

Abstract
The Holliday junction is a central intermediate in genetic recombination. It contains four strands of DNA that are paired into four double helical arms flanking a branch point. In naturally-occurring Holliday junctions, the sequence flanking the branch point contains twofold (homologous) symmetry. As a consequence of this symmetry, the junction can undergo a conformational isomerization known as branch migration, which relocates the site of branching. In the absence of proteins and in the presence of Mg2+, the four arms are known to stack in pairs, forming two helical domains whose orientations are antiparallel. Nevertheless, the mechanistic models proposed for branch migration are all predicated on a parallel alignment of helical domains. Here, we have used antiparallel DNA double crossover molecules to demonstrate that branch migration can occur in antiparallel Holliday junctions. We have constructed a DNA double crossover molecule with three crossover points. Two adjacent branch points in this molecule are flanked by symmetric sequences. The symmetric crossover points are held immobile by the third crossover point, which is flanked by asymmetric sequences. Restriction of the helices that connect the immobile junction to the symmetric junctions releases this constraint. The restricted molecule undergoes branch migration, even though it is constrained to an antiparallel conformation.
Keywords: Branch migration, DNA double crossover molecules, Branched junction conformational isomerizations, Hydroxyl radical autofootprinting, DNA topology
The Holliday (1) junction is the most prominent DNA intermediate in genetic recombination. It is known to be involved in site specific recombination (2–4), and it is likely to be involved in homologous recombination (5). The life cycle of the Holliday junction in recombination is illustrated in Figure 1, which depicts the process in junctions with both parallel and antiparallel conformations. The first step, shown at the left of the drawing, is the formation of the junction (II) from two pieces of DNA containing homology (I). The branch point of the Holliday junction is flanked typically by regions of dyad (homologous) sequence symmetry; this symmetry enables the branch point to relocate through an isomerization known as branch migration (e.g., 6). Branch migration (III) is illustrated in the next step to the right of formation. Proceeding to the right, the Holliday junction may undergo another conformational change, known as crossover isomerization. This rearrangement reverses the helical and crossover strands (IV), and it is known to be spontaneous (7). The junctions may then be cleaved (V) by resolvases, such as endonuclease VII (8) or RuvC (9), and religated (VI). It is clear from Figure 1 that the extent of branch migration and the number of crossover isomerizations will influence the products of recombination; the extent of branch migration will determine the site of resolution, and (for a resolvase that cleaves the crossover strand) an even number of crossover isomerizations leads to a patch recombinant, whereas an odd number produces a splice recombinant.
Figure 1. The Formation and Resolution of the Holliday Structure in Genetic Recombination.

The process is illustrated both in the commonly shown parallel conformation, and also in the antiparallel conformation, suggested by physical data; it proceeds from the left to the right. Each of the possible stages is labeled with capital or small Roman numerals. In the first stage, I, two homologous double helices of DNA align with each other. The two strands of each duplex are indicated by the two pairs of lines terminated by half arrows, which indicate the 3′ ends of the strands. Strands are distinguished by their thickness. Each of these two homologous regions carries a flanking marker, A and B in the strands on the left, and α and β on the right. After the first step, the homologous pairs have formed a Holliday intermediate, II, by exchanging strands. Note that the two crossover strands are composite strands with both a thick and a thin portion formed through any of a number of possible mechanisms. The parallel representation of the Holliday junction is shown. The homologous twofold sequence symmetry of this structure permits it to undergo the iterative isomerization process, branch migration; movement in the direction indicated results in structure III. The Holliday intermediate may or may not undergo the crossover isomerization process to produce structure IV, in which the crossover and non-crossover strands are switched. Although indicated as separate, the crossover isomerization process could be a feature of branch migration (8). If crossover isomerization occurs an odd number of times, resolution by cleavage of the crossover strands yields structure V, but structure v results if crossover isomerization occurs an even number of times (including 0) before cleavage. Ligation of v generates a patch recombinant, vi; this is a pair of linear duplex DNA molecules containing heteroduplex DNA because of branch migration, but which have retained the same flanking markers. Ligation of VI yields splice recombinant molecules, that have exchanged flanking markers.
Much of our information about the physical properties of branched junctions (10,11) derives from the study of immobile DNA branched junctions (12); these are synthetic four-stranded complexes in which the sequence symmetry has been destroyed, thereby fixing the site of the branch point. The accepted structural model for the immobile junction in solution is that pairs of adjacent arms stack to form two helical domains (13). This structure leads to a molecule in which two ‘helical’ strands have a structure similar to strands in conventional double helical DNA, and the other two ‘crossover’ strands connect the domains. The helical domains are oriented about 60° from antiparallel to each other (14–16). Recent crystal structures (17–19) confirm the antiparallel relationship, although the angle observed is somewhat smaller.
The relative orientation of the helix axes is somewhat puzzling, because antiparallel helix axes are not in accord with models of recombinational intermediates derived from genetic experiments. In parallel conformations, homologous nucleotides would be in proximity everywhere, which seems likely for systems where DNA-DNA recognition is expected to occur by interactions of homologous bases. By contrast, in antiparallel junctions, homologous nucleotides are only in proximity near the branch point. Sigal and Alberts (20) modeled the Holliday junction as a parallel structure, and Meselson (21) used this structural model to derive a mechanistic model for branch migration, based on rotary diffusion.
