

Abstract
Clostridium difficile is currently the leading cause of hospital-acquired infections in the United States. Here, we observed increased interleukin 23 (IL-23) protein levels in human colon biopsy specimens positive for C. difficile toxins, compared with levels in negative controls (P = .008) We also investigated the role of IL-23 during C. difficile infection, using 2 distinct murine models. Mice lacking IL-23 signaling had a significant increase in survival (100% [12 mice]), compared with control mice (16.7%–50% [12 mice]). These data suggest a new potential drug target for human C. difficile treatment and indicate the first link between IL-23 and disease severity during murine infection.
Keywords: Clostridium difficile; colitis; interleukin 23 (IL-23); Th17 cytokines, enteric; Interleukin-10; lamina propria; nosocomial; mucosal immunity
Clostridium difficile is currently the leading cause of nosocomial infections in the United States, with an estimated cost of $4.8 billion to the healthcare industry annually [1]. The incidence of infection continues to rise in healthcare and community-settings, in part because of the emergence of a new hypervirulent strain, BI/NAP1 [2]. Despite therapy, strains with increased virulence have resulted in death within 30 day in up to 15% of patients [3] and disease recurrence in 20%–40% of cases [2].
The aim of this study was to evaluate the host inflammatory response to C. difficile infection. It has been hypothesized that the intensity of the host response and resulting inflammation may be correlated with disease severity. Understanding and targeting host-based mediators of inflammation may provide a target for more effective therapy. Interleukin-23 (IL-23) causes inflammation because of its ability to induce proinflammatory cytokine production by innate and immune cells, such as interleukin 17A (IL-17A), interleukin 17F (IL-17F), and interleukin 21 (IL-21), and its ability to neglect the induction and maintenance of the antiinflammatory cytokine interleukin 10 (IL-10) [4–7]. Previous studies have implicated IL-23 signaling as a precursor to colitis in models of inflammatory bowel disease (IBD) [8, 9]. This prompted us to examine its role in the development of C. difficile–associated pathology. Colon biopsy specimens from patients with C. difficile colitis demonstrated the presence of IL-23–producing infiltrating white blood cells in the lamina propria. We used the murine model of C. difficile colitis to test the role of IL-23 signaling during infection. Mice lacking IL-23 signaling had decreased disease severity, as manifest by clinical scoring and survival proportions. The implications of these studies for understanding the pathogenesis of C. difficile disease and potential host-targeted therapy are discussed.
MATERIALS AND METHODS
Animals
C57Bl/6 wild-type mice were purchased from Jackson Laboratory (Bar Harbor, ME) for use in the IL-23p19 neutralization study. IL-23p19−/− mice on a C57Bl/6 background were a generous gift from Dr. Daniel Cua (Merck, Palo Alto, CA). Mice used in the gene knockout study were bred for use in the University of Virginia vivarium. Mice were 8–16 weeks of age and housed under the same conditions, with free access to food and water. Protocols were approved by the Center of Comparative Medicine at the University of Virginia and were in agreement with National Institutes of Health and Institutional Animal Care and Use Committee standards.
Bacterial Culture
C. difficile strain VPI 10463 (ATCC 43255) was cultured overnight in anaerobic chopped meat medium (Anaerobe Systems, Morgan Hill, CA) at 37°C. It was then subcultured under the same conditions for 5 hours before infection.
Murine Model for C. difficile Challenge
BRIEF REPORTA murine C. difficile infection model has previously been established [10]. Briefly, mice were treated with a cocktail of 4 antibiotics prior to infection. Mice received antibiotic-containing water for 3 days, followed by autoclaved, antibiotic-free water for 2 days, with a subsequent intraperitoneal injection of clindamycin (10 mg/kg) 1 day prior to infection. IL-23p19−/− and C57BL/6 control mice were bred in the University of Virginia vivarium and were challenged by gavage with 105 colony-forming units (CFU) of vegetative C. difficile. C57BL/6 mice used in neutralization studies obtained directly from Jackson Laboratory were challenged by gavage with 104 CFU of C. difficile, because of a heightened susceptibility to challenge. Mice were observed twice daily, and survival and clinical scores were recorded. The clinical scoring system has been previously described [11] and is based on weight loss, coat ruffling, ocular discharge, activity level, posture, and diarrhea severity. Each parameter was scored on a scale from 0 to 3, with higher clinical scores indicative of more severe morbidity. Mice were euthanized if the score indicated intense morbidity (score >14) on any day of the experiment.
