

Abstract
Background
We have reported that minocycline (Mino) induced autophagic death in glioma cells. In the present study, we characterize the upstream regulators that control autophagy and switch cell death from autophagic to apoptotic.
Methods
Western blotting and immunofluorescence were used to detect the expressions of eukaryotic translation initiation factor 2α (eIF2α), transcription factor GADD153 (CHOP), and glucose-regulated protein 78 (GRP78). Short hairpin (sh)RNA was used to knock down eIF2α or CHOP expression. Autophagy was assessed by the conversion of light chain (LC)3-I to LC3-II and green fluorescent protein puncta formation. An intracranial mouse model and bioluminescent imaging were used to assess the effect of Mino on tumor growth and survival time of mice.
Results
The expression of GRP78 in glioma was high, whereas in normal glia it was low. Mino treatment increased GRP78 expression and reduced binding of GRP78 with protein kinase-like endoplasmic reticulum kinase. Subsequently, Mino increased eIF2α phosphorylation and CHOP expression. Knockdown of eIF2α or CHOP reduced Mino-induced LC3-II conversion and glioma cell death. When autophagy was inhibited, Mino induced cell death in a caspase-dependent manner. Rapamycin in combination with Mino produced synergistic effects on LC3 conversion, reduction of the Akt/mTOR/p70S6K pathway, and glioma cell death. Bioluminescent imaging showed that Mino inhibited the growth of glioma and prolonged survival time and that these effects were blocked by shCHOP.
Conclusions
Mino induced autophagy by eliciting endoplasmic reticulum stress response and switched cell death from autophagy to apoptosis when autophagy was blocked. These results coupled with clinical availability and a safe track record make Mino a promising agent for the treatment of malignant gliomas.
Keywords: apoptosis, autophagy, ER stress, glioma, minocycline
Malignant gliomas are the most frequent primary tumors of the brain and account for ∼70% of the 22 500 new cases diagnosed in the United States each year.1 The outlook for patients with malignant gliomas is poor. Median survival for patients with intermediate-grade malignant glioma (World Health Organization [WHO] grade III or anaplastic astrocytoma) is 3–5 years.1–3 For patients with the most severe, aggressive form of malignant glioma (WHO grade IV, or glioblastoma multiforme), median survival is 19–23 months.4,5 Treatment for malignant gliomas depends both on the degree of malignancy and the location of the tumor.6,7 If the tumor is localized around or near extremely important structures within the brain, surgical dissection is not suggested because of the risk to surrounding structures. In a majority of cases, with or without surgical excision, combination radiation treatment and chemotherapy are used to combat the malignancy. Despite improvements provided by cytoreductive surgery and primary chemotherapy, the prognosis for patients with malignant gliomas remains very poor. Therefore, development of novel strategies and discovery of new drugs for malignant glioma treatment are essential.
Endoplasmic reticulum (ER) is an organelle of cells in eukaryotic organisms where lipid synthesis, protein folding, and protein maturation take place. ER is the major signal-transducing organelle that senses and responds to changes of homeostasis.8 Conditions interfering with the function of ER, including those of cellular redox regulation, trigger an evolutionarily conserved response, termed the unfolded protein response (UPR).9 The UPR is activated when unfolded or misfolded proteins accumulate in the lumen of ER to alleviate ER stress by arresting general translation, upregulating chaperones and folding enzymes, and degrading misfolded protein.10,11 In mammalian cells, 3 transmembrane ER stress sensors—protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcriptional factor 6 (ATF6)—play the central role in ER stress signaling. PERK phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α), and IRE1 catalyzes the splicing of the mRNA encoding the transcription factor X-box binding protein 1 (XBP-1). These active transcription factors regulate the expression of ER chaperones such as glucose-regulated protein 78 (GRP78) and pro-death proteins such as cytidine-cytidine-adenosine-adenosine-thymidine (CCAAT)/enhancer binding homologous protein (CHOP). In general, intense or persistent ER stress induces apoptosis, resulting in cell death.12–14 Recent studies have shown that ER stress could also cause cell death via inducing autophagy.15,16 However, it was also noted that an ER stress response could trigger autophagy associated with cell survival under unfavorable conditions.17–19
Minocycline (7-dimethylamino-6-desoxytetracycline; Mino) is a semisynthetic tetracycline derivative with bacteriostatic activity against acne and rosacea.20,21 It is a small, highly lipophilic molecule that can be readily absorbed from the gut after oral ingestion and is capable of crossing the blood–brain barrier.20,22 We have previously shown that Mino induced cell death in glioma cells that were associated with the presence of autophagic vacuoles in the cytoplasm.23 Interestingly, apoptosis did not occur in Mino-treated glioma cells. Only when autophagy was inhibited by 3-methyladenine (3-MA) did Mino induce apoptosis. In the present study, we characterize the upstream regulators that control both autophagy and apoptosis induced by Mino in glioma cells.
Materials and Methods
Cell Culture and Reagents
The human glioma cell line U87 was kindly provided by Dr. Michael Hsiao (Genomics Research Center, Academia Sinica, Taiwan) and cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 2 mM l-glutamine (Invitrogen), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Invitrogen). Rat glioma C6 cells were kindly provided by Dr. Shun-Fen Tzeng (National Cheng-Kung University, Taiwan) and cultured in DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. Mino and 3-MA (Sigma-Aldrich) were dissolved in phosphate buffered saline (PBS). Temozolomide (Temodar), bafilomycin (BAF) A1, the pan-caspase inhibitor z-VAD, and rapamycin (Sigma-Aldrich) were dissolved in dimethyl sulfoxide.
Cell Viability Assay
For the cell viability assay, 2 × 103 glioma cells per well were seeded in 96-well plates and allowed to attach overnight at 37°C. Culture medium containing vehicle or drugs was added to the medium in each well, and cells were incubated at 37°C for indicated time points. Cytotoxicity assay by MTS (tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega) was used to measure cell viability as described previously.24 At indicated time points, cells in 96-well plates were incubated with MTS solution (20 μL/well) in growth medium for 1–4 h at 37°C. The absorbance of soluble formazan was measured at 490 nm with a microplate reader. Cell viability was presented as the percentage of survivors relative to the vehicle-treated control culture.
Clonogenic Survival Assay
To determine long-term effects, different cells were cultured overnight in a 6-well plate (1500 cells per well) and treated with Mino (50 μM) alone or with the addition of 3-MA (1 mM) and/or z-VAD (20 μM) for 24 h. Then the medium was replaced with drug-free medium, and cells were incubated for 10 days to form colonies. After 10 days, cells were fixed and stained with crystal violet (0.2%) to visualize cell colonies. Colonies were counted, and results were normalized to the colonies of the control cells. Each sample was repeated in 3 wells.
