Evans D, et al. Germline mutations of the LKB1 (STK11) gene in Peutz-Jeghers patients. J Med Genet. 1999;36:365–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 13.Papageorgiou T, Stratakis CA. Ovarian tumors associated with multiple endocrine neoplasias and related syndromes (Carney complex, Peutz-Jeghers Syndrome, von Hippel Lindau disease, Cowden's disease) Int J Gynecol Cancer. 2002;12:337–47. doi: 10.1046/j.1525-1438.2002.01147.x. [DOI] [PubMed] [Google Scholar]
- 14.Sarlós P, Király A, Nagy L. Family study in Peutz-Jeghers syndrome. Orv Hetil. 2007;148:255–8. doi: 10.1556/OH.2007.27640. [DOI] [PubMed] [Google Scholar]
- 15.Westerman AM, Entius MM, Boor PP, Koole R, de Baar E, Offerhaus GJ, et al. Novel mutations in the LKB1/STK11 gene in Dutch Peutz-Jeghers families. Hum Mutat. 1999;13:476–81. doi: 10.1002/(SICI)1098-1004(1999)13:6<476::AID-HUMU7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 16.Lindor NM, Greene MH. The concise handbook of family cancer syndromes. Mayo Familial Cancer Program. J Natl Cancer Inst. 1998;90:1039–71. doi: 10.1093/jnci/90.14.1039. [DOI] [PubMed] [Google Scholar]
- 17.Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: Its natural course and management. Johns Hopkins Med J. 1975;136:71–82. [PubMed] [Google Scholar]
- 18.Spigelman AD, Murday V, Phillips RK. Cancer and the Peutz- Jeghers syndrome. Gut. 1989;30:1588–90. doi: 10.1136/gut.30.11.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boardman LA, Pittelkow MR, Couch FJ, Schaid DJ, McDonnell SK, Burgart LJ, et al. Association of Peutz-Jeghers-like mucocutaneous pigmentation with breast and gynecologic carcinomas in women. Medicine. 2000;79:293–8. doi: 10.1097/00005792-200009000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–53. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 21.Srivatsa PJ, Keeney GL, Podratz KC. Disseminated cervical adenoma malignum and bilateral ovarian sex cord tumors with annular tubules associated with Peutz-Jeghers syndrome. Gynecol Oncol. 1994;53:256. doi: 10.1006/gyno.1994.1127. [DOI] [PubMed] [Google Scholar]
- 22.Young RH, Welch WR, Dickersin GR, Scully RE. Ovarian sex cord tumor with annular tubules. Review of 74 cases including 27 with Peutz-Jeghers syndrome and 4 with adenoma malignum of the cervix. Cancer. 1982;50:1384. doi: 10.1002/1097-0142(19821001)50:7<1384::aid-cncr2820500726>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 23.Srivatsa PJ, Keeney GL, Podratz KC. Disseminated cervical adenoma malignum and bilateral ovarian sex cord tumors with annular tubules associated with Peutz-Jeghers syndrome. Gynecol Oncol. 1994;53:256–64. doi: 10.1006/gyno.1994.1127. [DOI] [PubMed] [Google Scholar]
- 24.de Jong MM, Nolte IM, te Meerman GJ, van der Graaf WT, Oosterwijk JC, Kleibeuker JH, et al. Genes other than BRCA1 and BRCA2 involved in breast cancer susceptibility. J Med Genet. 2002;39:225–42. doi: 10.1136/jmg.39.4.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raju U, Vertes D. Breast papillomas with atypical ductal hyperplasia: A clinicopathologic study. Hum Pathol. 1996;27:1231–8. doi: 10.1016/s0046-8177(96)90320-2. [DOI] [PubMed] [Google Scholar]
- 26.Valdes EK, Tartter EI. Significance of papillary lesions at percutaneous breast biopsy. Ann Surg Oncol. 2006;13:480–2. doi: 10.1245/ASO.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Renshaw AA. Papillomas and atypical papillomas in breast core needle biopsy specimens: Risk of carcinoma in subsequent excision. Am J Clin Pathol. 2004;122:217–21. doi: 10.1309/K1BN-JXET-EY3H-06UL. [DOI] [PubMed] [Google Scholar]
- 28.Gutman H. Are solitary breast papillomas entirely benign? Arch Surg. 2003;138:1330–3. doi: 10.1001/archsurg.138.12.1330. [DOI] [PubMed] [Google Scholar]