As noted above, branch migration is a key step in Holliday-mediated recombination. It is known that this reaction may be spontaneous (e.g., 22), but it may also be enzyme-catalyzed (23 24). To date, Holliday junction branch migration has been studied only in systems containing a single crossover point (22, 25–28). In those molecules, the helical domains could re-orient themselves between migratory steps. Indeed, a re-orientation was a key feature of a model that simulated the kinetics of the process (29).
Here, we have asked whether branch migration can occur in a system constrained to be antiparallel. To examine this issue, we have used antiparallel DNA double crossover (DX) molecules (30), which are analogs of the parallel molecules known to be involved in meiosis (31) and in recombination involving double strand breaks. Antiparallel DX molecules are stable and stiff molecular complexes (30,24), and we have used them previously to demonstrate thermodynamic features of Holliday junctions (33, 34), enzyme susceptibilities (35), and conformational transitions (7). In addition, DX molecules have been used recently to produce a nanomechanical device (36) and two-dimensional arrays with predictable patterns (37,38). Here, we exploit the constrained orientation of the helix axes, to demonstrate conclusively that branch migration is a phenomenon that can occur in an antiparallel context.
MATERIALS AND METHODS
DNA Sequence Design, Synthesis and Purification
Sequences have been designed with the program SEQUIN (39), to minimize sequence symmetry. All DNA molecules used in this study have been synthesized on an Applied Biosystems 380B automatic DNA synthesizer, removed from the support, and deprotected using routine phosphoramidite procedures (40). All strands have been purified by polyacrylamide gel electrophoresis.
Formation of the Experimental Molecule
The strands of the three-junction molecule (50 pmoles) are dissolved to a concentration of 5 μM in 10 μL of a solution containing 40 mM Tris.HCl, pH 8.0, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate (TAEMg) and heated to 90 °C for 5 minutes, and the cooled to 65 °C for 15 minutes, 37 °C for 15 minutes, room temperature (ca. 22 °C) for 20 minutes, and then 4 °C for 20 minutes.
Streptavidin Bead Treatment
Streptavidin beads (50 μL) (Promega), stored at 4 °C in a solution containing phosphate-buffered saline (PBS), 1 mg/ml BSA, and 0.02% sodium azide, are put into a siliconized tube on a magnetic stand and allowed to settle for 5–10 min. The buffer is then removed, and the beads are rinsed three times with 50 μL of fresh PBS buffer. The beads are then rinsed twice with a solution containing NEBuffer 2. The biotinylated DNA, which has been restricted, is added to the beads, mixed well, and allowed to sit at 4 °C for 30 min. The solution is separated from beads, by allowing it to settle on the magnetic stand. Streptavidin particles are supplied as a 1 mg/ml suspension with a binding capacity of 0.75–1.25 nmol/mg.
Hydroxyl Radical Analysis
Individual strands of the complexes are radioactively labeled, and are additionally gel purified from a 15% denaturing polyacrylamide gel. Each of the labeled strands (approximately 10 pmol in TAEMg) is annealed to a fourfold excess of the unlabeled complementary strand, or it is annealed to the complex, as described above, or it is left untreated as a control, or it is treated with sequencing reagents (41) for a sizing ladder. The samples are annealed as described above. Hydroxyl radical cleavage of the double-strand and junction samples for all strands takes place for 2 min (42), with modifications noted by Churchill, et al. (13). The reaction is stopped by addition of thiourea and is then precipitated with ethanol. Where necessary, radioactive labels are freed from internal positions on cyclic strands by restriction, as done previously (35); in addition to the enzymes mentioned below, the Sca I site (Figure 2) is used for this purpose. The sample is dried, dissolved in a formamide/dye mixture, and loaded directly onto a 14% polyacrylamide/8.3M urea sequencing gel. Autoradiograms are analyzed on a BioRad GS-525 Molecular Imager.
Figure 2. The Experiment That Demonstrates Antiparallel Branch Migration.

The top portion of this drawing illustrates the molecule used in the work described here. The upper drawing illustrates the strand topology, including the junction names, A, B and C. The spacing between A and B is a DAO structure, because there are three double helical half-turns between crossovers, and that between B and C is a DAE structure, because there are four. The three strands of the catenane are drawn with lines of different thickness. Below this schematic is the sequence of the molecule (‘B’ represents a biotinylated residue), drawn with unwound helices, indicating strand numbers, restriction sites and strand numbering. The bottom portion of the drawing shows the new strand numbers, new nucleotide numbers, and new junction names for the molecule following the restrictions indicated. The restricted molecule is shown as having migrated to its designated ending position.
Polyacrylamide Gel Electrophoresis
Conventional Denaturing Gels
These gels contain 8.3 M urea and are run at 55 °C. Gels contain 5% acrylamide (19:1, acrylamide:bisacrylamide). The running buffer consists of 100 mM Tris.HCl, pH 8.3, 89 mM Boric acid, 2 mM EDTA (TBE). The sample buffer consists of 10 mM NaOH, 1 mM EDTA, containing 0.1% Xylene Cyanol FF tracking dye. Gels are run on a Hoefer SE-600 gel electrophoresis unit at 35 Volts/cm, dried onto Whatman 3MM paper and imaged by using a BioRad GS-250 Molecular Imager.