Western Blot Analysis
Drug- or vehicle-treated cells were lysed in a lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) with complete protease inhibitor cocktail (Roche). Lysates were centrifuged at 13 000 rpm for 30 min. Supernatants were collected, subjected to electrophoresis on 15% (for light chain [LC]3 antibody), 7% (for PERK and GRP78/Bip antibody), or 10% SDS-polyacrylamide gel, and transferred to Immobilon-P membranes (Millipore). The blot was incubated in 5% nonfat dry milk for 60 min and reacted overnight at 4°C with the following primary antibodies: mouse monoclonal anti-microtubule-associated protein 1 LC3 (MBL International); mouse monoclonal anti-GRP78/Bip (BD Biosciences); rabbit polyclonal anti-ATG5, phospho-PERK, PERK and mouse monoclonal anti-phospho-IRE1α (IRE1α, CHOP from Abcam); rabbit polyclonal anti-phospho–mammalian target of rapamycin (mTOR), phospho-p70S6K, p70S6K, phospho-eIF2α, eIF2α, and caspase 3 (Cell Signaling Technology); and mouse monoclonal anti–α-tubulin (Sigma-Aldrich). The blot was then incubated with horseradish peroxidase–conjugated secondary antibodies (Santa Cruz Biotechnology) at room temperature for 1 h. Immunoreactivity was detected by using the western blot chemiluminescence reagent system (Perkin-Elmer). Films were exposed at different time points to ensure the optimum density, but not saturated. Three replicates were performed for each experiment.
XBP-1 Reverse Transcriptase PCR Splicing Assay
Total RNA was extracted by Trizol reagent (Sigma-Aldrich) and reverse transcribed using MultiScribe reverse transcriptase (RT; ABI) and oligo-deoxythymine primers. Primer sequences used to amplify rat XBP-1 were 5′-TTA CGA GAG AAA ACT CAT GGG C-3′ and 5′-GGG TCC AAC TTG TCC AGA ATG C-3′. Rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers were 5′-ACA GCA ACA GGG TGG TGG AC-3′ and 5′-TTT GAG GGT GCA GCG AAC TT-3′. The thermal cycle consisted of 94°C for 10 min; 28 cycles of 94°C for 1 min, 60°C for 1 min, 72°C for 1 min, and 72°C for 10 min. RT-PCR products were resolved on a 2% agarose gel and visualized using ethidium bromide. The size of unspliced XBP-1 was 289 bp, spliced XBP-1 was 263 bp, and GAPDH was 252 bp.
Cell Transfection and GFP-LC3 Dot Assay
Cells were transiently transfected with green fluorescent protein (GFP)–LC3 vector kindly provided by Dr Noboru Mizushima (Tokyo Medical and Dental University) using Fugene 6 transfection reagent (Roche). After 48 h, cells were treated with 50 μM Mino for 24 h, fixed with 4% paraformaldehyde, and examined under a fluorescence microscope. To quantify autophagic cells after Mino treatment, we counted the number of autophagic cells demonstrating GFP-LC3 dots (≥10 dots/cell) among 200 GFP-positive cells.
Immunofluorescent Staining
In 24-well plates, 2 × 104 cells were seeded on poly-d-lysine–coated 12-mm glass coverslips and allowed to attach for 24 h at 37°C. Culture medium containing Mino or saline was added, and cells were incubated at 37°C. At indicated time points, cells were fixed in 4% paraformaldehyde in PBS for 30 min. After being permeabilized by 0.1% Triton X-100 in 1X PBS for 10 min, cells were blocked in 1% normal goat serum for 1 h. The cells were immunostained for ER stress with rabbit polyclonal anti–protein disulfide isomerase (PDI; Abcam) and mouse monoclonal anti-CHOP (Abcam) and then stained with appropriate secondary antibodies conjugated with Texas Red for 1 h. Nuclei were stained with Hoechst 33342 for 10 min (0.5 μg/mL; Sigma-Aldrich). Fluorescence images were detected by a confocal laser scanning microscope (FV1000, Olympus).
Annexin V–Fluorescein Isothiocyanate /Propidium Iodide Staining
To evaluate whether glioma cells treated with drugs underwent apoptosis or necrosis by flow cytometry, annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining was performed to detect early or late apoptosis. Translocation of phosphatidylserine to the outer membrane that disrupts membrane asymmetry of membrane is a characteristic in early apoptosis in which the cell membrane remains intact. Annexin V has a high binding affinity to phosphatidylserine, thus annexin V was used to detect the early apoptotic cells. Seeded in 10-cm plates, 3 × 105 cells were incubated at least 24 h at 37°C. Glioma cells were treated with medium containing different drugs and incubated at 37°C. At indicated time points, cells were suspended with 0.05% trypsin-EDTA, and 1 × 105 cells were stained with annexin V–FITC (5 μL; BD Biosciences) and PI (1 μg/mL; Sigma-Aldrich). Analyzed in a FACScan flow cytometer (BD Biosciences) were the percentages of the apoptotic or necrotic cells (intact cell, FITC−/PI−; early apoptotic cell, FITC+/PI−; late apoptotic or necrotic cell, FITC+/PI+).
RNA Interference With Short Hairpin RNA
Short hairpin (sh)ATG5 (CCA GAT ATT CTG GAA TGG AAA), sheIF2α (CCT CAG AAT ATG AAA CCA CAA), and shCHOP (GCC AAT GAT GTG ACC CTC AAT) were conjugated in the lentiviral vector of pLKO.1 with a puromycin resistant region (National RNAi Core Facility, Academia Sinica). Glioma cells were plated and infected with lentiviruses expressing shATG5, sheIF2α, shCHOP, and sh-luciferase (Luc) as control for 24 h, followed by puromycin selection (2 μg/mL; Sigma-Aldrich). Western blotting was performed to validate knockdown efficiency, and cells were split for different assays.
GFP-Luciferase Stable Glioma Cell Establishment and Bioluminescence Imaging
U87 and C6 glioma cells were transduced with a lentiviral vector expressing GFP and firefly Luc. GFP-overexpressing infected cells (U87-GL and C6-GL) were sorted out for further passages (FACS-Aria, BD Biosciences). U87-GL and C6-GL cells were used to establish intracranial tumors that were monitored by longitudinal bioluminescence imaging, for which mice were injected with 100 μg luciferin (Caliper), simultaneously anesthetized with isoflurane, and subsequently imaged with a cooled charge-coupled device camera (IVIS-200, Xenogen). Tumor light output was quantitated using the Living Image 2.5 software package (Xenogen).
Intracranial Xenograft Model and Drug Therapy
For orthotopic glioma tumorigenesis, nude mice were anesthetized with chlorohydrate and securely placed on a stereotactic frame. Using aseptic surgical procedures, an incision was made in the scalp and a small burr hole was drilled 2.5 mm lateral to the bregma. U87-GL or C6-GL cells (1 × 105 in 2 μL PBS) were implanted 2.5 mm into the left striatum using a Hamilton syringe. Ten days after implantation, 200 μL of Mino (100 mg/kg in saline) or vehicle control (saline) was i.p. injected once per day for 10 days.
