

Summary
We have previously demonstrated that viral-mediated overexpression of corticotropin-releasing factor (CRF) within the central nucleus of the amygdala (CeA) reproduces many of the behavioral and endocrine consequences of chronic stress. The present experiment sought to determine whether administration of the selective serotonin reuptake inhibitor (SSRI) escitalopram reverses the adverse effects of CeA CRF overexpression. In a 2 × 2 design, adult male rats received bilateral infusions of a control lentivirus or a lentivirus in which a portion of the CRF promoter is used to drive increased expression of CRF peptide. Four weeks later, rats were then implanted with an Alzet minipump to deliver vehicle or 10 mg/kg/day escitalopram for a 4-week period of time. The defensive withdrawal (DW) test of anxiety and the sucrose-preference test (SPT) of anhedonia were performed both before and after pump implantation. Additional post-implant behavioral tests included the elevated plus maze (EPM) and social interaction (SI) test. Following completion of behavioral testing, the dexamethasone/CRF test was performed to assess HPA axis reactivity. Brains were collected and expression of HPA axis-relevant transcripts were measured using in situ hybridization. Amygdalar CRF overexpression increased anxiety-like behavior in the DW test at week eight, which was only partially prevented by escitalopram. In both CRF-overexpressing and control groups, escitalopram decreased hippocampal CRF expression while increasing hypothalamic and hippocampal expression of the glucocorticoid receptor (GR). These gene expression changes were associated with a significant decrease in HPA axis reactivity in rats treated with escitalopram. Interestingly, escitalopram increased the rate of weight gain only in rats overexpressing CRF. Overall these data support our hypothesis that amygdalar CRF is critical in anxiety-like behavior; because the antidepressant was unable to reverse behavioral manifestations of CeA CRF-OE. This may be a potential animal model to study treatment-resistant psychopathologies.
Keywords: Corticotropin-releasing factor, Amygdala, Escitalopram, Anxiety, Hypothalamic-pituitary-adrenal (HPA) axis
1. Introduction
Mood and anxiety disorders are a leading cause of disability and increasing burden on society in terms of direct medical costs and lost productivity (Alonso et al., 2011; Birnbaum et al., 2010; Levinson et al., 2010). In the United States, the lifetime prevalence rate is 28.8% for anxiety disorders and 20.8% for mood disorders (Kessler et al., 2005a). The 12-month prevalence for anxiety and mood disorders is 18.1% and 9.5%, respectively, with comorbid cases having the greatest severity (Kessler et al., 2005b).
Symptoms of depression and anxiety are most commonly treated with selective serotonin reuptake inhibitors (SSRIs) and SSRI use is increasing worldwide (Lockhart and Guthrie, 2011). Although the precise mechanisms of action of the many therapeutic effects of SSRIs are unclear, one postulated pathway to symptom improvement in a significant subgroup of individuals is via normalization of dysregulated central corticotropin-releasing factor (CRF) circuits.
CRF is a 41 amino acid peptide discovered for its role in regulating hypothalamic-pituitary-adrenal (HPA) axis activity (Vale et al., 1981) and it has since been identified as a key mediator of the endocrine, autonomic, and behavioral response to stress. Dysregulation of CRF circuits and the stress response systems they coordinate have been implicated in the pathophysiology of mood and anxiety disorders, most notably major depressive disorder (MDD) and post-traumatic stress disorder (PTSD). For example, many MDD and PTSD patients exhibit state-dependent disruptions in HPA axis reactivity, perhaps in part from altered expression of CRF within the paraventricular nucleus of the hypothalamus (PVN), and exhibit elevated CRF concentrations within cerebrospinal fluid (CSF), attributable to overexpression of CRF from extrahypothalamic sources such as the central amygdala (CeA). The role of CRF in the pathogenesis of mood and anxiety disorders has been extensively reviewed (Claes, 2004; Gold et al., 1995; Holsboer and Ising, 2008; Kasckow et al., 2001; Lloyd and Nemeroff, 2011; Owens and Nemeroff, 1991; Reul and Holsboer, 2002; Risbrough and Stein, 2006; von Bardeleben and Holsboer, 1988).
SSRIs have previously been shown to normalize HPA axis reactivity in patients with depressive and anxiety disorders (Lenze et al., 2011; Manthey et al., 2011; Nikisch et al., 2005b); this normalization is associated with improved clinical outcome (Hinkelmann et al., 2012; Paslakis et al., 2010). Similarly, in depressed patients, normalization of CSF CRF concentration is associated with improvements in the Hamilton Depression Rating Scale (HAM-D) following treatment with the SSRI escitalopram (Nikisch et al., 2005a) or fluoxetine (De Bellis et al., 1993). These changes also roughly follow the time course of symptom resolution, supporting the hypothesis that normalization of CRF neurotransmission plays a causal role in the mechanism of action of antidepressant drugs (Brothers et al., 2012). In animal models, antidepressants, anxiolytics, and mood stabilizers have been shown to reduce the overall responsiveness of the HPA axis, the activity of hypothalamic and extrahypothalamic CRF neurons, and to alter CRF mRNA expression and CRF concentrations as well as type-1 CRF receptor (CRF1) mRNA expression and binding (Gilmor et al., 2003; Grigoriadis et al., 1989; Skelton et al., 2000; Stout et al., 2001; Valentino and Curtis, 1991).