Immunohistochemistry (IHC) Staining
Human biopsy specimens were obtained from the University of Virginia Biorepository and Tissue Research Facility. Biopsy samples were identified as positive or negative for C. difficile through a toxin A/B enzyme-linked immunosorbent assay. Only female patients with the closest age match were chosen, to ensure that the population consisted of a single sex. Samples were stained for IL-23 from 6 C. difficile–positive patients and 9 C. difficile–negative patients. Immunochemistry staining was performed using the DAKO Autostainer Universal System (Dako, Denmark) with a primary antibody directed against IL-23p19 (Sigma Prestige) at a 1:200 dilution. Scoring was done by 4 independent blinded scorers and was based on intensity and abundance of IL-23p19 stain on lamina propria cell infiltrates. The staining scale was 0–2, with 1 point given for a high intensity of staining and 1 point given for high abundance of staining. IL-23p19–stained thyroid tissue was used as a positive control.
Statistical Analysis
All data are expressed as mean values, and the analysis was performed as a 2-tailed Student t test, using Excel software (Microsoft). P values of < .05 were statistically significant.
RESULTS
To ascertain the relevance of IL-23 in human C. difficile infection, intestinal biopsy specimens from patients with C. difficile colitis were stained for the presence of IL-23–positive infiltrating immune cells. Biopsy specimens were taken from female patients who tested either positive or negative for C. difficile toxins. Negative tissues were taken from patients who had suspected various inflammatory disease or were in remission from IBD but who had confirmed normal pathologic findings upon biopsy. We observed increased levels of IL-23p19 staining in lamina propria cell infiltrates from patients testing positive for disease, compared with negative controls (Figure 1). Biopsy specimens were scored by 4 independent blinded scorers on a scale from 0 to 2 on the basis of staining intensity and abundance. C. difficile–positive tissues had an average score (±standard error of the mean [SEM]) of 1.33 ± 0.30 (n = 6), and C. difficile–negative control tissues had an average score (±SEM) of 0.7 ± 0.29 (n = 9; P = .008; Figure 1).
Figure 1.
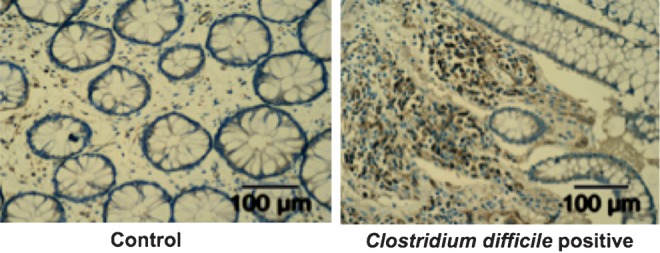
Interleukin 23 (IL-23) is upregulated in the colon of patients with Clostridium difficile colitis. Immunohistochemical staining for IL-23p19 in a representative colon biopsy specimen from a patient without (left) and a patient with (right) C. difficile colitis demonstrating increased lamina propria IL-23–positive infiltrating immune cells in C. difficile infection.