Brain Histology
Two weeks after treatment with Mino or vehicle, mice were anesthetized with chlorohydrate and perfused transcardially with 4% paraformaldehyde in PBS. Whole brains were removed and postfixed overnight in 4% paraformaldehyde in PBS. The brains were coronally sectioned into 5 slices, and these were embedded in paraffin. Ten-micrometer-thick tissue sections were cut and stained with hematoxylin-eosin reagent.
Human Tumor Samples
Six glioma specimens from patients who underwent surgery were collected from National Cheng-Kung University Hospital, Tainan, from September 2010 to October 2011. The clinical evaluation and use of tumor samples in each patient were approved by the institutional review board of National Cheng-Kung University Hospital. All patients did not receive radiation or chemotherapy before surgical resection. Each sample was cut into 2 parts. One part was promptly frozen in liquid nitrogen for western blot and the other immediately fixed with 4% paraformaldehyde for immunohistochemistry. Two normal human brain lysates (Abcam) were used to compare GRP78/Bip expression with tumor samples.
Coimmunoprecipitation
For detecting GRP78/Bip and PERK interaction, Mino- or vehicle-treated C6 cells were lysed with lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, and protease inhibitor cocktail) on ice for 1 h. After centrifuging at 12 000 rpm for 20 min, lysates (1–2 mg) were incubated with anti-GRP78/Bip antibody (2 μg; Abcam) overnight at 4°C on a rotatory wheel. Immunoprecipitates were collected by incubating with 20 μL protein A magnetic beads (Millipore) for 2 h at 4°C. Then the immunoprecipitates were washed 5 times with PBS and resuspended and boiled in 30 μL sample loading buffer. Samples were subjected to electrophoresis, and GRP78/Bip and PERK (Abcam) were detected by immunoblotting.
Statistical Analysis
Data are presented as mean ± SEM. Independent experiments were pooled when the coefficient of variance could be assumed identical. One-way ANOVA was used to analyze the effects of group. Survival data were presented using Kaplan–Meier plots and analyzed using a log-rank test. P < .05 was considered statistically significant.
Results
Minocycline Induces ER Stress Response
We examined whether Mino induced ER stress response and found that Mino induced phosphorylation of PERK and IRE1 in time- and dose-dependent manners, respectively (Fig. 1A and C). Figure 1B shows a transient increase of eIF2α phosphorylation by Mino (F(6,14) = 4.66, n = 3 in each group, P < .01). Newman–Keuls tests revealed that the increase was significant at 30 min, peaked at 2 h, and returned to baseline at 8 h after treatment with Mino. By contrast, the expression of CHOP began at 2 h after treatment with Mino and was sustained for at least 24 h (F(7,16) = 15.08, n = 3 in each group, P < .001). The effects of Mino on eIF2α phosphorylation and CHOP expression were also exhibited in a dose-dependent manner (Fig. 1D). A downstream target of IRE1 activation is the splicing of XBP-1 mRNA. Figure 1E shows that treatment of C6 glioma cells with Mino (50 μM) increased levels of spliced mRNA forms of XBP-1 in a time-dependent manner. PDI is an enzyme in ER in eukaryotes that catalyzes thiol–disulphide exchange, thus facilitating disulphide bond formation and rearrangement reactions.25 Immunostaining showed that PDI accumulated in cells treated with Mino, suggesting that ER stress occurred (Fig. 1F). Furthermore, Hoechst staining of CHOP revealed that Mino induced CHOP expression in the nuclei (Fig. 1G).
Fig. 1.

Minocycline induces ER stress-related proteins in C6 glioma cells. C6 glioma cells were treated with 50 μM Mino or vehicle (control) for different times. Cell lysates were harvested at the indicated time after incubation with Mino and were resolved in SDS-polyacrylamide gel electrophoresis and probed with specific antibodies against p-PERK, PERK p-IRE1α, IRE1α (A), p-eIF2α, eIF2α, and CHOP (B). Relative levels of phosphorylated eIF2α to total eIF2α and CHOP to α-tubulin are indicated in the graphs. Data were quantified using ImageJ software (mean ± SEM, n = 3). (C) Concentration-dependent effect of Mino on the phosphorylation of PERK and IRE1α. (D) Concentration-dependent effect of Mino on the phosphorylation of eIF2α and CHOP expression. (E) Effect of Mino on the level of spliced mRNA forms of XBP-1. uXBP1, unspliced XBP-1; sXBP-1, spliced XBP-1. (F) C6 glioma cells were treated with Mino (50 μM) for 1 h, and the distribution of PDI (red) in the cytoplasm was observed by confocal microscopy. ***P < .001 vs control. (G) Hoechst and CHOP co-staining revealed that Mino induced CHOP expression in the nuclei. ***P < .001 vs control.
GRP78 Is Upregulated and Released by Mino in Glioma Cells
GRP78 is a molecular chaperone that resides in ER and is induced under certain stress conditions, such as glucose starvation, hypoxia, and oxidative stress.26,27 We examined GRP78 expression from tumor specimens of 6 patients and 2 nontumor brain tissues of epilepsy patients. We found that GRP78 was upregulated in tumor specimens compared with specimens from control brains (Fig. 2A). We next compared the levels of GRP78 expression among human glioma cell lines, rat glioma cell lines, and human normal glia. As shown in Fig. 2B, the expression of GRP78 in human normal glia was low. In contrast, higher levels of GRP78 were observed in both human glioma cell lines and rat glioma cell lines. In addition, treatment with Mino increased GRP78 expression (Fig. 2C). As a positive control, we found that temozolomide increased GRP78 expression in a time-dependent manner (Fig. 2C). Taken together, the induction of representative UPR markers GRP78 and CHOP indicates that Mino is an inducer of the ER stress response. GRP78 normally binds with PERK and inhibits its phosphorylation. When unfolded proteins increase in the ER lumen, GRP78 switches its binding to the unfolded proteins, allowing PERK to be phosphorylated.28,29 We determined whether Mino induced GRP78 dissociation from its client protein PERK. Protein lysates were immunoprecipitated with anti-PERK antibody and then analyzed by Western blot for GRP78. Figure 2D shows that a substantial amount of GRP78 was bound with PERK, and this binding became quite weak after treatment with Mino.
Fig. 2.