We have previously shown that lentiviral vector-mediated chronic overexpression of CRF from the CeA (CeA CRF-OE) increases anxiety-like behavior and HPA axis reactivity in rats (Flandreau et al., 2012). The following experiment was designed to assess whether chronic administration of the SSRI escitalopram can reverse the behavioral effects of CeA CRF-OE and prevent CeA CRF-OE-induced HPA axis hyperactivity. To better understand the mechanism of any escitalopram and CRF-OE interactions, we also examined central expression patterns of HPA axis-relevant transcripts. Given that amygdalar CRF is more closely associated with the behavioral stress response and hypothalamic CRF more closely associated with the endocrine stress response, we hypothesized that escitalopram would reverse CeA CRF-OE-induced HPA axis hyperreactivity but may have limited efficacy on behavioral measures because it is unable to decrease virally mediated overexpression of CeA CRF. Alternatively, escitalopram attenuation of CeA CRF-OE behavioral effects may occur if SSRI effects are downstream of or at the same level as postsynaptic CRF receptors.
2. Materials and methods
2.1. Animals
Animal protocols were approved by the Emory University Institutional Animal Care and Use Committee (IACUC) and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Resources, 1996). All efforts were made to minimize animal suffering and to reduce the number of animals used. Adult male Wistar rats (approximately PND70) were purchased from Charles River Laboratories (Burlington, MA) and pair housed. In compliance with Emory University biosafety requirements for lentiviral vectors, rats were housed in the animal BSL-2 quarantine facility for 3 days following LV infusion surgery then transferred to the Psychiatry Department facility. In the ABSL-2 quarantine facility, the cage rack was enclosed in one cubicle within the room. In the Psychiatry Department facility, several cage racks are housed together in a larger room. Both facilities were maintained on a 0700/1900 light/dark cycle with food and water available ad libitum.
2.2. Virus production and titering
Production and testing of the LVCRFp3.0CRF construct is described in detail in Flandreau et al. (2012). Virus-production procedures have been described in detail (Chhatwal et al., 2006; Rattiner et al., 2004) and follow from procedures initially outlined by Verma and co-workers (Miyoshi et al., 1998; Naldini et al., 1996; Pfeifer et al., 2001; Zufferey et al., 1998). In brief, VSV-G pseudotyped HIV vectors were generated by Lipofectamine-mediated transient co-transfection of the CRFp1.3Cre or CRFp3.0Cre expression plasmid, VSV-G pseudotyping construct, and the packaging construct pDelta8.91 into HEK293T cells. Serum containing the packaged virus was collected from the cells over a period of 5 days post-transfection, concentrated using ultracentrifugation then resuspended in sterile phosphate-buffered saline (PBS)/1% bovine serum albumin. The resulting titer was assessed in HEK293Tcells, and the observed titer of the virus used here was at least 5 × 108 infectious particles per ml.
2.3. Stereotaxic surgery
Rats were anesthetized with ketamine, xylazine, and ace-promazine maleate (Welberg et al., 2006). 1 μl of control (LVCMVGFP) or CRF-overexpressing (LVCRFp3.0CRF) virus was injected bilaterally into the CeA as described previously (Flandreau et al., 2012). Coordinates from Bregma: A/P −2.1; M/L ± 4.0; D/V −8.0 (Paxinos and Watson, 2005).
2.4. Escitalopram administration and assessment
Escitalopram free base was prepared as described (McConathy et al., 2007). Alzet osmotic minipumps containing escitalopram or vehicle (10% ethanol in polyethylene glycol 300 warmed to 37 °C) were implanted subcutaneously. Dose was calculated to deliver 10 mg (free base)/kg/day over a four week period of time based on estimated average weight. Plasma escitalopram concentrations were measured in a fully validated LC–MS/MS assay which measures both citalopram and its hydroxyl metabolite (there is little or no interconversion of escitalopram to R-citalopram in vivo so the assay is measuring escitalopram). The assay is highly specific and sensitive to 0.2 ng/ml (see Supplemental Methods for additional details). Final plasma escitalopram concentrations ranged between 13 and 45 ng/ml and represent clinically relevant exposure. There were no differences between the LVCMVGFP and LVCRFp3.0CRF groups (Control = 33.31 ± 2.165 ng/ml; LVCRFp3.0CRF = 31.45 ± 1.935 ng/ml p > 0.05).
2.5. Behavior
Because lentiviral vectors reach maximum expression 7–10 days after lentivirus infusion, behavioral testing began at least 14 days after lentivirus-infusion surgery. To minimize effects of multiple testing, tests were ordered from least to most stressful, and cagemates were tested simultaneously or on consecutive days (McIlwain et al., 2001; Paylor et al., 2006). Detailed methods for the elevated plus maze (EPM), defensive withdrawal (DW) and social interaction (SI) tests are included as supplemental material. The timeline of the individual tests is shown in Fig. 1.
Figure 1.