Sequencing Denaturing Gels
The samples are phenol extracted, ethanol-precipitated, dried, dissolved in a formamide/dye mixture, and loaded directly onto a 14% polyacrylamide sequencing gel containing 8.3M urea. Gels are run on an IBI Model STS 45 electrophoresis unit at 70 watts (50 v/cm), constant power. Autoradiograms are quantitated using a BioRad GS-250 Molecular Imager.
Nondenaturing Gels
Gels contain 5% acrylamide (19:1, acrylamide:bisacrylamide). DNA is suspended in 10–25 μL of TAEMg and the solution is hybridized as described above. Samples are then brought to a final volume of 20 μL and a concentration of 1 μM, with a solution containing TAEMg, 50% glycerol and 0.02% each of Bromophenol Blue and Xylene Cyanol FF tracking dyes. Gels are run on a Hoefer SE-600 gel electrophoresis unit at 11 Volts/cm at 4 °C, and stained with Stainsall dye.
Enzymatic Reactions
A. Phosphorylation
1 μg of an individual strand of DNA is dissolved in 10 μL of a solution containing 66 mM Tris.HCl, pH 7.6, 1 mM spermidine, 10 mM MgCl2, 15 mM dithiothreitol (DTT), 0.2 mg/mL nuclease free bovine serum albumin (BSA) (BRL), and mixed with 1–2 μL of 1.25 mM γ-32P-ATP (10 μCi/μl) and 3 units of polynucleotide kinase (US Biochemical) for 2 hrs at 37 °C. The reaction is stopped by phenol extraction and ethanol precipitation of DNA.
B. Ligations
Ligations are performed in the same buffer as phosphorylations. 10 units of T4 polynucleotide ligase (U.S. Biochemical) are added, and the ligation proceeds at 16 °C for 18 hrs. The reaction is stopped by phenol extraction and ethanol precipitation of DNA. Phosphorylated and ligated fragments are purified from denaturing 5% polyacrylamide gels.
C. Restriction Endonuclease Digestions
Restriction enzymes are purchased from New England Biolabs, and used in buffers suggested by the supplier. To release to symmetric junctions, the complex (500 nM) is digested at 4°C for 24 h with 100 units Hae III and 100 units of Pvu II in 50 μL of NEBuffer 2. For characterization of the closed structure, the complex (100 nM) is digested at 37°C for 2 h with 10 units of the appropriate restriction enzyme.
RESULTS
Experimental Design
A system in which it possible to demonstrate branch migration in a constrained conformation requires several features: [1] The helical domains must be held tightly in the conformation of interest. [2] One must be able to constrain the molecule to a ‘starting set’ of branch points. [3] While constrained, one must be able to demonstrate that the branch point is restricted to the starting set. [4] It must be possible to release the molecule from branch migratory constraint without releasing it from conformational constraint. [5] If it can migrate, the molecule must migrate to a fixed ‘final set’ of branch points, so that its new position can be characterized. [6] The range of migration (extent of homology) must be low enough that the molecule assembles only to the designated starting set, and no other position.
The molecule shown in Figure 2 satisfies all of these conditions. This drawing depicts a DNA double crossover (DX) molecule containing three crossover points. Regarding condition [1], the DNA double crossover molecule (30) has been shown to be a very stiff molecule (32). We have also shown that antiparallel DX molecules do not undergo crossover isomerization (7) The criteria for condition [2] are satisfied easily by the use of symmetric immobile junctions (43). Symmetric immobile junctions are DX molecules in which one branch point is immobile because it is asymmetric, and the other branch point is flanked by sequence symmetry. If the two branch points are close enough (1.5 turns has proved effective), the torsional coupling between them keeps the symmetric junction immobile. The ability of this design to hold symmetric junctions immobile has been demonstrated by their use to characterize the thermodynamics of symmetric crossover isomers (33) and of branch migratory minima (34).
Hydroxyl radical autofootprinting has been used to characterize a number of unusual DNA motifs, such as branched junctions (13), antijunctions and mesojunctions (44, 45), partially mobile junctions (46), double (30) and triple (47) crossover molecules, Bowtie junctions (48), and symmetric immobile junctions (43). The site of the branch point is a readily apparent from a distinct protection pattern, relative to a double helical control. We have used this form of analysis to establish the sites of the branch points, thereby satisfying condition [3]. In a previous study (7), we have separated the two Holliday junctions of a DX molecule by restriction of the helical domains between them. We have used this method to release the symmetric molecule from the asymmetric molecule, and thus satisfy condition [4]. Figure 2 shows that restriction of this molecule can lead to branch migration.
Once released from a strong constraint, a branch migratory system is free to migrate to any of the sites within the region of sequence symmetry. Such polydispersity is hard to characterize, because it will generate a mixed and weak signal in the hydroxyl radical pattern. To satisfy condition [5], we have included mispairing in the starting set that is eliminated when the system migrates to the final set. Thus, the final set is a sink that absorbs molecules capable of branch migration. Condition [6] entails making sure that there are no possible crossover positions besides those designated by the design. This can occur if the length of the symmetric region is long enough so that crossover points are not designated unambiguously. It is possible to satisfy this condition by limiting the possible range of migration to four steps in a parallel system, but fortunately this problem does not exist for symmetric immobile antiparallel junctions; the same torsional constraints that limit mobility in this system also prevent the wrong site from being the position of the crossover.