GRP78 expression is upregulated in glioma tumors, and Mino treatment reduced binding of GRP78 with PERK. (A) Western blotting analysis of GRP78 expression in tumor specimens of 6 patients and 2 nontumor brain tissues obtained from epilepsy patients. (B) Western blotting analysis of GRP78 expression in human glioma cell lines, rat glioma cell lines, and human normal glia. (C) C6 glioma cells were treated with Mino (50 μM) for the times indicated, and GRP78 expression was analyzed with western blotting. Temozolomide (TMZ, 100nM) is an ER-stress inducer as positive control. (D) C6 glioma cell was treated with Mino for 1 h, and GRP78 was immunoprecipitated from the resulting lysates and then immunoblotted with PERK antibody. Abbreviation: WCL, whole cell lysate.
The PERK/eIF2α Signaling Pathway Is Required for Mino-induced Autophagy in C6 Glioma Cells
Specific shRNA directed against eIF2α was utilized in the C6 and U87 glioma cells. Cells were transfected with shLuc or sheIF2α. In the shLuc-transfected cells, Mino led to a marked increase in the levels of CHOP to 379.0 ± 19.3% (n = 4) of control. In the sheIF2α-transfected cells, the baseline levels of phospho-eIF2α and eIF2α were largely reduced, and importantly, Mino-induced upregulation of CHOP was significantly attenuated (188.3 ± 8.7% of control, n = 4, P < .001 vs shLuc-transfected) (Fig. 3A). Similar results were observed in the effect of Mino on the conversion of LC3-I to LC3-II in the C6 and U87 glioma cells. Knockdown of eIF2α inhibited the effect of Mino-induced LC3-II conversion in C6 glioma cells. In shLuc-transfected cells, Mino led to a marked increase in the levels of LC3-II to 175.2 ± 7.1% (n = 4) of control, whereas in the sheIF2α-transfected cells, Mino-induced LC3-II conversion was significantly attenuated (125.6 ± 4.4%, n = 4, P < .001).
Fig. 3.

ER stress precedes autophagy and apoptosis in glioma cells. (A) C6 and U87 glioma cells transfected with sheIF2α or shLuc (control) were treated with Mino (50 μM) for 24 h and then harvested for protein analysis. Relative levels of phosphorylated eIF2α to total eIF2α and CHOP to α-tubulin are indicated in the graphs. Expression of eIF2α and p-eIF2α was markedly reduced in sheIF2α-transfected cells. In addition, Mino-induced upregulation of CHOP and LC3-II conversion were significantly attenuated in the sheIF2α-transfected cells. Data were quantified using ImageJ software (mean ± SEM, n = 4). (B) Knockdown of CHOP inhibited the effect of Mino-induced increased conversion of LC3-I to LC3-II in C6 and U87 glioma cells. Relative levels of CHOP to α-tubulin, LC3-II to α-tubulin, and LC3-II to LC3-I ratio are indicated in the graphs. Data were quantified using ImageJ software (mean ± SEM, n = 4). (C) Nuclear staining of CHOP was not seen after transfection with shCHOP. Bar, 10 μm. (D) Knockdown of CHOP blocked Mino-induced punctuate patterns of GFP-LC3 and the puncta were quantified from random fields (mean ± SEM, 3 independent counts of 100 cells each). Bar, 10 μm. (E) Mino-induced cell death was significantly attenuated after knockdown of CHOP in C6 and U87 glioma cells. (F) Clonogenic activity of C6 glioma cells after treatment with Mino (50 μM) for 24 h. After washing, cells were incubated with fresh medium for 10 days, fixed with methanol and stained with 0.2% crystal violet. Clusters of cells greater than 50 were counted as a colony. Mino-induced reduction of colony formation was attenuated in shCHOP-transfected cells.
We next determined whether knockdown of CHOP affected Mino-induced LC3-II conversion. The efficacy of knockdown CHOP was confirmed by western blotting (Fig. 3B) and immunostaining (Fig. 3C), where the nuclear staining of CHOP was not seen after transfection with shCHOP. As illustrated in Fig. 3B, Mino led to a marked increase in the levels of LC3-II to 176.7 ± 10.6% (n = 4) of control in shLuc-transfected cells, whereas in shCHOP-transfected cells, Mino failed to increase the conversion of LC3-I to LC3-II (95.6 ± 4.3%, n = 4, P < .001 vs shLuc-transfected). A GFP-LC3 plasmid was transfected into C6 glioma cells. Microtubule-associated LC3 protein is used as a biomarker of autophagy.30 This protein normally exhibits diffused cytosolic distribution but is processed and localized to autophagosomes during autophagy. As shown in Fig. 3D, diffused distribution of GFP-LC3 in the basal state was observed in the whole cells. After treatment with Mino, punctuate patterns of GFP-LC3 representing autophagic vacuoles were formed in the cytoplasm. Knockdown of CHOP blocked Mino-induced punctuate patterns of GFP-LC3. Furthermore, the cytotoxic effect of Mino was significantly reduced after knockdown of CHOP (n = 4, P < .001 vs shLuc-transfected) (Fig. 3E). We also determined the long-term effect of Mino on the clonogenic activity of C6 glioma cells. As shown in Fig. 3F, Mino (50 μM) reduced colony formation by 47.7 ± 1.6% in shLuc-transfected cells, whereas in shCHOP-transfected cells, Mino reduced colony formation by 24.1 ± 3.2% (P < .01). These results indicate that downregulation of ER stress signal pathways attenuates Mino-induced autophagy and cell death.
Inhibition of Autophagy Does Not Affect Mino-induced ER Stress and Cell Death
We determined whether knockdown of ATG5 affected Mino-induced CHOP expression and cell death. Efficacy of knockdown ATG5 and subsequent autophagy was confirmed by western blotting (Fig. 4A). Mino led to an increase in the levels of LC3-II to 181.3 ± 6.7% (n = 4) of control in shLuc-transfected cells, whereas in shATG5-transfected cells, Mino-induced conversion of LC3-I to LC3-II was significantly attenuated (121.8 ± 6.3%, n = 4, P < .001 vs shLuc-transfected). Consistent with this observation, knockdown of ATG5 blocked Mino-induced punctuate patterns of GFP-LC3 (Fig. 4B). However, in marked contrast to LC3-II conversion, in shATG5-transfected cells, Mino still increased the expression of CHOP to 328.5 ± 18.3% (n = 4) of control, which was not different from that in shLuc-transfected cells (307.8 ± 17.6%, n = 4, P > .1). In addition, immunostaining showed that Mino did not affect nuclear staining of CHOP in the shATG5-transfected cells (Fig. 4C).
Fig. 4.

