

BN/CC isosterism has recently emerged as a viable strategy to increase structural diversity.[1, 2] In particular, the chemistry of 1,2-dihydro-1,2-azaborines (hereafter abbreviated as 1,2-aza-borines), which are BN isosteres of the family of arenes, has attracted attention as novel aromatic compounds relevant to biomedical research and materials science. In the past decade, significant advances have been made in the synthesis and characterization of 1,2-azaborines.[3, 4] However, virtually no information is available on the behavior of 1,2-azaborines in a biological context. We previously demonstrated that N-ethyl-1,2-azaborine and the parent 1,2-azaborine bind inside the hydrophobic pocket of the L99A mutant of T4 lysozyme.[5] The demonstration of a biochemically active role of 1,2-azaborines, for example, as substrate or inhibitor of an enzyme, has to date remained elusive. In view of the dominance of arenes in pharmaceuticals, the study of the biochemical reactivity of 1,2-azaborines is significant: the new chemical space made available by 1,2-azaborines could open up opportunities in drug discovery and biomedical research. Herein, we reveal that the BN isosteres of ethylbenzene, N- and B-ethyl-1,2-azaborine, are actually strong inhibitors for the hydroxylation of ethylbenzene by ethylbenzene dehydrogenase (EbDH), providing the proof of concept that BN/CC isosterism can lead to novel biochemical behavior (Scheme 1). Ethylbenzene dehydrogenase is a molybdenum enzyme catalyzing the oxygen-independent hydroxylation of ethylbenzene to (S)-1-phenylethanol, initiating anaerobic ethylbenzene mineralization in the denitrifying betaproteo-bacterium “Aromatoleum aromaticum”.[6–10] Oxygen-independent hydroxylation of a non-activated hydrocarbon is an unusual and unique feature of this enzyme.[8] A potential reaction mechanism involving consecutive radical and carbocation derivatives of the substrates as reaction intermediates was derived from quantum chemical calculations and experimental data.[11–16] The enzyme hydroxylates an extraordinarily broad spectrum of alkylated aromatic and heteroaromatic compounds in an enantioselective manner,[12] making it a very suitable system to investigate 1,2-azaborines as potential substrates.
Scheme 1.
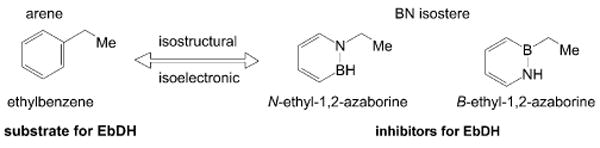
Effect of BN/CC isosterism on enzyme reactivity.
Herein, we explore N-ethyl-1,2-azaborine and B-ethyl-1,2-azaborine as substrates for EbDH (Scheme 1). Somewhat surprisingly, we did not detect enzymatic turnover for either of the 1,2-azaborines by photometric assay or by GC-MS analysis of liquid-phase extractions of reactions incubated for up to 12 h. However, we were able to identify both N-ethyl-1,2-azaborine and B-ethyl-1,2-azaborine as very strong inhibitors of EbDH (see Figure 1a). In particular, N-ethyl-1,2-azaborine turned out to be a very efficient inhibitor with an IC50 value of 2.8 μm whereas B-ethyl-1,2-azaborine had an IC50 value of 100 μm. The nature of inhibition with N-ethyl-1,2-azaborine was investigated by a more detailed inhibitory kinetic analysis (Figure 1b). We determined N-ethyl-1,2-azaborine to be a mixed-type inhibitor with a very low competitive inhibition constant (Kic = 0.55 μm), which is basically identical to the Km of ethylbenzene (0.45 μm).[11] The uncompetitive inhibition constant (Kiu = 9 μm) is 16-fold higher than that value, suggesting the presence of a second binding site. The experimental observations are consistent with the very similar molecular/geometric structures between N-ethyl-1,2-azaborine and the native substrate ethylbenzene, which should make it hard for the enzyme to distinguish the two compounds.
Figure 1.