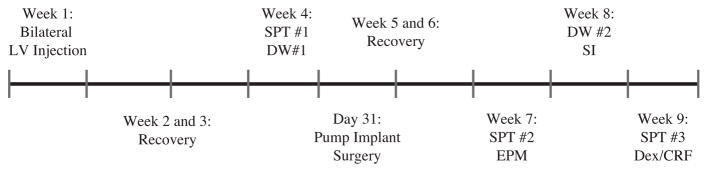
Experimental timeline.
2.6. Dex/CRF test
From each cage, one rat was randomly assigned to the Dex/CRF condition and the other to the Sal/Sal condition. At 1200 h, rats received an intraperitoneal injection of 20 μg/kg dexamethasone at a concentration of 40 μg/ml or an equivalent volume of saline. 90 min later, rats received an IV (tail vein) injection of 0.5 μg/kg rat/human CRF, diluted to 2 μg/ml, or an equivalent volume of saline. Tail vein injections were performed by trained personnel; the rats were not anaesthetized, but were lightly restrained. Rats were killed by decapitation 25 min later and trunk blood collected and brains frozen on dry ice. Trunk blood was collected in cold polypropylene centrifuge tubes for corticosterone and glass EDTA-treated tubes for ACTH (BD Vacutainer®). Quantifications of ACTH and corticosterone were performed by the Emory University Hospital Core and Toxicology Laboratory. ACTH was measured by immunoradiometric assay according to the manufacturer’s instructions (Dia Sorin, Stillwater, MN). Corticosterone was measured using ImmuChemTM Double Antibody Radioimmunoassay (MP Biomedicals, Orangeburg, NY).
Dexamethasone concentrations were measured as described previously (Ritchie et al., 1990). Final dexamethasone concentration did not differ between virus and drug groups.
2.7. Gene-expression analysis
Riboprobe and oligoprobe in situ hybridization were performed and analyzed as previously described (Flandreau et al., 2012). See supplemental methods for additional information.
2.8. Statistical analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad Software). Unpaired Student’s t-test or analysis of variance (ANOVA) was used where appropriate. Specific analyses are described in individual figure captions. Data are shown as mean ± SEM.
3. Results
3.1. Gene-expression changes following CeA CRF-OE and escitalopram administration
As expected, infusion of LVCRFp3.0CRF into the CeA, as a main effect, increased CeA CRF transcript (F(1,76) = 10, p = 0.0022), Fig. 2a and b. Contrary to our previous report, CeA CRF-OE failed to increase PVN CRF or AVP transcript. However, there was a significant drug x virus interaction on PVN AVP expression; escitalopram increased PVN AVP transcript only in CeA CRF-OE rats (F(1,73) = 7.039, p = 0.0098) Fig. 2c and d. In contrast to our previous findings, decreased expression of hippocampal MR in CeA CRF-OE rats failed to reach significance (see supplemental results). Main effects of escitalopram included decreased expression of CRF within the hippocampus (DG: F(1,73) = 7.186, p = 0.0091; CA1/2: F(1,73) = 4.309, p = 0.041; CA3: F(1,73) = 4.272, p = 0.042) Fig. 2e–g. Escitalopram also significantly increased GR transcript in both the PVN and hippocampus of CeA CRF-OE and control rats Fig. 2h–j (PVN: F(1,78) = 8.59, p = 0.0044; Dentate Gyrus: F(1,77) = 11.27, p = 0.0012; CA1/2: F(1,77) = 9.443, p = 0.0029). There were no effects of virus or drug on tyrosine hydroxylase (TH) expression in the locus ceruleus or BDNF expression in the hippocampus or the PVN (Supplemental Figures 2 and 3). CRF1 receptor binding in the anterior pituitary gland was also measured and no differences were observed (Supplemental Figure 4).
Figure 2.

Gene-expression changes following CeA CRF-OE and escitalopram administration. As expected, LVCRFp3.0CRF significantly increased CeA CRF transcript (a, representative ISH image; b, semi quantitative analysis of ISH). Escitalopram increased AVP transcript in the PVN (c, representative ISH image; d, semi quantitative analysis of ISH). Escitalopram decreased CRF transcript in the hippocampal DG (e), CA1/2 (f) and CA3 (g). Escitalopram increased GR transcript in the PVN (h), hippocampal DG (i) and CA1/2 (j). Data are displayed as mean + SEM; p-values reflect results of two-factor ANOVA followed by post hoc Bonferroni tests (n = 16–23/experimental group; *p < 0.05; **p < 0.01; ***p < 0.001).
3.2. HPA axis reactivity following CeA CRF-OE and escitalopram administration
In the Sal/Sal injected group, escitalopram significantly decreased plasma ACTH (F(1,37) = 4.452, p = 0.04) and decreased plasma corticosterone (F(1,37) = 4.985, p = 0.032) concentrations (Fig. 3). In the Dex/CRF injected group, there was an apparent increase in plasma ACTH in vehicle-treated CeA CRF-OE rats, however this increase did not reach significance (F(1,36) = 2.354, p = 0.13). Any differences in corticosterone in the Dex/CRF injected group were masked by marked individual variability. Adrenal glands were also removed and weighed. However, no differences were observed (Supplemental Figure 5).