Inhibition of autophagy does not affect Mino-induced ER stress and cell death. (A) C6 glioma cells transfected with shATG5 or shLuc (control) were treated with Mino (50 μM) for 24 h and then harvested for protein analysis. Relative levels of ATG5 and CHOP to α-tubulin are indicated in the graphs. Expression of ATG5 and LC3-II were largely reduced in shATG5-transfected cells. By contrast, Mino-induced upregulation of CHOP was not affected in the shATG5-transfected cells. It is important to note that Mino induced cleavage of caspase 3 in shATG5-transfected cells. Data were quantified using ImageJ software (mean ± SEM, n = 4). (B) Knockdown of ATG5 blocked Mino-induced punctuate patterns of GFP-LC3, and the puncta were quantified from random fields (mean ± SEM, 3 independent counts of 100 cells each). Bar, 10 μm. (C) Nuclear staining of CHOP was not affected after transfection with shATG5. Bar, 10 μm. (D) Mino-induced cell death was not affected after knockdown of ATG5 in C6 and U87 glioma cells. (E) Clonogenic activity of C6 glioma cells after treatment with Mino (50 μM) for 24 h. Mino-induced reduction of colony formation was exacerbated in shATG5-transfected cells.
Comparing the cytotoxic effect of Mino between shLuc-transfected and shATG5-transfected cells revealed no difference in both C6 and U87 glioma cells. In C6 glioma cells, the survival rates were 43.2 ± 4.2% (n = 4) in shLuc-transfected cells and 39.2 ± 5.2% (n = 4, P = .108) in shATG5-transfected cells after treatment with Mino (Fig. 4D). Similarly, in U87 glioma cells, the survival rates were 60.4 ± 6.3% (n = 4) in shLuc-transfected cells and 54.1 ± 3.7% (n = 4, P > .1) in shATG5-transfected cells after treatment with Mino. However, Mino (50 μM) reduced C6 glioma cell colony formation by 52.3 ± 1.6% (n = 4) in shLuc-transfected cells, whereas in shATG5-transfected cells, Mino reduced colony formation by 75.9 ± 3.2% (n = 4, P < .01) (Fig. 4E). These results suggest that inhibition of autophagy by downregulation of ATG5 does not affect Mino-induced CHOP expression and cell death. However, knockdown of ATG5 enhances Mino-induced reduction of colony formation, suggesting that autophagy could be cytoprotective, and its inhibition could reveal a potential to die by apoptosis.
Minocycline Induces Apoptosis in C6 Glioma Cells
We have previously shown that the conversion of LC3-I to LC3-II was significantly attenuated in the presence of the autophagy inhibitor 3-MA. However, 3-MA did not attenuate Mino-induced cell death that could be due to 3-MA–mediated exacerbation of Mino-induced apoptosis.23 To examine the effect of Mino on cell viability, glioma cells were treated with Mino (50 μM) or 3-MA (1 mM) + Mino. Mino-induced cell death is not attenuated by 3-MA. Mino reduced cell viability to 41.2 ± 3.8% of control. In the presence of 3-MA, Mino reduced cell viability to 45.7 ± 3.3% of control (P > .1). The caspase inhibitor z-VAD alone did not influence Mino-induced cell death (41.4 ± 2.6% of control, P > .1) but rescued Mino-induced cell death in the presence of 3-MA (79.3 ± 2.7% of control, P < .01) (Fig. 5A). We also determined the effect of Mino on the clonogenic activity of C6 glioma cells. As shown in Fig. 5C, Mino (50 μM) reduced colony formation to 43.8 ± 5.3% of control. In the presence of 3-MA, Mino further reduced colony formation to 16.9 ± 3.1% of control (P < .05). Alone, z-VAD did not influence the effect of Mino (41.7 ± 7.1% of control, P > .1) but rescued Mino-induced reduction in colony formation in the presence of 3-MA (71.9 ± 7.6% of control, P < .05) (Fig. 5C).
Fig. 5.

Effects of 3-MA or BAF A1 on Mino-induced cell death and activation of caspase 3. (A) C6 glioma cells were pretreated with 3-MA (1 mM) and/or z-VAD (20 μM) 1 h before exposure to Mino (50 μM). Mino-induced LC3-II conversion was blocked by 3-MA. However, in the presence of 3-MA, Mino activated caspase 3, which was blocked by z-VAD. Pretreatment with 3-MA did not affect Mino-induced cell death. z-VAD alone did not influence Mino-induced cell death but rescued Mino-induced cell death in the presence of 3-MA. (B) LC3 turnover assay. C6 glioma cells were treated with Mino (50 μM) for 12 h and then 40 nM BAF A1 was added for a further 4 h. LC3 protein was detected by western blot. Mino still induced cell death in the presence of BAF A1, which likely resulted from the activation of caspase 3. (C) Clonogenic activity of C6 glioma cells after treatment with Mino (50 μM) for 24 h. Pretreatment with 3-MA further exacerbated Mino-induced reduction of clonogenic activity, and the exacerbation by 3-MA was blocked by z-VAD (20 μM). z-VAD alone did not influence Mino-induced reduction of clonogenic activity but rescued Mino-induced reduction of clonogenic activity in the presence of 3-MA. DMSO, dimethyl sulfoxide. (D) Fluorescence-activated cell sorting analysis for annexin V/PI. C6 glioma cells were pretreated with 3-MA (1 mM) 1 h before exposure to Mino (50 μM). Annexin V/PI staining was then done to assess apoptosis/necrosis. Annexin V labeling (bottom right quadrants) represent the population undergoing apoptosis. Annexin V and PI double labelings (top right quadrants) represent cells that have already died by apoptosis. The percentage of FITC+/PI− (early apoptosis) cell population was increased in the 3-MA + Mino-treated group, and administration of z-VAD attenuated the increase.
Mino induced LC3-II conversion, which was blocked by 3-MA. However, in the presence of 3-MA, Mino activated caspase 3, which was blocked by z-VAD (Fig. 5A). To confirm the autophagy flux, we performed an LC3 turnover assay. When C6 glioma cells were treated with BAF A1, a vacuolar H+-ATPase inhibitor preventing the fusion between autophagosomes and lysosomes, endogenous LC3-II was increased (Fig. 5B). Addition of Mino to BAF A1 further enhanced LC3-II protein levels compared with BAF A1 alone, indicating that Mino enhanced autophagy flux. In theory, by blocking the fusion between autophagosomes and lysosomes, BAF A1 should completely prevent Mino-induced cell death if Mino-induced cell death was solely due to induction of autophagy. In contrast to the expectation, Mino still induced cell death in the presence of BAF A1, which likely resulted from the activation of caspase 3 (Fig. 5B).
To further demonstrate that Mino induced apoptosis in the presence of 3-MA, C6 glioma cells were treated with Mino (50 μM), 3-MA (1 mM) + Mino, or 3-MA + Mino + z-VAD for 24 h. Cells were collected and stained with PI and annexin V–FITC and were analyzed by flow cytometry. Figure 5D shows that the percentage of FITC+/PI− (early apoptosis) cell population was increased in the 3-MA + Mino-treated group (25.5 ± 2.9%, n = 5) and administration of z-VAD attenuated the increase (14.5 ± 0.8%, n = 8, P < .001). Together, these results suggest that when autophagy is inhibited by 3-MA or BAF A1, Mino still induces cell death through induction of apoptosis.