Molecular Features of the Experimental Molecule
As noted above, the molecule shown at the top of Figure 2 contains three crossover sites, labeled A, B and C. Antiparallel DX molecules can exhibit two different topologies, DAE, with an even number of double helical half-turns between crossovers, and DAO, with an odd number (30). The distance between junctions A and B is one and a half turns, leading to a DAO topology, and the distance between junctions B and C is two turns, corresponding to a DAE topology. Junctions A and B have symmetric sequences, and C is a fixed junction with the J1 (49) sequence. After removing junction C by Pvu II and Hae III restriction, the DAO molecule on the left is free to migrate through the symmetric sequences. The symmetric sequences are flanked by mispairs (drawn in outline in Figure 2) in the unmigrated state that produce perfect pairs after migration. Hence migration is designed to stop when it moves five positions to reach that point. We have designated the two junctions that result from migration of junctions A and B as junctions A′ and B′, respectively. We have used an estimate of the sequence dependence of branch point free energy (26) to ensure that in this system the gradient of free energy for migration will be downhill. We have added biotin-containing hairpins to the ends of the outside four arms to produce a topologically closed structure that is readily purified from a denaturing gel. In addition to the Pvu II and Hae III sites, we have included other sites that aid in characterization of the molecule.
Formation of the Experimental Molecule
The first issue to be addressed with a new DNA motif is whether it forms in a satisfactory fashion. The molecule at the top of Figure 2 is a triple catenane of DNA, whose individual circles, numbered I, II and III, contain 158, 42 and 122 nucleotides. Figure 3 shows a denaturing autoradiogram of a 5% gel containing the restriction analysis of the purified molecule on a denaturing gel. Digestion with EcoR V cleaves strand 2, digestion with Pvu II cleaves strands 1 and 2, and digestion with BsrB I cleaves strand 3. The leftmost lane contains linear markers and the rightmost lane contains cyclic markers. Undigested triple catenane material is shown to the right of the linear markers. The central circle (strand I) is labeled in the next three lanes to the right. Cleavage with EcoR V releases cyclic strand I, and digestion by Pvu II releases a small amount of linear strand I; the more rapid mobility of the 42-mer circle has been verified elsewhere (data not shown). Removal of strand III by BsrB I produces the catenane of 42 and 158 nucleotide circles; the relative mobilities of the catenane and the 158-mer circle have been established elsewhere (data not shown). In the next three lanes, strand II is labeled: Digestion with EcoR V produces the expected linear 132-mer, and weak digestion with Pvu II generates a nicked 158-mer linear molecule. Again, removal of strand 3 by BsrB I produces the 42 + 158 catenane. In the last three lanes, strand III is labeled. Cleavage both with EcoR V and with Pvu II decatenates strand III from the other strands, generating a 122-mer circle. BsrB I cleavage leads to the expected linear product, a 101 nucleotide linear molecule.
Figure 3. Characterization of the Experimental Molecule.
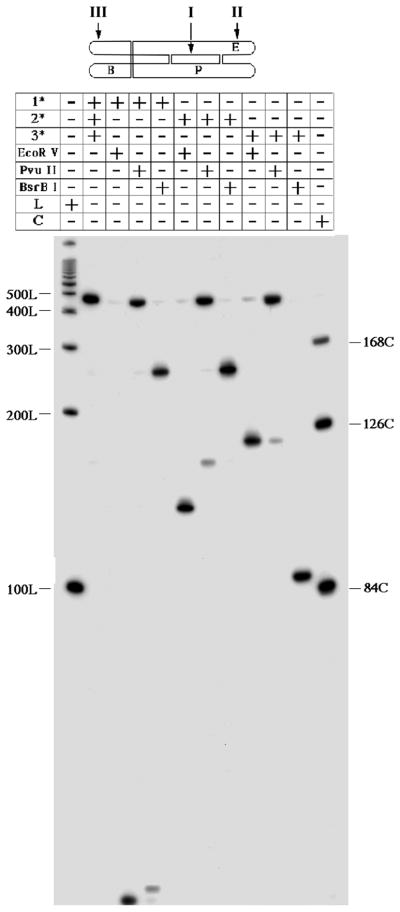
An autoradiogram of a 5% denaturing gel. The molecule is shown at the top, with the strand numbers and first letters of the relevant restriction sites indicated. The chart at the top describes the contents of each lane. The notations 1*, 2* and 3* indicate the labeled strand for a particular experiment. Digestion by a particular restriction enzyme is noted. ‘L’ and ‘C’ indicate the presence in the lane of linear and circular markers, respectively.
Release of the Symmetric Junctions
The immobile experimental molecule requires cleavage to convert it to a molecule free to undergo branch migration. Cleavage at the Hae III and Pvu II sites are all that are necessary to remove the blockage to migration. However, it is an important control to demonstrate that strand exchange does not occur during our analysis (7); to perform this experiment, we also restrict at the Dra I and BsrB I sites (see below). Incomplete restriction could lead to ambiguity in the interpretation of the results. To solve this problem, biotin groups have been incorporated on the hairpin loops of the molecule (Figure 2). After restriction treatment, those biotin groups that remain are tagging incompletely restricted molecules. The system can be purified of these contaminants by treatment with streptavidin beads, as performed previously (52).