Figure 3.

HPA axis reactivity following CeA CRF-OE and escitalopram administration. In Sal/Sal treated subjects, escitalopram significantly decreased concentrations of plasma ACTH (a) and corticosterone (b). In Dex/CRF treated rats, no significant differences in ACTH (c) or corticosterone (d) were revealed. Data are displayed as mean + SEM; p-values reflect results of two-factor ANOVA followed by post hoc Bonferroni tests (n = 6–13/experimental group; *p < 0.05; **p < 0.01; ***p < 0.001).
3.3. Behavioral effects of CeA CRF-OE and escitalopram
3.3.1. Defensive withdrawal test
Rats were moved into a holding room at least 1 h prior to lights out and the DW tests were performed 2 h after lights-out under red light conditions.
The first DW test was performed in week four, two weeks following lentivirus infusion. While there were trends toward increased anxiety-like behavior (e.g. Latency to Emerge was 56.72 ± 11.13 (s) for control rats and 105.1 ± 26.09 (s) for CeA CRF-OE rats (p = 0.09), Student’s t-test), no significant differences were observed. However, one subset of rats were excluded from this analysis after the lights failed to turn off, as such the rats in group 1 experienced at least 1 h of extra light. The DW #1 data from this latter group of rats were analyzed separately and revealed an interesting effect of this altered light cycle; rats overexpressing CeA CRF exhibited a significant increase in both horizontal (number of squares crossed per minute exploring = 13.58 ± 3.26 for control rats (n = 6) and 23.25 ± 1.34 (n = 7) for CeA CRF-OE rats; p = 0.013, Student’s t-test) and vertical (total rears per minute exploring = 4.14 ± 0.67 for control rats; 6.601 ± 0.56 for CeA CRF-OE rats; p = 0.016, Student’s t-test) locomotor activity. DW #1 data are shown in Fig. 4.
Figure 4.

Behavioral activity following CeA CRF-OE. In DW#1, week four, CeA CRF-OE rats had a trend toward increased latency to emerge (a) and had a decrease in rearing behavior (b). CeA CRF-OE rats accidentally exposed to an additional hour of light during the dark cycle had increased horizontal (c) and vertical (d) locomotion. Data are displayed as mean + SEM; p-values reflect results of two-tailed T-test analysis (n = 33–34/experimental group (a) and (b); n = 6–7/experimental group (c) and (d); *p < 0.05; **p < 0.01; ***p < 0.001).
The second DW test was performed in week eight, three weeks after Alzet minipump implantation and five weeks after lentivirus infusion. Two-factor ANOVA followed by Bonferroni post hoc analysis revealed a significant anxiogenic-like main effect of virus on total time withdrawing (F(1,63) = 6.4, p = 0.014), percentage of center squares crossed (F(1,63) = 5.9, p = 0.018), and an overall decrease in horizontal (F(1,63) = 4.8, p = 0.032) and vertical (F(1,63) = 6.2, p = 0.016) locomotion. DW #2 data are shown in Fig. 5d–g. A subset of rats (n = 6 control LVCMVGFP (2 escitalopram) and n = 6 LVCRFp3.0CRF (2 escitalopram) were excluded from DW #2 analysis due to an accidental activation of a motion-sensing light during the dark cycle. This momentary light exposure also seemed to alter exploratory activity; however the number of subjects was too small for a two-factor ANOVA.
Figure 5.

Behavioral activity following CeA CRF-OE and escitalopram administration. In the EPM, week seven, there was vehicle-treated CeA CRF-OE subjects exhibited a trend toward decreased time in the open arm (a) and decreased number of open arm entries (b) without altering total arm entries (c) relative to LVCMVGFP subjects. In DW#2, week eight, CeA CRF-OE subjects exhibited a significant increase in total time withdrawing (d) and a decrease in horizontal locomotion (e), which was not reversed by escitalopram. CeA CRF-OE also decreased the number of center squares crossed (f) but this was reversed by escitalopram. CeA CRF-OE tended to decrease vertical locomotion as in the first DW test; however this decrease was only significant when comparing the escitalopram-treated rats in the two virus groups (g). In the SI test, week eight, escitalopram significantly decreased time interacting in the LVCMVGFP and LVCRFp3.0CRF groups (h). In SPT #3, week eight, escitalopram significantly increased consumption of a 0.1% sucrose solution in both virus groups (i). Data are displayed as mean + SEM; p-values reflect results of two-factor ANOVA followed by post hoc Bonferroni tests (n = 15–23/experimental group; *p < 0.05; **p < 0.01; ***p < 0.001; †p < 0.05 vs. LVCMVGFP–escitalopram group).
3.3.2. Elevated plus maze
The EPM was performed during week seven, two weeks after pump implantation. In contrast to the DW test, two-factor ANOVA followed by post hoc Bonferroni tests revealed no differences in total arm entries or total number of rears. The percentage of open arm entries was reduced by CeA CRF-OE in vehicle-treated subjects relative to vehicle-treated rats in the LVCMVGFP group but this reduction did not reach significance (F(1,77) = 3.3, p = 0.074). There was also an apparent decrease in time in the open arm in CeA CRF-OE rats, which was prevented in the escitalopram-treated LVCRFp3.0CRF subjects; however this decrease was also not significant (F(1,77) = 2.5, p = 0.11). One rat was excluded due to falling off the maze. EPM data are shown in Fig. 5a–c.