Mutual Suppression of the Akt/mTOR/p70S6K Pathway by Mino and Rapamycin Combination Induces Synergistic Glioma Cell Cytotoxicity
Knockdown of eIF2α or CHOP largely attenuated Mino-induced punctuate patterns of GFP-LC3 and cell cytotoxicity (Figs 2C and 3D), suggesting mediation by ER stress. We have previously reported that Mino reduced activation of the Akt/mTOR/p70S6K pathway, and both the reduction of this pathway and the expression of CHOP could be linked to autophagy and apoptosis. We therefore tested the effect of 3-MA on Mino-induced reduction of the phosphorylation of mTOR and p70S6K. As shown in Fig. 6, mTOR phosphorylation at position Thr2481 and p70S6K phosphorylation at position Thr389 were significantly decreased by Mino. Importantly, 3-MA counteracted this effect of Mino, suggesting that reduction of the Akt/mTOR/p70S6K pathway did not play a major role in the switch from autophagy to apoptosis. Thus, it is likely that in the normal condition, Mino induced glioma cell death through ER stress with a smaller contribution from reduction of the Akt/mTOR/p70S6K pathway. If this hypothesis is correct, then inhibition of the Akt/mTOR/p70S6K pathway by rapamycin in combination with Mino would produce a synergistic effect on the glioma cells.
Fig. 6.

Effects of Mino and rapamycin alone or in combination on Akt/mTOR/p70S6K cascades, LC3-II conversion, and cell survival. (A) Cells were treated with Mino (50 μM) and rapamycin (10 nM) alone or in combination for 24 h and subjected to western blotting. Cells were pretreated with 3-MA (1 mM) 1 h before adding Mino and/or rapamycin to test the effect of 3-MA on Mino-induced reduction of the phosphorylation of mTOR and p70S6K. Relative levels of phosphorylated mTOR to total mTOR and phosphorylated p70S6K to total p70S6K were indicated in the graphs. Data were quantified using ImageJ software (mean ± SEM, n = 3). (B) Western blot analysis of LC3-I and LC3-II expression in C6 glioma cells treated with Mino (50 μM) and rapamycin (10 nM) alone or in combination for 24 h. Cell were pretreated with 3-MA (1 mM) 1 h before exposure to Mino. Mino and rapamycin combination enhanced LC3-II conversion, which was abolished by 3-MA. (C) 3-MA did not affect rapamycin enhancement of Mino-induced cell death.
Figure 6A shows that suppression of the mTOR pathway by rapamycin increases LC3-II conversion. Rapamycin in combination with Mino produces a synergistic effect on the LC3 conversion that was attenuated by 3-MA. Interestingly, rapamycin in combination with Mino produces a synergistic effect on the reduction of the Akt/mTOR/p70S6K pathway and the glioma cell death that were not affected by 3-MA.
Minocycline Inhibits Intracranial Growth of C6 Glioma Cells
We determined whether Mino exhibited an antitumor effect under in vivo conditions using an intracranial tumor model. To monitor intracranial tumor growth, we infected Luc-expressing C6 glioma cells with the lentiviruses carrying the expression vector containing shATG5. Transduced C6 glioma cells were i.c. injected into athymic mice, and tumor growth was studied using the IVIS-200 imaging system. At day 10 after i.c. injection of tumor cells, Mino (100 mg/kg in saline) or saline was i.p. administered once per day for 10 days, and tumor growth was observed for 30 days after cessation of treatment. Ten days after the cessation of drug injection, Mino significantly inhibited tumor growth and increased the survival of the experimental mice (Fig. 7A). Kaplan–Meier analysis of the survival data demonstrated a statistically significant difference (P < .001) in median survival between Mino- and saline-treated mice. Control mice receiving saline succumbed to disease within 42 days. Interestingly, mice receiving shATG5 did not show improved survival and died within the same time frame. Control mice receiving Mino treatment survived significantly longer, succumbing to disease between 58 and 70 days. Expression of shATG5 did not influence the effects of Mino on tumor growth or survival (Fig. 7B). Histological examination (Fig. 7C) confirmed slower tumor development after Mino treatment in both control and shATG5-transfected mice. In order to investigate whether in vivo drug treatment would activate the ER stress response, we collected tumor tissues from shATG5 or vector-control glioma-bearing mice. The efficiency of shRNA-mediated knockdown of ATG5 was confirmed by western blot analysis. As shown in Fig. 7D, Mino-induced CHOP expression was not affected in tumor tissue from shATG5 glioma-bearing mice, whereas Mino-induced conversion of LC3-I to LC3-II was attenuated. Furthermore, Mino-induced activation of caspase 3 was markedly enhanced in shATG5 glioma-bearing mice. The observation that Mino failed to induce autophagy but caused obvious accumulation of cleaved caspase 3 in shATG5 glioma-bearing mice is consistent with the idea that Mino induces apoptosis when autophagy is inhibited.
Fig. 7.

Mino-induced inhibition of intracranial tumor growth is unaffected after knockdown of ATG5. (A) C6 glioma cells expressing luciferase were infected with lentiviruses carrying the expression vector containing shATG5. Transduced C6 glioma cells were i.c. injected into athymic mice, and tumor growth was studied using the IVIS-200 imaging system. At day 10 after i.c. injection of tumor cells, Mino (100 mg/kg in saline) or saline was i.p. administered once per day for 10 days and tumor growth was observed for 30 days after the cessation of treatment. Mino significantly inhibited tumor growth and increased the survival of the experimental mice (8 mice per group). (B) Expression of shATG5 did not influence the effects of Mino on tumor growth or survival (***P = .0007 for vector/Mino; **P = .0014 for shATG5/Mino by log-rank analysis). (C) Histological examination confirmed slower tumor development after Mino treatment in both shATG5 or vector-control glioma-bearing mice. (D) Tumor tissues were collected from shATG5 or vector-control glioma-bearing mice. The efficiency of shRNA-mediated knockdown of ATG5 was confirmed by western blot analysis. Mino-induced CHOP expression was not affected in tumor tissue from shATG5 glioma-bearing mice, whereas Mino-induced conversion of LC3-I to LC3-II was attenuated. Furthermore, Mino-induced activation of caspase 3 was markedly enhanced in shATG5 glioma-bearing mice.