To control for molecular dissociation during the experiment, an extra strand is added to the solution containing the experimental molecule. This extra strand is a version of strand iii (bottom of Figure 2), containing extra nucleotides on each end, which leads to a band of lower mobility if it replaces strand iii (data not shown). The extra strand is added to the restriction digest, and after restriction the molecules are allowed to equilibrate for two days at 4 °C (Figure 4) or at room temperature (not shown but identical to Figure 4), before treatment with streptavidin beads.
Figure 4. Restriction and Purification of the Experimental Molecule.

Shown is an autoradiogram of a 5% non-denaturing gel. Two experiments are shown, one performed with a concentration of 500 nM DNA, and the other with a concentration of 100 nM DNA. The restriction is only partial, but streptavidin bead treatment removes incompletely restricted material. The extra strand is a control to ensure that no dissociation has occurred.
In both experiments, it is clear that the final molecule can be prepared cleanly, and that it does not dissociate under the conditions shown. The sequence of the extra strand is: 5′-CTAGACAGTA.TGACTAAAGA.CGTGGCATTG.GTGTATCGCA.GAGCAGGCG G.TACTACACAT.TCCGTAGCCG.CAGGTCACTT.CGGTC-3′.
Figure 4 illustrates both the efficacy of restriction and the absence of dissociation. This is an autoradiogram of a 5% non-denaturing gel, illustrating experiments run at two different concentrations, 500 nM (left) and 100 nM (right). In agreement with the partial digestion seen in Figure 3, particularly for Pvu II, a relatively small quantity of completely restricted material results from treatment by the four restriction enzymes. However, all of the partially restricted material is removed cleanly by the streptavidin bead treatment. The final molecule shown at the bottom of Figure 2 contains 78 nucleotide pairs (39 in each of its helical domains), and Figure 4 shows that it migrates on the gel somewhat more rapidly than a 100 nucleotide pair marker, in agreement with previous observations (data not shown). The lanes indicating the presence of the extra strand are identical to the ones without it. Thus, any phenomenon noted on the experimental molecule is a consequence of its behavior as an intact molecular complex, rather than as a group of continually shifting complexes like the couples in Arthur Schnitzler’s La Ronde (50) or the protons in the Grotthus mechanism of proton mobility in water (e.g., 51).
Hydroxyl Radical Autofootprinting
The autofootprinting experiment with unusual motifs compares the quantitative hydroxyl radical cleavage pattern of each strand paired with its duplex complement to the pattern of the same strand complexed in the unusual motif. The duplex pattern serves to control for sequence effects, and provides a baseline of known structure with which to compare the pattern in the unusual motif. If the cleavage pattern of the strand in the two complexes is the same, the inference is that the strand has adopted a helical structure in the unusual motif. The nucleotides flanking crossover points characteristically evince protection from cleavage, relative to the double helical control; sometimes this protection extends one nucleotide in the 5′ direction (13).
Figure 5 illustrates the pattern of the experimental molecule before digestion by restriction enzymes. Junction A shows the characteristic pattern discussed above. The crossover strands, Strand II (residues) [46, 47] and strand III [85, 86] are protected, relative to the double helical baseline structure. By contrast, there is no significant difference between the DX pattern and the duplex pattern for the helical positions, residues [14, 15], and residues [46, 47] on strand III. In addition to demonstrating which nucleotides are involved in the crossover, the pattern shows that the site of the branch point is well localized at the residues designated. From these data, we cannot exclude the possibility of a small percentage of the molecules having their branch point shifted one position in the direction of symmetry, but certainly this population does not dominate the distribution. The other symmetric immobile junction, B, has a similar pattern, wherein crossover residues [30, 31] of strand III and [33, 34] of strand I are protected, but helical residues [30, 31] and [62, 63] of strand II are unprotected. Junction C is an immobile junction, and it serves as a control for the patterns seen there. Crossover residues [123, 124] of strand II are protected, as are residues [12, 13] of strand I, but helical residues [9, 10], and [83, 84] of strand II show no protection. These data confirm that the molecule constructed has the conformation indicated at the top of Figure 2, and it is poised to branch migrate following restriction.
Figure 5. Hydroxyl Radical Autofootprinting of the Experimental Molecule Before Restriction.

Each of the junctions is shown, A, B, and C, moving from top to bottom. The appropriate residues are shown in a clockwise fashion as one proceeds around the junction; see the top of Figure 2 for strand numbering. Each panel contains two intensity traces, one for the indicated residues when incorporated into the double crossover (DX) and a second for the same strand when paired with its traditional Watson-Crick complement (DS). The two nucleotides that flank the junction are indicated by a pair of arrows, regardless of whether the nucleotides are designated to be on the crossover strands or on the helical strands. The helical strands are in the first and third panels, from left to right, and the crossover strands are in the second and fourth panels. In each case, the pattern at the junction is similar between the DX and DS molecules for the helical strands, but marked protection is visible on the crossover strands.