3.3.3. Social interaction test
The SI test was performed during week eight, three weeks after pump implantation. Two-factor ANOVA followed by post hoc Bonferroni tests revealed a major impact of escitalopram administration; escitalopram, as a main effect, decreased time interacting (including chasing, following, leading, sitting and grooming together) in both CeA CRF-OE rats and control rats (F(1,75) = 16.8, p = 0.0001) (Fig. 5h).
3.3.4. Sucrose preference test
The SPT was performed three times. SPT #1 was performed two weeks following lentivirus infusion. No significant differences in preference for a 1% sucrose solution were observed (LVCMVGFP = 93.72 ± 0.655%; LVCRFp3.0CRF = 92.79 ± 1.047 p > 0.05). SPT #2 was performed four weeks after lentivirus infusion surgery and two weeks following subcutaneous implantation of vehicle or escitalopram-containing Alzet minipumps and, again, no differences in preference for a 1% sucrose solution were observed. SPT #3 was performed during week eight and, to combat the influence of multiple testing, used a 0.1% sucrose solution rather than the 1% solution used in the first two tests. This time, two-factor ANOVA revealed a significant increase in preference for 0.1% sucrose in rats treated with escitalopram regardless of virus treatment (F(1,33) = 4.7, p = 0.037), Fig. 5i.
3.4. Weight gain
Body weight was recorded over the course of the experiment and an unexpected interaction between CeA CRF-OE and escitalopram was observed. The final body weight is significantly greater in escitalopram-treated rats overexpressing CeA CRF relative to vehicle-treated CeA CRF-OE rats and relative to escitalopram-treated rats infused with control virus (F(1,78) = 10.0, p = 0.0023). Interestingly, while the other three virus x drug groups exhibited a slight flattening of weight gain during behavioral testing, the LVCRFp3.0CRF × escitalopram group seemed immune to any slight effects of behavioral testing on body weight gain (Fig. 6). Because food intake was not recorded, it is unknown if this effect is behavioral or metabolic in nature.
Figure 6.

Body weight gain. (a) Rate of weight gain over the course of the experiment. Pump implantation occurred at day 31. Behavioral testing was between days 47 and 54. (b) Final body weight is elevated in CeA CRF-OE rats treated with escitalopram. Data are displayed as mean ± SEM (a) or mean + SEM (b) (n = 18–23/experimental group; **p < 0.01 vs. LVCRFp3.0CRF–vehicle group; ††p < 0.01 vs. LVCMVGFP–escitalopram group).
4. Discussion
A burgeoning literature supports the hypothesis that dysregulation of CRFergic circuitry, including hypothalamic and extra-hypothalamic components, plays a seminal role in the etiology of affective and anxiety disorders in a significant subgroup of individuals. Considerable data has also demonstrated a role for CRF in the mechanism of action of anti-depressant therapies. Current antidepressants, despite exerting their primary pharmacological effects on monoaminergic systems, all reduce the overall responsiveness of the HPA axis and the activity of hypothalamic and extrahypothalamic CRF neurons. This research sought to explicitly understand the differential roles of chronic CRF overexpression within the central amygdala and systemic chronic SSRI treatment.
4.1. Gene-expression changes following CeA CRF-OE and escitalopram administration
Our previous work revealed that CeA CRF-OE produced an increase in PVN CRF and AVP transcript and a decrease in hippocampal MR expression (Flandreau et al., 2012). In the present study, CeA CRF-OE produced a trend toward decreased hippocampal MR expression, which did not attain statistical significance. The previously observed changes in PVN CRF and AVP following CeA CRF-OE were also, unexpectedly, not replicated. The magnitude of CeA OE in this study was comparable to the previous experiment (approximately 140% increase in CRF transcript). However, the baseline CeA CRF expression was not the same; in the previous experiment CeA CRF in LVCMVGFP-treated rats was 180.15 nCi/g but was 235.13 (vehicle) or 240.96 (escitalopram) in the present study. While inconsistent with our previous study (Flandreau et al., 2012), the lack of increased PVN CRF transcripts in the present study is congruent with the lack of significant effect of CeA CRF-OE on HPA axis reactivity. Interestingly, there was an increase in PVN AVP only in CeA CRF-OE subjects treated with escitalopram. A previous study found a similar paradoxical increase in AVP mRNA following restraint and citalopram administration (Hesketh et al., 2005). Also consistent with our data, Hesketh et al. (2005) found no effect of chronic citalopram on PVN CRF transcript. Therefore, in a heightened state of central amygdala CRF expression, SSRI administration may act to alter the expression of both CRF and AVP to tune the pituitary response to stress.