Luc-expressing C6 glioma cells were infected with lentiviruses carrying the expression vector containing shCHOP. In contrast to what we observed in shATG5-infected cells, infection with shCHOP blocked the effect of Mino on the tumor growth (Fig. 8A and B). Expression of shCHOP also blocked the effects of Mino on survival time (Fig. 8B). Histological examination (Fig. 8C) confirmed slower tumor development after Mino treatment was blocked in shCHOP glioma-bearing mice. The efficiency of shRNA-mediated knockdown of CHOP was confirmed by western blot analysis (Fig. 8D). Mino-induced conversion of LC3-I to LC3-II was attenuated by shCHOP transfection, whereas Mino-induced activation of caspase 3 was slightly increased. These results coupled with our previous report suggest that Mino induces autophagy by eliciting ER stress response and suppressing Akt/mTOR/p70S6K cascades. When autophagy was genetically or pharmacologically blocked by shATG5 transfection or by 3-MA, both effects switched cell death from autophagy to apoptosis.
Fig. 8.

Block of Mino-induced inhibition of intracranial tumor growth in shCHOP glioma-bearing mice. (A) Mino significantly inhibited tumor growth and increased the survival of the experimental mice, and these effects were attenuated in shCHOP glioma-bearing mice (8 mice per group). (B) Expression of shCHOP blocked the effects of Mino on tumor growth or survival (P = .0897 for shCHOP/Mino by log-rank analysis). (C) Histological examination confirmed slower tumor development after Mino treatment in control mice, and this effect was not observed in shCHOP glioma-bearing mice. (D) Tumor tissues were collected from shCHOP or vector-control glioma-bearing mice. The efficiency of shCHOP-mediated knockdown of CHOP was confirmed by western blot analysis. Mino-induced conversion of LC3-I to LC3-II was attenuated by shCHOP transfection, whereas Mino-induced activation of caspase 3 was inhibited.
Discussion
Mino is a neuroprotective agent against many neurodegenerative diseases but exerts antitumor activity toward glioma cells. Mechanisms underlying Mino-induced cytotoxicity are beginning to be defined. The present study demonstrates activation of ER stress responses in malignant glioma cells exposed to Mino. This is supported by the following evidence. First, PDI is an enzyme in ER in eukaryotes that aids wrongly folded proteins to reach a correctly folded state.31,32 PDI accumulates when ER stress occurs.33 We detected the expression of PDI and found that PDI accumulated in cells after treatment with Mino (Fig. 1F). Secondly, GRP78 is an ER chaperone protein, which is upregulated by ER stress. When cells were treated with Mino, GRP78 protein expression was significantly increased. Thirdly, Mino increased PERK and eIF2α phosphorylation and subsequently the phosphorylation of IRE1α and the expression of CHOP. Fourthly, knockdown of eIF2α or CHOP using specific shRNA attenuated Mino-induced LC3-II conversion and glioma cell death, suggesting that induction of ER stress led to autophagy, which was responsible for Mino-induced cell death. Finally, in agreement with cell experiments, in an intracranial tumor model, Mino significantly inhibited tumor growth and increased survival of the experimental mice. Furthermore, Mino's tumor inhibitory effect was attenuated in shCHOP glioma-bearing mice. To the best of our knowledge, this is the first demonstration that Mino-induced cell death involves ER stress response.
GRP78 is primarily involved in the folding and assembly of newly synthesized polypeptides in ER and chaperoning on improperly folded proteins.34,35 In unstressed cells, GRP78 binds to the lumen domain of the 3 documented ER transmembrane receptors (PERK, IRE1α, and ATF6) and keeps them inactive. Accumulation of misfolded proteins in ER results in the dissociation of GRP78 from receptors and leads to ER stress response.36,37 Our observation that GRP78 was expressed at low levels in normal brain but was significantly elevated in malignant glioma specimens and malignant glioma cell lines is consistent with the role of GRP78 in maintaining ER homeostasis.38 Furthermore, coimmunoprecipitation assay revealed that the binding of GRP78 with PERK was decreased by Mino, suggesting that it initiated ER stress response by causing dissociation of GRP78 from PERK.
Mino-induced cell death of glioma cells was associated with the induction and processing of the autophagy marker LC3 (Fig. 5A). Mino-induced LC3-II conversion was blocked by 3-MA. However, when autophagy was inhibited by 3-MA, Mino still induced cell death through the activation of caspase 3. Thus, neither 3-MA nor the caspase inhibitor z-VAD alone influenced the effect of Mino in C6 glioma cells, but in combination they rescued Mino-induced cell death. Interestingly, in a colony formation assay, Mino caused a greater inhibition in the presence of 3-MA, suggesting that autophagy may be a cytoprotective mechanism.
In an intracranial tumor model, we demonstrated that knockdown of ATG5 attenuated Mino-induced LC3-II conversion without influencing the effects of Mino on tumor growth or survival. Tumor tissues collected from shATG5 glioma-bearing mice revealed that Mino-induced activation of caspase 3 was markedly enhanced in shATG5 glioma-bearing mice compared with shLuc glioma-bearing mice. These results confirmed the experiments in glioma cells that under normal conditions, Mino killed glioma cells by inducing autophagy and that programmed cell death switched from autophagy to apoptosis if autophagy was inhibited. A previous study using in situ and in vivo glioma mouse models has shown that Mino interfered with tumor growth and expansion by attenuating microglial membrane type 1–matrix metalloproteinase expression.39
We found that knockdown of ATG5 did not affect cell survival and tumor growth (Figs 4 and 7), consistent with a previous report.40 It is likely that the basal autophagy of C6 glioma cells is low, so that shATG5 alone does not affect tumor growth but significantly inhibits Mino-induced autophagy.
How could cell death be switched from autophagy to apoptosis? Mino has 2 major effects on glioma cells related to autophagy and apoptosis: suppression of the Akt/mTOR/p70S6K pathway and induction of ER stress. Since LC3-II conversion and cell death were largely attenuated after transfection with sheIF2α or shCHOP, it was likely that induction of ER stress was primarily responsible for Mino-induced autophagic cell death. When autophagy was genetically or pharmacologically blocked by shATG5 transfection or by 3-MA, Mino induced activation of caspase 3, suggesting a switch from autophagic to apoptotic cell death. Consistent with these results, rapamycin in combination with Mino produces a synergistic effect on LC3-II conversion, reduction of the Akt/mTOR/p70S6K pathway, and glioma cell death. Rapamycin's effects on LC3-II conversion but not on the reduction of the Akt/mTOR/p70S6K pathway or glioma cell death was attenuated by 3-MA. These results suggest that Mino induces autophagic cell death primarily by eliciting the ER stress response. Inhibition of autophagy switches cell death to apoptosis due to Mino's suppression of the Akt/mTOR/p70S6K cascades.