To evaluate the role of IL-23 in murine C. difficile infection, we used both genetic knockout and monoclonal antibody neutralization techniques to disrupt signaling in mice. Survival and clinical scores of IL-23p19 gene knockout mice (IL-23p19−/−) and wild-type C57BL6/J were compared during C. difficile infection. Both groups were treated with an identical regimen of antibiotics and were challenged with 105 CFU of bacteria. Mice were observed twice daily for 2 days after infection and evaluated for clinical score and survival. Clinical scoring was based on parameters previously established [11]. IL-23p19−/− mice were protected from morbidity and mortality associated with infection, compared with controls (Figure 2A): IL-23p19−/− mice survival was 100% throughout the infection time course, whereas wild-type mice survival decreased to 16.7% by day 2 of infection. Similarly, clinical scores for IL-23p19−/− mice were less severe than those of wild-type mice. On day 2, IL-23p19−/− mice had a maximum score (±SEM) of 6.33 ± 1.36 (n = 6), and wild-type mice had a significantly greater maximum score (±SEM) of 14.00 ± 1.55 (n = 6; P = .0087). Wild-type mice reached a severe clinical score (>14) on day 2 and were euthanized for ethical reasons. We also performed a second 8-day study comparing the survival of IL-23p19−/− and wild-type mice to confirm that IL-23p19−/− mice do not have delayed onset of disease. Mice from both groups did not exhibit a decrease in survival after day 3, indicating that 100% IL-23p19−/− mice survived through day 8 infection (Supplementary Figure 1).
Figure 2.

Mice that lack interleukin 23 (IL-23) signaling are protected from mortality and morbidity associated with Clostridium difficile. Mice infected on day 0 with 104–105 colony-forming units of C. difficile after 3 days of antibiotic pretreatment. IL-23p19−/− knockout mice (A) and anti–IL-23p19 monoclonal neutralized mice (B) had decreased disease severity, as manifested by enhanced survival and decreased clinical scores. *P < .05, **P < .01. Abbreviations: IgG, immunoglobulin G; mAb, monoclonal antibody; WT, wild type.
To further investigate the pathogenic role for IL-23 seen in gene knockout mice, we used a second murine model, in which IL-23p19 was neutralized using a monoclonal antibody directed toward the p19 subunit of IL-23. Wild-type mice were given the same regimen of antibiotics described above and treated 18 hours prior to infection with either 100 µg of IgG isotype control antibody or 100 µg of anti-IL-23p19 monoclonal antibody. Mice were given a subsequent dose of respective antibodies 2 days later. Mice obtained from Jackson Laboratory were challenged with 104 CFU. Mice were evaluated twice daily for clinical score and survival and euthanized on day 3. Analogous to our previous observation, mice lacking IL-23 signaling were protected from morbidity and mortality associated with infection (Figure 1B). IL-23p19–neutralized mice had a 100% survival rate (n = 6), whereas isotype control survival was 50% by day 3 (n = 6). Concurrently, isotype controls had a significantly increased clinical score as compared to IL-23p19–neutralized mice on day 2. At this time point, control mice had an average maximum score (±SEM) of 9.00 ± 2.00, while neutralized mice had an average maximum score (±SEM) of 3.17 ± 0.54 (P = .018).
DISCUSSION
Our present study suggests the relevance of IL-23 in human C. difficile–associated colitis and disease. We have demonstrated that IL-23p19 protein levels are elevated in colon biopsy specimens from patients with C. difficile infection, compared with negative controls. C. difficile–positive biopsy specimens have upregulated expression of IL-23p19–positive cell infiltrates to the lamina propria, suggesting that IL-23 may be responsible for mediating the enhanced inflammation responsible for tissue injury and severe disease. IL-23 is a known pathogenic mediator in IBD-associated colitis [8]. Likewise, IL-23R messenger RNA levels are upregulated in lamina propria CD4+ cells from patients with IBD, compared with controls [9]. Active clinical trials indicate that ustekinumab, a monoclonal antibody directed toward IL-23 and IL-12, is effective in increasing remission rates in some patients with Crohn disease [12]. Although these diseases use differing initiators in pathogenesis, our observations for IL-23 in C. difficile infection closely resemble the data described for the pathogenic role of IL-23 in colitis. The effectiveness of anti–IL-23p19 as a therapy for IBD supports the plausibility of our proposal for IL-23 as a potential target in human C. difficile disease treatment.
Our study also supports the observations seen in human tissue by using 2 distinct murine models that demonstrate that deficient IL-23 signaling is protective against mortality and morbidity associated with C. difficile infection. Mice lacking IL-23 signaling through genetic knockout or monoclonal antibody neutralization had enhanced overall survival and clinical health, compared with wild-type controls.