A main effect of escitalopram on several genes of interest in the present study likely contributed to the effect of this SSRI on HPA axis activity. Escitalopram increased GR transcript in the hypothalamus and hippocampus of CeA CRF-OE and control subjects. Previous studies have demonstrated the potential for antidepressants to increase GR. For example, long-term administration of the SSRI fluoxetine selectively increases GR mRNA and protein in primary hippocampal cultures (Lai et al., 2003) and in vivo (Yau et al., 2004). Tricyclic antidepressants also directly increase hippocampal GR after 14-day exposure via upregulation of GR expression (Okugawa et al., 1999). Furthermore, chronic escitalopram administration has been shown to restore the stress-induced decrease in GR expression (Uys et al., 2006). Although other studies have failed to observe SSRI-induced increased GR expression or binding (Durand et al., 1999; Yau et al., 2001), the explanation may reside in differences in the duration (acute vs. chronic effects), dose, and mechanism of administration; single daily injections (e.g. Durand et al., 1999) produce transient spikes and periods of no drug exposure while, depending upon the specific medication, oral administration (Yau et al., 2004) or minipumps, as in the present study, provide a more reliable steady-state concentration similar to that experienced by humans.
Escitalopram also decreased hippocampal CRF transcript in CeA CRF-OE and control subjects. CRF is expressed in hippocampal interneurons (Yan et al., 1998), stress increases hippocampal CRF, which activates receptors on pyramidal neurons (Chen et al., 2004), contributing to dendritic atrophy and cognitive deficits following stress exposure (Chen et al., 2010; Ivy et al., 2010). Escitalopram has previously been shown to restore hippocampal function in a rat model of depression (Bhagya et al., 2011). Whether this was due to changes in CRF or GR expression was not explored.
4.2. HPA axis reactivity following CeA CRF-OE and escitalopram administration
We have previously reported that overexpression of CRF from the CeA (Flandreau et al., 2012; Keen-Rhinehart et al., 2009) but not BNST (Sink et al., 2012) increases HPA axis reactivity. However, other studies have failed to observe HPA axis disruption following CeA CRF-OE (Regev et al., 2011). Escitalopram has previously been shown to normalize HPA axis reactivity in depressed patients (Bschor et al., 2012; Nikisch et al., 2005b) and in animal models of stress (Uys et al., 2006). We hypothesized that CeA CRF-OE would elevate HPA axis reactivity via indirect mechanisms, which would be normalized by escitalopram administration.
In the present study, rats infused with LVCRFp3.0CRF exhibited an increase in ACTH and corticosterone in the Dex/CRF test. While this increase is consistent with what is observed in depressed patients (Heuser et al., 1994), it did not attain statistical significance (Fig. 3). Interestingly, escitalopram decreased plasma concentrations of ACTH and corticosterone in the control and CeA CRF-OE groups. However, this effect only reached significance in the Sal/Sal group.
The ability of escitalopram to alter HPA axis reactivity in both control and CeA CRF-OE rats suggests that the control rats were also experiencing elevated stress relative to control subjects in other studies in which the effects of escitalopram are exclusive to the group exposed to stress (Hesketh et al., 2005; Uys et al., 2006). However, escitalopram has previously been demonstrated to decrease expression of proopiomelanocortin (POMC), a precursor to ACTH production, in rats in the absence of stress exposure (Jensen et al., 1999). This supports a role for escitalopram in HPA axis regulation in “control” subjects.
While the main effect of escitalopram on GR expression almost certainly contributed to the decrease in HPA axis reactivity seen in escitalopram-treated rats, it is unlikely that changes in hippocampal CRF mediated this effect. However, secondary changes in hippocampal structure following a decrease in CRF transcript may play a role but were not specifically examined in the present study. Additional studies are needed to clarify the interactions between CRF and GR on hippocampal structure following chronic antidepressant administration.
4.3. Behavioral effects of CeA CRF-OE and escitalopram
We have previously shown that CeA CRF-OE reproduced some of the behavioral consequences often observed following chronic stress paradigms. For example, CeA CRF-OE decreased open arm exploration in an EPM six weeks following LVCRFp3.0CRF infusion. Additionally, seven weeks after LVCRFp3.0CRF infusion, CeA CRF-OE increased latency to emerge and total withdrawal time in the DW test (Flandreau et al., 2012). Because previous work has shown that the SSRIs citalopram or escitalopram can reverse the behavioral effects of chronic stress (e.g. Bhagya et al., 2011; Bondi et al., 2008; Jayatissa et al., 2006; Overstreet et al., 2004; Reed et al., 2009), we sought to determine whether escitalopram could reverse the behavioral consequences of CeA CRF-OE. We hypothesized that CeA CRF-OE would directly elevate behavioral measures of anxiety and that, without the ability to decrease CeA CRF expression, escitalopram would have limited efficacy unless escitalopram was able to act downstream of postsynaptic CRF neurons.