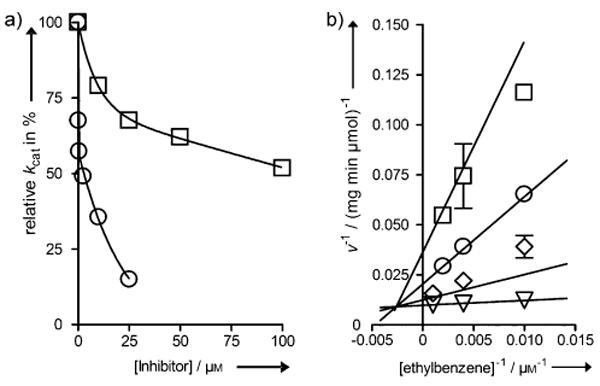
A: Nonlinear plots of the remaining ethylbenzene-dependent activity of EbDH when preincubated with different concentrations of N-ethyl-1,2-azaborine (circles) and B-ethyl-1,2-azaborine (squares). B: Double reciprocal plot of ethylbenzene-dependent enzymatic activity obtained when preincubated with different concentrations of N-ethyl-1,2-azaborine: 0 μm (triangles), 2.5 μm (diamonds), 10 μm (circles) 25 μm (squares). The kinetic data were fitted using the model for mixed inhibition[17] (see also Supporting Information).
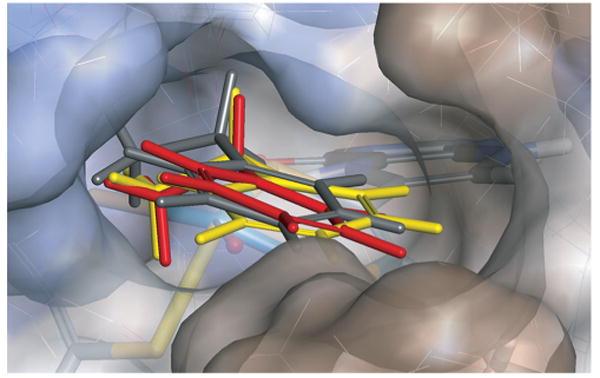
We further performed a MM (molecular mechanics) modeling (see Supporting Information) of the 1,2-azaborines inside the EbDH substrate binding pocket and of bound ethylbenzene to probe this hypothesis (Figure 2 and Supporting Information). Indeed, both 1,2-azaborines fit nicely in the pocket. In contrast to N-ethyl-1,2-azaborine, which is almost congruent with ethylbenzene, B-ethyl-1,2-azaborine seems to bind in a slightly different orientation to the active site. As can be seen from Figure 1a, B-ethyl-1,2-azaborine is a less-efficient inhibitor than N-ethyl-1,2-azaborine. Our modeling suggests that the lower inhibitory effect of B-ethyl-1,2-azaborine in comparison to N-ethyl-1,2-azaborine is a result of weaker binding through less-favorable van der Waals interactions with the hydrophobic residues in the active site (see Supporting Information for details).
Figure 2. Overlay of N-ethyl-1,2-azaborine (red), B-ethyl-1,2-azaborine (yellow), and ethylbenzene (gray) modeled into the EbDH active site. The protein surface is given in grades of hydrophobicity (brown= hydrophobic, blue=hydrophilic). The view leads from the substrate channel towards the molybdenum cofactor. Note that force-field parameters had to be estimated for modeling the 1,2-azaborines, which may cause some deviations from the actual binding mode (see Supporting Information).
All the known substrates of EbDH are alkylaromatic compounds substituted at the C atoms of the aromatic ring.[11,12] To demonstrate that the relatively strong inhibitory behaviors of 1,2-azaborines originate from the combination of geometrical similarity and electronic structure differences between ethylbenzene and the corresponding 1,2-azaborines (BN/CC isosterism) rather than simple electronic structure changes through alkyl substitution at a heteroatom, we investigated the reactivity of EbDH with N-ethylimidazole and N-ethylpyrrole as other heteroatom-substituted hetero-cyclic substrate analogues. In doing so, we found that N-ethylimidazole is another inhibitory compound rather than a substrate, showing albeit a very weak inhibitory effect (IC50 value of 2200 μm) compared to N- and B-ethyl-1,2-azaborine. On the other hand, we detected clear enzymatic activity with N-ethylpyrrole, although it had a threefold lower rate than ethylbenzene (kcat: 0.16 s−1 and 0.4 s−1, respectively) and showed a relatively low affinity to EbDH (Km(app) value: 130 μm). The hydroxylation of N-ethylpyrrole proceeded with an observed enantiomeric excess (ee) of 60% for the S-enantiomer of the product 1-(1 H-pyrrol-1-yl)ethanol, determined by chiral GC coupled with mass spectrometry (see Supporting Information). We also detected a dehydrogenated side product (1-vinyl-1 H-pyrrole), derived from a carbocation intermediate postulated to be generated by EbDH.[12] 1-Vinyl-1 H-pyrrole may react non-enzymatically with water, resulting in a racemic mixture of the 1-(1 H-pyrrol-1-yl)ethanol enantiomers, which would explain the relatively low stereoselectivity observed for N-ethylpyrrole hydroxylation.