Figure 6 contains the hydroxyl radical autofootprints of the restricted and purified molecule shown at the bottom of Figure 2. Figure 6a shows the results for the experiment using a 100 nM concentration of the experimental molecule, and Figure 6b illustrates the same results for a concentration of 500 nM. There is a new strand numbering for the restricted molecule. In this new numbering, the original pre-restriction crossover residues for junction A were [25, 26] on strand i and [9, 10] on strand iv. It is evident that protection is seen at residues [30, 31] on strand i and [14, 15] of strand iv, corresponding to junction A′ (Figure 2); nothing remarkable has occurred on the helical strands. Thus, the junction has migrated the entire range of the symmetry, five positions in the 3′ direction on the crossover strands, and it has stopped at the position where the original mispairs now form proper pairs.
Figure 6. Hydroxyl Radical Autofootprinting of the Restricted and Purified Experimental Molecule.

The same conventions apply as in Figure 5. (a) The Experiment Performed at 100 nM Molecular Concentration. It is evident that the pattern at the junction residues follows the same pattern seen in Figure 5. However, the positions have now shifted to the residues that correspond to the designated endpoint of migration, rather than the beginning. (b) The Experiment Performed at 500 nM Molecular Concentration. The same findings are visible here as in (a): The branch point has migrated from the starting point to the designated ending point.
A similar result is apparent for junction B′. In the new numbering scheme, the crossover the crossover at junction B occurred before restriction at [14, 15] on strand ii and at [30, 31] on strand iii. Figure 6a shows that the key protection on strand ii has shifted to [9, 10] and the major protection on strand iii has shifted to [25, 26]; these crossover sites correspond to junction B′ (Figure 2). Thus, this junction has also migrated 5 positions through the symmetric region; however, the crossover strands have migrated in the 5′ direction, rather than the 3′ direction, as in junction A′. No changes are notable on the helical strands at sites [14, 15] or [46, 47] of strand i, that flank the branch point. Figure 6b reinforces the same conclusion derived from Figure 6a: The same protections are seen at the same sites, indicating that this result is independent of strand concentration.
DISCUSSION
Mobility in Antiparallel Junctions and Double Crossover Molecules
The results above demonstrate unambiguously that antiparallel double crossover molecules are capable of undergoing spontaneous branch migration. Two junctions, whose migration is tightly coupled together (43), have been shown to migrate to a designated stopping position favored by a lower free energy. This finding suggests strongly that individual antiparallel junctions are also capable of branch migration. Thus, such problems as may appear to be involved in branch migration in an antiparallel context seem not to exist in a practical sense. It remains an open question as to whether parallel double crossover molecules can branch migrate, but our demonstration of branch migration in an antiparallel double crossover molecule shows that the coordinated movement necessary for this isomerization is possible.
Does this result tell us anything about the mechanism of branch migration? An early simulation of the system (29) suggested that structures far from the Sigal-Alberts structure might undergo a slow isomerization to produce those structures, thereby enabling the Meselson mechanism to occur. The success of that model suggests that there may well be a slow conformational step in branch migration, but it appears unlikely that a Sigal-Alberts-like structure is necessary for branch migration to occur. The double-crossover molecule is known to be quite stiff (32), and at 4 °C, even over a long time period, it seems improbable that the extreme fluctuations needed to produce that structure are likely to occur. Of course, other fluctuations, even unlikely fluctuations such as strand dissociation at the junction, may be required to enable the system to migrate.
The essence of branch migration is movement of the strands about the dyad axis of sequence symmetry. There is only a single axis of symmetry in the individual Holliday junction, parallel or antiparallel; the parallel double crossover molecule also has a single axis of symmetry. Here, we have required the molecule to move about two different axes of symmetry in a fashion that seems to require coordination. Owing to the DAO topology, the helical strands of one junction are also one of the crossover strands of the other. These strands each have moved a few steps that resemble the movement of the treads of a bulldozer or a battle tank. Of course the strand motion is along a helical pathway, rather than confined to a single plane. It is important that the motion of the crossover strands at the two junctions is in opposite directions. The migration about junction A′ is in the 5′ direction, and the migration about junction B′ is in the 3′ direction. Thus, there is no bias regarding migratory direction that reduces the generality of our result.
Features of the Molecular Design
The cyclic nature of strand I proved to be important to ensure that the symmetric immobile junctions were fixed at their designated positions. In early experiments the strand was left nicked, as prepared on the synthesizer, and it was impossible to form the starting molecule: The strands assembled at the ending position. Likewise, initial designs of the system included a pair of mismatched base pairs for both junctions, A′ and B′, rather than just for junction A′. This structure was unstable, and again it formed only at the ending position, rather than at the designated starting position. These two findings indicate that the system we have designed here is quite delicate. Although this delicacy may affect the starting position when the complex is assembled from single strands, we see no reason to expect that it affects the stability of the structural features of the restricted experimental molecule that constrain it to an antiparallel orientation.
Intramolecular Nature of the Experiment
There is a difference between observing an intramolecular event in an isolated molecule, say in a cell, and observing it in a laboratory vessel that contains 1012 molecules or more. One must exclude the influence of intermolecular interactions, both those that involve dissociation of the molecule and those that involve molecular collisions. We have controlled for dissociation by adding the extra strand to the system in the experiments described above. The fact that it is not detectable as part of the complex (Figure 4) suggests that we are dealing with an intramolecular phenomenon. The independence of the results to concentration suggests that intermolecular collisions have no impact on the system.