IL-23 was originally implicated as an essential driver in the differentiation of the CD4+ T-cell subset categorized by the production IL-17, T-helper 17 (Th17) cells. It is now understood that IL-23 does not drive differentiation but instead enhances proliferation of Th17 cells and the maintenance of IL-17 production in both an innate and adaptive capacity [6, 7, 13]. IL-17A and IL-17F are key mediators of inflammation by inducing proinflammatory cytokines (tumor necrosis factor, interleukin 1, interleukin 6 (IL-6), and granulocyte macrophage colony-stimulating factor) and chemokines (CXCL1-2, CXCL5, IL-8, CCL2, and CCL7), which are critical in granulopoiesis and the recruitment of neutrophils to the site of infection [14]. In a previous study, IL-6 and transforming growth factor β stimulation were shown to drive IL-17 production and low amounts of IL-10 production by Th17 cells. Interestingly, the addition of IL-23 as a stimulus neglected IL-10 production and skewed the cell subset toward pathogenicity [4]. IL-23 has since been implicated in the inhibition of IL-10 production by not only Th17 CD4+ cells, but also through the prevention of Foxp3+ inducible T-regulatory cell differentiation [5]. IL-23 signaling also has been shown to play a vital role in IL-17A, IL-17F, interleukin 22, and IL-21 production in other T-cell subsets, such as natural killer T cells and γδ T cells, as well as in the non–T-cell innate lymphoid cells [15].
We hypothesize that IL-23 leads to increased susceptibility following C. difficile infection by signaling to both T cells and non-T cells to enhance the recruitment of immune cells to the site of infection and create a robust inflammatory response. Resulting inflammation will be sustained at the site through the ability of IL-23 to maintain downstream cytokines and chemokines capable of continually attracting neutrophils. Additionally, recruitment of immune cells will be dysregulated by the restriction of anti-inflammatory cytokine IL-10 production. These changes will result in enhanced intestinal injury and pseudomembranous colitis and in increased disease severity. We hypothesize that, in the absence of IL-23, immune cells are still capable of responding to infection and clearing the bacterium but that acute colitis and severe disease is avoided through regulatory IL-10 responses.
In conclusion, our study suggests a pathogenic role for IL-23 signaling during C. difficile infection in both human and murine models. We plan to elucidate mechanisms by which IL-23 drives pathogenesis in both models, and we believe that the neutralization of IL-23 may ultimately serve as a potential therapy for C. difficile disease.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Financial Support. This work was supported by the National Institutes of Health (Middle Atlantic Regional Center for Excellence for Biodefense and Emerging Infectious Diseases grant U54 AI057168, grant AI-26649, and Interdisciplinary Training in Immunology grant 5T32 AI07496).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Dubberke ER, Olsen MA. Burden of Clostridium difficile on the Healthcare System. Clin Infect Dis. 2012;55:S88–S92. doi: 10.1093/cid/cis335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ananthakrishnan AN. Clostridium difficile infection: epidemiology, risk factors and management. Nat Rev Gastroenterol Hepatol. 2010;8:17–26. doi: 10.1038/nrgastro.2010.190. [DOI] [PubMed] [Google Scholar]
- 3.Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile–associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–9. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 4.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain Th-17 cell–mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 5.Ahern PP, Schiering C, Buonocore S, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–88. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 7.Langrish CL. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell–mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi T, Okamoto S, Hisamatsu T, et al. IL-23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut. 2008;57:1682–9. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Katchar K, Goldsmith JD, et al. A mouse model of Clostridium difficile–associated disease. Gastroenterology. 2008;135:1984–92. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Warren CA, Van Opstal E, Ballard TE, et al. Amixicile, a novel inhibitor of pyruvate: ferredoxin oxidoreductase, shows efficacy against Clostridium difficile in a mouse infection model. Antimicrob Agents Chemother. 2012;56:4103–11. doi: 10.1128/AAC.00360-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandborn WJ, Gasink C, Gao L, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012;367:1519–28. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 13.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–89. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 14.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Buonocore S, Ahern PP, Uhlig HH, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–5. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.