In the first DW test, during week four, CeA CRF-OE rats exhibited a trend toward increased latency to emerge from the DW box and had decreased rears per minute exploring. Interestingly, after accidental exposure to light during the dark cycle just prior to the DW test, rats overexpressing CeA CRF exhibited a significant increase in horizontal and vertical locomotion. This suggests that rats overexpressing CeA CRF demonstrate enhanced sensitivity to environmental changes. In the second DW test, during week eight, rats overexpressing CeA CRF exhibited significantly increased total time withdrawing, decreased horizontal locomotion, and decreased vertical locomotion. These effects were not prevented by escitalopram. In fact, the effect of CeA CRF-OE on rearing behavior was significant only in the escitalopram-treated groups. LVCRFp3.0CRF also decreased activity in the center of the open area in DW#2 and this effect was no longer significant in the escitalopram group, which is consistent with previous studies showing an improvement in anxiety-like behavior following chronic escitalopram administration (e.g. Burghardt et al., 2004). The lack of effect of escitalopram on the total withdrawal and locomotor activity supports our hypothesis that overexpression of amygdalar CRF is a critical component in modulating anxiety- and depression-like behavior; because the anti-depressant was unable to reverse the CRF overexpression, the behavior was also not reversed. Given these findings, overexpression of CRF in the CeA may be a potential animal model to study treatment resistant depression.
Consistent with our previous experiment (Flandreau et al., 2012), LVCRFp3.0CRF failed to alter anhedonia in the sucrose preference test (SPT) during week four, seven, or eight. Although escitalopram had no effect on consumption of a 1% sucrose solution at week seven, two weeks following pump implantation, escitalopram increased preference for a 0.1% sucrose solution in SPT#3 at the end of week eight. This result is consistent with previous studies showing that escitalopram can decrease anhedonia in rat models of depression or chronic stress (Bah et al., 2011; Strekalova et al., 2006). That CeA CRF-OE did not produce anhedonia suggests that this source of CRF is less critically involved in anhedonia in the SPT than in anxiety in the DW test. Perhaps escitalopram-induced sucrose preference in both groups suggests that the control rats were also experiencing some stress during the behavioral testing. Escitalopram-induced changes in hippocampal CRF and/or GR transcript may also have contributed to the increased preference for 0.1% sucrose in escitalopram-treated rats; previous work has suggested that the effects of escitalopram on sucrose preference are mediated by hippocampal changes (Jayatissa et al., 2006).
A main effect of escitalopram was also observed in the social interaction test, which took place in week eight; rats exposed to escitalopram exhibited a significant decrease in time interacting regardless of CRF-OE status. Although this effect is counterintuitive, previous studies have also demonstrated a decrease in social activity following escitalopram administration in rats in the absence of stress (Dekeyne et al., 2000; Tonissaar et al., 2008). However, other studies have shown an increase in social interaction in a genetically sensitive rat line, the Flinders rats (Overstreet et al., 2004). The interpretation of this result is certainly complex given that (es)citalopram is associated with improved social functioning in humans treated for depression (Denninger et al., 2011) or dysthymic disorder (Hellerstein et al., 2010).
The clinical literature supports the hypothesis that a significant subgroup, but not all, subjects with mood and anxiety disorders may have a dysregulated CRF system and identification of these individuals is paramount to gaining real clinical utility of CRF1 antagonists as novel medications. Further study is warranted to examine whether there is a correlation between weight gain during escitalopram exposure and measures suggestive of dysregulated CRF systems (Fig. 6) and whether some noninvasive measure may be utilized to select appropriate individuals for potential CRF1 antagonist treatment.
5. Summary
Together these data support a complex mechanism through which escitalopram alters HPA axis reactivity and behavior, and that chronic CRF overexposure may render the organism resistant to SSRI response in some behaviors but not others. The influence of escitalopram on behavioral anxiety caused by CeA CRF-OE was less robust than in chronic stress studies, suggesting that a reduction in CeA CRF (which was prevented here) is necessary for normalization of these behavioral measures. In contrast, we also observed main effects of escitalopram on preference for a 0.1% sucrose solution, a measure that was not affected by CeA CRF-OE. This result could suggest that escitalopram increases hedonic reactions to very low concentrations of sucrose but could also reflect an increase in anhedonia in all subjects having been exposed to a potentially stressful battery of tests prior to SPT#3. Main effects of escitalopram on hippocampal CRF and, especially, hippocampal and hypothalamic GR transcript demonstrate the ability of this drug to modify central regulation of the HPA axis, even when the CeA is overexpressing CRF. Further understanding of the separate and interacting roles of the HPA and serotonergic systems will advance our appreciation of the mechanisms underlying the pathophysiology of depression and other stress-related disorders as well as mechanisms of treatment and prevention.
Supplementary Material
Acknowledgments
Role of the funding sources
This research was supported by DK 26741, F31MH079667A, Howard Hughes Medical Institute (HHMI), MH-42088, T32 DK007044-30, T32 ES012870, and T32 MH18399-25.
These funding sources had no further role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
We thank Christopher Horoszko for slicing brains and in situ hybridization, Catie Cappello for tail-vein injections and brain removal, Nicola Hanson for adrenal gland removal, Susan Plott for pituitary gland CRF binding assay, and Swati Rogers for escitalopram extraction.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.psyneuen.2012.11.020.