Nanotechnological Applications
Double crossover molecules are used prominently in nanotechnological applications (36–38, 53). Previously, we have described a system in which it is possible to control the migration of a single branch point through DNA torsion (54). The restriction system described here could be used as a trigger for a onetime event to create a small displacement between the ends to two helices. This movement could be used to transduce the occurrence of the restriction, through a change in a FRET signal (36). Likewise transduction could be done by enabling protein binding through the exposure of a helix. For example, the lower helix in the restricted DAO of Figure 2 could contain a binding site on its left end, and the upper helix could have one on its right end, although more DNA would be needed than shown there.
Acknowledgments
This research has been supported by grants GM-29554 from the National Institute of General Medical Sciences, N00014-98-1-0093 from the Office of Naval Research, NSF-CCR-97-25021 from the National Science Foundation/DARPA and F30602-98-C-0148 from the Information Directorate of the Air Force Research Laboratory located at Rome NY.
ABBREVIATIONS USED
- BSA
Bovine serum albumin
- DAE
Antiparallel double crossover with an even number of double helical half-turns between crossovers
- DAO
Antiparallel double crossover with an odd number of double helical half-turns between crossovers
- DTT
Dithiothreitol
- DX
Double crossover
- FRET
Fluorescence resonance energy transfer
- PBS
Phosphate buffered saline
- TAEMg
A solution containing 40 mM Tris.HCl, pH 8.0, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate
- TBE
A solution containing 100 mM Tris.HCl, pH 8.3, 89 mM Boric acid, 2 mM EDTA
References
- 1.Holliday R. Genet Res. 1964;5:282–304. [Google Scholar]
- 2.Hoess R, Wierzbicki A, Abremski K. Proc Nat Acad Sci USA. 1987;84:6840–6844. doi: 10.1073/pnas.84.19.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitts PA, Nash HA. Nature. 1987;329:346–348. doi: 10.1038/329346a0. [DOI] [PubMed] [Google Scholar]
- 4.Nunes-Duby SE, Matsumoto L, Landy A. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 5.DasGupta C, Wu AM, Kahn R, Cunningham RP, Radding CM. Cell. 1981;25:507–516. doi: 10.1016/0092-8674(81)90069-6. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh P, Panyutin IG. DNA branch migration. In: Eckstein F, Lilley DMJ, editors. Nucleic Acids & Mol Biol. Vol. 9. Springer-Verlag; Berlin: 1995. pp. 42–65. [Google Scholar]
- 7.Li X, Wang H, Seeman NC. Biochemistry. 1997;36:4240–4247. doi: 10.1021/bi9629586. [DOI] [PubMed] [Google Scholar]
- 8.Mueller JE, Kemper B, Cunningham RP, Kallenbach NR, Seeman NC. Proc Nat Acad Sci USA. 1988;85:9441–9445. doi: 10.1073/pnas.85.24.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinagawa H, Iwasaki H. Trends in Biochem Sci. 1996;21:107–111. [PubMed] [Google Scholar]
- 10.Lilley DMJ, Clegg RM. Ann Rev Biophys & Biomol Str. 1993;22:299–328. doi: 10.1146/annurev.bb.22.060193.001503. [DOI] [PubMed] [Google Scholar]
- 11.Seeman NC, Kallenbach NR. Ann Rev Biophys Biomol Struct. 1994;23:53–86. doi: 10.1146/annurev.bb.23.060194.000413. [DOI] [PubMed] [Google Scholar]
- 12.Seeman NC. J Theor Biol. 1982;99:237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 13.Churchill MEA, Tullius TD, Kallenbach NR, Seeman NC. Proc Nat Acad Sci (USA) 1988;85:4653–4656. doi: 10.1073/pnas.85.13.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murchie AIH, Clegg RM, von Kitzing E, Duckett DR, Diekmann S, Lilley DMJ. Nature. 1989;341:763–766. doi: 10.1038/341763a0. [DOI] [PubMed] [Google Scholar]
- 15.Eis P, Millar DP. Biochem. 1993;32:13852–13860. doi: 10.1021/bi00213a014. [DOI] [PubMed] [Google Scholar]
- 16.Mao C, Sun W, Seeman NC. J Am Chem Soc. 1999;121:5437–5443. [Google Scholar]
- 17.Ortiz-Lombardia M, Gonzalez A, Eritja R, Aymami J, Azorin F, Coll M. Nature Str Biol. 1999;6:913–917. doi: 10.1038/13277. [DOI] [PubMed] [Google Scholar]
- 18.Nowakowski J, Shim PJ, Prasad GS, Stout CD, Joyce GF. Nature Str Biol. 1999;6:151–156. doi: 10.1038/5839. [DOI] [PubMed] [Google Scholar]
- 19.Eichman BF, Vargason JM, Mooers BHM, Ho PS. Proc Nat Acad Sci (USA) 2000;97:3971–3976. doi: 10.1073/pnas.97.8.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sigal N, Alberts B. J Mol Biol. 1972;71:789–793. doi: 10.1016/s0022-2836(72)80039-1. [DOI] [PubMed] [Google Scholar]