To correlate the enzyme kinetic parameters of substrate conversion and enzyme inhibition to the molecular and electronic properties of the heteroatom-substituted compounds, calculations of the stabilities of corresponding radical and carbocation intermediates relative to those of ethylbenzene were performed (Table 1).[14] The data for N-ethyl-pyrrole show that radical formation is slightly less favored than for ethylbenzene. However, carbocation formation is energetically more favorable than for ethylbenzene, consistent with the involvement of a carbocation intermediate in the oxidation of N-ethylpyrrole by EbDH. Because substrate activation by radical formation is believed to be the rate limiting step,[16] these values may also explain the lower enzymatic activity with N-ethylpyrrole than with ethylbenzene. For the inhibitory compounds including the 1,2-azabor-ines, both calculated energies for radical and carbocation formation are substantially higher than those for ethylbenzene (Table 1). Therefore, it appears plausible that the energy barriers for forming the radical and carbocation intermediates cannot be overcome with these compounds, leading to inhibition by formation of non-productive enzyme–ligand complexes. These new data are consistent with the model for the catalytic mechanism of EbDH and will help to refine and expand the quantitative structure–activity relationship (QSAR) analysis for EbDH.[11]
Table 1.
Relative calculated energy levels for radical and carbocation formation from different ethylbenzene analogues.
| Compound[a] | ΔΔG radical [kJ mol−1] | ΔΔG carbocation [kJ mol−1] |
|---|---|---|
| N-ethyl-1,2-azaborine | 23.69 | 12.28 |
| B-ethyl-1,2-azaborine | 15.41 | 24.43 |
| N-ethylimidazole | 23.0 | 37.9 |
| ethylbenzene | 0 | 0 |
| N-ethylpyrrole | 18.56 | −14.46 |
Compounds in bold are substrates, the other compounds are inhibitors of EbDH.
In summary, this study provides the first evidence that non-natural 1,2-azaborine compounds can influence enzyme activity as structural mimics of aromatic compounds. The differences in electronic structures between ethylbenzene and its BN isosteres result in a role change from substrate to inhibitor in EbDH-mediated hydroxylation reactions. Control experiments with other heteroatom-substituted alkylarenes suggest that the combined effects of geometric similarity and electronic structure differences provided by BN/CC isosterism are responsible for the potent inhibition. The data reported herein demonstrate the proof-of-concept that BN/ CC isosterism can lead to novel behavior in a biological context. The extended chemical space made available by azaborines should provide chemists with new tools to explore structure–activity relationships in biomedical research and medicinal chemistry.
Supplementary Material
Footnotes
We acknowledge the financial support of the priority program 1319 of the German Research Foundation (DFG), the excellence program LOEWE/Synmikro from the state of Hessen, the Polish Ministry of Science and Higher Education under the grant N N204 269038 and MNiSW/SGI3700/PAN/121/2006. Support for this research has been provided by the National Institutes of Health (Grant R01-GM094541). Support for G.P.H has been provided by the Arnold and Mabel Beckman Foundation and the Camille & Henry Dreyfus Foundation. We thank Jesse Jenkins for some early synthetic contributions.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201208351.
Contributor Information
Daniel H. Knack, Laboratory for Microbial Biochemistry, Philipps University of Marburg, 35043 Marburg (Germany)
Jonathan L. Marshall, Department of Chemistry, University of Oregon, Eugene, OR, 97403-1253 (USA)
Gregory P. Harlow, Department of Chemistry, University of Oregon, Eugene, OR, 97403-1253 (USA)
Agnieszka Dudzik, Jerzy Haber Institute for Catalysis and Surface Chemistry, Polish Academy of Sciences, 30-239 Kraków (Poland).
Dr. Maciej Szaleniec, Jerzy Haber Institute for Catalysis and Surface Chemistry, Polish Academy of Sciences, 30-239 Kraków (Poland)
Prof. Dr. Shih-Yuan Liu, Email: lsy@uoregon.edu, Department of Chemistry, University of Oregon, Eugene, OR, 97403-1253 (USA).
Prof. Dr. Johann Heider, Email: heider@biologie.uni-marburg.de, Laboratory for Microbial Biochemistry, Philipps University of Marburg, 35043 Marburg (Germany).