It was noted that rapamycin not only inhibited mTOR activity and downstream events but also reduced mTOR phosphorylation. These results were consistent with previous studies41,42 suggesting that binding of the rapamycin–FK506 binding protein 12 complex to TOR directly inhibits mTOR kinase activity or induces conformational changes in TOR that displace substrates from the catalytic domain.43
In summary, we have shown that Mino induced glioma autophagic cell death and inhibited glioma growth primarily through induction of ER stress (Fig. 9). The activation of autophagy may offer cell protection. However, depending on the length and/or strength, autophagy leads to glioma cell death. Even when autophagy was inhibited, Mino-induced ER stress and suppression of the Akt/mTOR/p70S6K pathway led glioma cells to apoptosis. The identification of this new route will help us to link ER stress to autophagy and apoptosis. These results coupled to clinical availability and a safe track record make Mino a promising agent for the treatment of malignant gliomas.
Fig. 9.
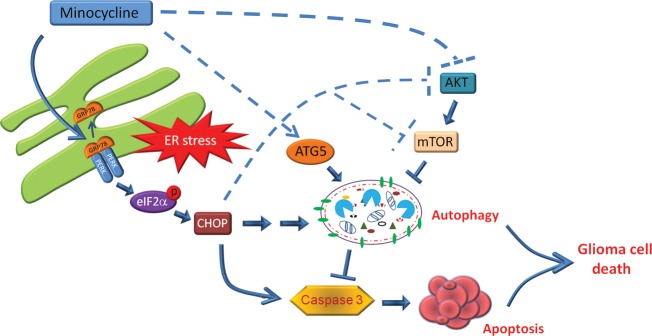
A schematic diagram illustrating the proposed mechanism of Mino-induced cell death. Mino treatment disturbs the interaction of GRP78 with PERK in ER and activates ER stress. Activated PERK phosphorylates eIF2α and sequentially increases the expression of CHOP. Consequently, the PERK/eIF2α/CHOP pathway induces autophagic cell death. When the autophagy is inhibited, the ER stress switches to induce apoptotic cell death through activation of caspase 3.
Funding
This study was supported by grants NSC101-2320-B-006-040-MY3 from the National Science Council and NHRI-EX101-10117NI from the National Health Research Institute and Aim for the Top University Project of the National Cheng-Kung University.
Acknowledgments
The authors thank Dr. Min-Der Lai for critical comments on the manuscript, Dr. Shun-Fen Tzeng for providing C6 glioma cells and critical scientific discussion, and Dr. Michael Hsiao for providing human glioma cell lines.
Conflict of interest statement. None declared.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Davis FG, Freels S, Grutsch J, Barlas S, Brem S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: an analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973–1991. J Neurosurg. 1998;88(1):1–10. doi: 10.3171/jns.1998.88.1.0001. [DOI] [PubMed] [Google Scholar]
- 3.Shrieve DC, Alexander E, 3rd, Black PM, et al. Treatment of patients with primary glioblastoma multiforme with standard postoperative radiotherapy and radiosurgical boost: prognostic factors and long-term outcome. J Neurosurg. 1999;90(1):72–77. doi: 10.3171/jns.1999.90.1.0072. [DOI] [PubMed] [Google Scholar]
- 4.Patil CG, Nuno M, Elramsisy A, et al. High levels of phosphorylated MAP kinase are associated with poor survival among patients with glioblastoma during the temozolomide era. Neuro Oncol. 2013;15(1):104–111. doi: 10.1093/neuonc/nos272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark AJ, Lamborn KR, Butowski NA, et al. Neurosurgical management and prognosis of patients with glioblastoma that progresses during bevacizumab treatment. Neurosurgery. 2012;70(2):361–370. doi: 10.1227/NEU.0b013e3182314f9d. [DOI] [PubMed] [Google Scholar]
- 6.Nieder C, Grosu AL, Astner S, Molls M. Treatment of unresectable glioblastoma multiforme. Anticancer Res. 2005;25(6C):4605–4610. [PubMed] [Google Scholar]
- 7.Stupp R, Pavlidis N, Jelic S. ESMO Minimum Clinical Recommendations for diagnosis, treatment and follow-up of malignant glioma. Ann Oncol. 2005;16(suppl 1):i64–i65. doi: 10.1093/annonc/mdi834. [DOI] [PubMed] [Google Scholar]
- 8.Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol. 2001;205:149–214. doi: 10.1016/s0074-7696(01)05004-5. [DOI] [PubMed] [Google Scholar]
- 9.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8(9–10):1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 10.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18(6):716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65(6):862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281(11):7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 14.Puthalakath H, O'Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 15.Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14(9):1576–1582. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 16.Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr Mol Med. 2008;8(3):157–172. doi: 10.2174/156652408784221324. [DOI] [PubMed] [Google Scholar]
- 17.Galavotti S, Bartesaghi S, Faccenda D, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32(6):699–712. doi: 10.1038/onc.2012.111. [DOI] [PubMed] [Google Scholar]
- 18.Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–1783. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan QW, Cheng C, Hackett C, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3(147):ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seukeran DC, Eady EA, Cunliffe WJ. Benefit-risk assessment of acne therapies. Lancet. 1997;349(9060):1251–1252. doi: 10.1016/s0140-6736(05)62445-2. [DOI] [PubMed] [Google Scholar]
- 21.Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3(12):744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- 22.Saivin S, Houin G. Clinical pharmacokinetics of doxycycline and minocycline. Clin Pharmacokinet. 1988;15(6):355–366. doi: 10.2165/00003088-198815060-00001. [DOI] [PubMed] [Google Scholar]
- 23.Liu WT, Lin CH, Hsiao M, Gean PW. Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy. 2011;7(2):166–175. doi: 10.4161/auto.7.2.14043. [DOI] [PubMed] [Google Scholar]
- 24.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3(7):207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 25.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31(8):455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16(4):343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6(1):45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 28.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 29.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 30.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699(1–2):35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 32.Hatahet F, Ruddock LW. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007;274(20):5223–5234. doi: 10.1111/j.1742-4658.2007.06058.x. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y, Yu H, Qin H, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012;314(2):232–243. doi: 10.1016/j.canlet.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 34.Dudek J, Benedix J, Cappel S, et al. Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci. 2009;66(9):1556–1569. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11(9):2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 37.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 38.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67(20):9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 39.Markovic DS, Vinnakota K, van Rooijen N, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun. 2011;25(4):624–628. doi: 10.1016/j.bbi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Gargini R, Garcia-Escudero V, Izquierdo M. Therapy mediated by mitophagy abrogates tumor progression. Autophagy. 2011;7(5):466–476. doi: 10.4161/auto.7.5.14731. [DOI] [PubMed] [Google Scholar]
- 41.Puli S, Jain A, Lai JC, Bhushan A. Effect of combination treatment of rapamycin and isoflavones on mTOR pathway in human glioblastoma (U87) cells. Neurochem Res. 2010;35(7):986–993. doi: 10.1007/s11064-010-0142-2. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Sun SY, Owonikoko TK, et al. Rapamycin induces Bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther. 2012;11(1):45–56. doi: 10.1158/1535-7163.MCT-11-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]