- 21.Meselson M. J Mol Biol. 1972;71:795–798. doi: 10.1016/s0022-2836(72)80040-8. [DOI] [PubMed] [Google Scholar]
- 22.Thompson BJ, Camien MN, Warner RC. Proc Nat Acad Sci USA. 1976;73:2299–2303. doi: 10.1073/pnas.73.7.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lloyd RG, Sharples GJ. EMBO J. 1993;12:17–22. doi: 10.1002/j.1460-2075.1993.tb05627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsaneva IR, Müller B, West SC. Proc Nat Acad Sci USA. 1993;90:1315–1319. doi: 10.1073/pnas.90.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner RC, Fishel RA, Wheeler FC. Cold Spring Harbor Symp Quant Biol. 1979;43(Pt 2):957–968. doi: 10.1101/sqb.1979.043.01.105. [DOI] [PubMed] [Google Scholar]
- 26.Gellert M, O’Dea MH, Mizuuchi K. Proc Natl Acad Sci (USA) 1983;80:5545–5549. doi: 10.1073/pnas.80.18.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson RD, Symington LS. J Mol Biol. 1993;229:812–820. doi: 10.1006/jmbi.1993.1087. [DOI] [PubMed] [Google Scholar]
- 28.Panyutin IG, Hsieh P. Proc Natl Acad Sci (USA) 1994;91:2021–2025. doi: 10.1073/pnas.91.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson BH, Seeman NC. Biophysical Journal. 1987;51:611–626. doi: 10.1016/S0006-3495(87)83386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu TJ, Seeman NC. Biochem. 1993;32:3211–3220. doi: 10.1021/bi00064a003. [DOI] [PubMed] [Google Scholar]
- 31.Schwacha A, Kleckner N. Cell. 1995;83:783–791. doi: 10.1016/0092-8674(95)90191-4. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Yang X, Qi J, Seeman NC. J Am Chem Soc. 1996;118:6131–6140. [Google Scholar]
- 33.Zhang S, Seeman NC. J Mol Biol. 1994;238:658–668. doi: 10.1006/jmbi.1994.1327. [DOI] [PubMed] [Google Scholar]
- 34.Sun W, Mao C, Liu F, Seeman NC. J Mol Biol. 1998;282:59–70. doi: 10.1006/jmbi.1998.1991. [DOI] [PubMed] [Google Scholar]
- 35.Fu TJ, Kemper B, Seeman NC, Biochemistry. 1994;33:3896–3905. doi: 10.1021/bi00179a015. [DOI] [PubMed] [Google Scholar]
- 36.Mao C, Sun W, Shen Z, Seeman NC. Nature. 1999;397:144–146. doi: 10.1038/16437. [DOI] [PubMed] [Google Scholar]
- 37.Winfree E, Liu F, Wenzler LA, Seeman NC. Nature. 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 38.Liu F, Sha R, Seeman NC. J Am Chem Soc. 1999;121:917–922. [Google Scholar]
- 39.Seeman NC. J Biomol Str & Dyns. 1990;8:573–581. doi: 10.1080/07391102.1990.10507829. [DOI] [PubMed] [Google Scholar]
- 40.Caruthers MH. Science. 1985;230:281–285. doi: 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]
- 41.Maxam AM, Gilbert W. Proc Nat Acad Sci (USA) 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tullius TD, Dombroski B. Science. 1985;230:679–681. doi: 10.1126/science.2996145. [DOI] [PubMed] [Google Scholar]
- 43.Zhang S, Fu TJ, Seeman NC. Biochemistry. 1993;32:8062–8067. doi: 10.1021/bi00083a002. [DOI] [PubMed] [Google Scholar]
- 44.Du SM, Zhang S, Seeman NC. Biochem. 1992;31:10955–10963. doi: 10.1021/bi00160a003. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Seeman NC. Biochem. 1995;34:920–929. doi: 10.1021/bi00003a026. [DOI] [PubMed] [Google Scholar]
- 46.Chen JH, Churchill MEA, Tullius TD, Kallenbach NR, Seeman NC. Biochem. 1988;27:6032–6038. doi: 10.1021/bi00416a031. [DOI] [PubMed] [Google Scholar]
- 47.LaBean TH, Yan H, Kopatsch J, Liu F, Winfree E, Reif JH, Seeman NC. J Am Chem Soc. 2000;122:1848–1860. [Google Scholar]
- 48.Sha R, Liu F, Bruist MF, Seeman NC. Biochemistry. 1999;38:2832–2841. doi: 10.1021/bi9823479. [DOI] [PubMed] [Google Scholar]
- 49.Seeman NC, Kallenbach NR. Biophys J. 1983;44:201–209. doi: 10.1016/S0006-3495(83)84292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schnitzler A. In: Four Plays. Mueller CR, translator. Smith & Kraus; North Stratford, NH: 1999. 224 pages. [Google Scholar]
- 51.Oxtoby DW, Nachtrieb NH. Principles of Modern Chemistry. Saunders Publishing; New York: 1985. pp. 513–514. [Google Scholar]
- 52.Qi J, Li X, Yang X, Seeman NC. J Am Chem Soc. 1996;118:6121–6130. [Google Scholar]
- 53.Fahlman RP, Sen D. J Am Chem Soc. 1999;121:11079–11085. [Google Scholar]
- 54.Yang X, Vologodskii AV, Liu B, Kemper B, Seeman NC. Biopolymers. 1998;45:69–83. doi: 10.1002/(SICI)1097-0282(199801)45:1<69::AID-BIP6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]