References
- 1.Liu Z, Marder TB. Angew Chem. 2008;120:248–250. Angew Chem. Int. Ed.2008, 47, 242–244. [Google Scholar]
- 2.Zhou HB, Nettles KW, Bruning JB, Kim Y, Joachimiak A, Sharma S, Carlson KE, Stossi F, Katzenellenbogen BS, Greene GL, Katzenellenbogen JA. Chem Biol. 2007;14:659–669. doi: 10.1016/j.chembiol.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 3.For a Review of azaborine chemistry, see:; a Campbell PG, Marwitz AJV, Liu SY. Angew Chem. 2012;124:6178–6197. Angew. Chem. Int. Ed.2012, 51, 6074–6092. [Google Scholar]; b Bosdet MJD, Piers WE. Can J Chem. 2009;87:8–29. [Google Scholar]; c Ashe AJ., III Organometallics. 2009;28:4236–4248. [Google Scholar]
- 4.a Abbey ER, Zakharov LN, Liu SY. J Am Chem Soc. 2008;130:7250–7252. doi: 10.1021/ja8024966. [DOI] [PubMed] [Google Scholar]; b Marwitz AJV, Matus MH, Zakharov LN, Dixon DA, Liu SY. Angew Chem. 2009;121:991–995. doi: 10.1002/anie.200805554. Angew. Chem. Int. Ed.2009, 48, 973–977. [DOI] [PubMed] [Google Scholar]; c Campbell PG, Abbey ER, Neiner D, Grant DJ, Dixon DA, Liu SY, Am J. Chem Soc. 2010;132:18048–18050. doi: 10.1021/ja109596m. [DOI] [PubMed] [Google Scholar]; d Abbey ER, Zakharov LN, Liu SY. J Am Chem Soc. 2011;133:11508–11511. doi: 10.1021/ja205779b. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lamm AN, Garner EB, Dixon DA, Liu SY. Angew Chem. 2011;123:8307–8310. Angew. Chem. Int. Ed.2011, 50, 8157–8160. [Google Scholar]; f Chrostowska A, Xu S, Lamm AN, Mazière A, Weber CD, Dargelos A, Baylère P, Graciaa A, Liu SY. J Am Chem Soc. 2012;134:10279–10285. doi: 10.1021/ja303595z. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Marwitz AJV, Abbey ER, Jenkins JT, Zakharov LN, Liu SY. Org Lett. 2007;9:4905–4908. doi: 10.1021/ol702383u. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Marwitz AJ, Matthews BW, Liu SY. Angew Chem. 2009;121:6949–6951. doi: 10.1002/anie.200903390. Angew. Chem. Int. Ed.2009, 48, 6817–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson HA, Pelletier DA, Spormann AM. J Bacteriol. 2001;183:4536–4542. doi: 10.1128/JB.183.15.4536-4542.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson HA, Spormann AM. J Bacteriol. 1999;181:5662–5668. doi: 10.1128/jb.181.18.5662-5668.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kniemeyer O, Heider J. J Biol Chem. 2001;276:21381–21386. doi: 10.1074/jbc.M101679200. [DOI] [PubMed] [Google Scholar]
- 9.Rabus R, Kube M, Beck A, Widdel F, Reinhardt R. Arch Microbiol. 2002;178:506–516. doi: 10.1007/s00203-002-0487-2. [DOI] [PubMed] [Google Scholar]
- 10.Kloer DP, Hagel C, Heider J, Schulz GE. Structure. 2006;14:1377–1388. doi: 10.1016/j.str.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Szaleniec M, Hagel C, Menke M, Nowak P, Witko M, Heider J. Biochemistry. 2007;46:7637–7646. doi: 10.1021/bi700633c. [DOI] [PubMed] [Google Scholar]
- 12.Knack DH, Hagel C, Szaleniec M, Dudzik A, Salwinski A, Heider Jm. Appl Environ Microbiol. 2012;78:6475–6482. doi: 10.1128/AEM.01551-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szaleniec M, Borowski T, Schuhle K, Witko M, Heider J. J Am Chem Soc. 2010;132:6014–6024. doi: 10.1021/ja907208k. [DOI] [PubMed] [Google Scholar]
- 14.Szaleniec M, Witko M, Heider J. J Mol Catal A. 2008;286:128–136. [Google Scholar]
- 15.Szaleniec M, Witko M, Tadeusiewicz R, Goclon J. J Comput Aided Mol Des. 2006;20:145–157. doi: 10.1007/s10822-006-9042-6. [DOI] [PubMed] [Google Scholar]
- 16.Szaleniec M, Salwinski A, Borowski T, Heider J, Witko M. Int J Quantum Chem. 2012;112:1990–1999. [Google Scholar]
- 17.Cornish Bowden A. Analysis of Enzyme Kinetic Data. Oxford University Press; Oxford: 1995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.