Footnotes
Conflict of interest
Dr. Flandreau has received no financial support or compensation from any individual or corporate entity for research or professional service and has no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Dr. Ressler has received no financial support or compensation from any individual or corporate entity for research or professional service and has no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Dr. Bourke has received no financial support or compensation from any individual or corporate entity for research or professional service and has no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Dr. Owens has research grants from the NIH, Lundbeck A/S, Cyberonics, Ortho-McNeil Janssen, AstraZeneca, Dainippon Sumitomo Pharma, SK Life Sciences, Sunovion Pharmaceuticals. He is a consultant with H. Lundbeck A/S, R.J. Reynolds and has a patent: Method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters (US 7,148,027B2).
Dr. Nemeroff has research grants from the National Institutes of Health (NIH), and Agency for Healthcare Research and Quality (AHRQ). Is a consultant with Xhale, Takeda, SK Pharma, Shire, Roche, and Lilly. He holds stock or other interests in CeNeRx BioPharma, PharmaNeuroBoost, Revaax Pharma, Xhale, NovaDel Pharma and serves on the scientific advisory boards for American Foundation for Suicide Prevention (AFSP), CeNeRx BioPharma, National, Alliance for Research on Schizophrenia and Depression (NARSAD), Xhale, PharmaNeuroBoost, Anxiety Disorders Association of America (ADAA), Skyland Trail, and AstraZeneca Pharmaceuticals (2009). He has served as board of directors for AFSP, Mt. Cook Pharma (2010), NovaDel (2011), Skyland Trail, Gratitude America, ADAA. The following provide income sources of $10,000 or more: AstraZeneca Pharmaceuticals, PharmaNeuroBoost, CeNeRx BioPharma, NovaDel Pharma, Reevax Pharma, American Psychiatric Publishing, Xhale. Dr. Nemer-off also holds patents: Method and devices for transdermal delivery of lithium (US 6,375,990B1) and Method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters by ex vivo assay (US 7,148,027B2).
Contributor Information
Elizabeth I. Flandreau, Email: eimartin@ucsd.edu.
Chase H. Bourke, Email: cbourke@emory.edu.
Kerry J. Ressler, Email: kressle@emory.edu.
Charles B. Nemeroff, Email: cnemeroff@med.miami.edu.
Michael J. Owens, Email: mowens@emory.edu.
References
- Alonso J, Petukhova M, Vilagut G, Chatterji S, Heeringa S, Ustun TB, et al. Days out of role due to common physical and mental conditions: results from the WHO World Mental Health surveys. Mol Psychiatry. 2011;16 (12):1234–1246. doi: 10.1038/mp.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bah TM, Benderdour M, Kaloustian S, Karam R, Rousseau G, Godbout R. Escitalopram reduces circulating pro-inflammatory cytokines and improves depressive behavior without affecting sleep in a rat model of post-cardiac infarct depression. Behav Brain Res. 2011;225 (1):243–251. doi: 10.1016/j.bbr.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Bhagya V, Srikumar BN, Raju TR, Rao BS. Chronic escitalopram treatment restores spatial learning, monoamine levels, and hippocampal long-term potentiation in an animal model of depression. Psychopharmacology (Berl) 2011;214 (2):477–494. doi: 10.1007/s00213-010-2054-x. [DOI] [PubMed] [Google Scholar]
- Birnbaum HG, Kessler RC, Kelley D, Ben-Hamadi R, Joish VN, Greenberg PE. Employer burden of mild, moderate, and severe major depressive disorder: mental health services utilization and costs, and work performance. Depress Anxiety. 2010;27 (1):78–89. doi: 10.1002/da.20580. [DOI] [PubMed] [Google Scholar]
- Bondi CO, Rodriguez G, Gould GG, Frazer A, Morilak DA. Chronic unpredictable stress induces a cognitive deficit and anxiety-like behavior in rats that is prevented by chronic antidepressant drug treatment. Neuropsychopharmacology. 2008;33 (2):320–331. doi: 10.1038/sj.npp.1301410. [DOI] [PubMed] [Google Scholar]
- Brothers S, Wahlestedt C, Nemeroff C. Modulation of HPA axis function for treatment of mood disorders. In: Rankovic Z, Bingham M, Hargreaves R, Nestler E, editors. Drug Discovery for Psychiatric Disorders. 2012. [Google Scholar]
- Bschor T, Ising M, Erbe S, Winkelmann P, Ritter D, Uhr M, et al. Impact of citalopram on the HPA system. A study of the combined DEX/CRH test in 30 unipolar depressed patients. J Psychiatr Res. 2012;46 (1):111–117. doi: 10.1016/j.jpsychires.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Burghardt NS, Sullivan GM, McEwen BS, Gorman JM, LeDoux JE. The selective serotonin reuptake inhibitor citalopram increases fear after acute treatment but reduces fear with chronic treatment: a comparison with tianeptine. Biol Psychiatry. 2004;55 (12):1171–1178. doi: 10.1016/j.biopsych.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004;126 (3):533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Rex CS, Rice CJ, Dube CM, Gall CM, Lynch G, et al. Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci USA. 2010;107 (29):13123–13128. doi: 10.1073/pnas.1003825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9 (7):870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes SJ. Corticotropin-releasing hormone (CRH) in psychiatry: from stress to psychopathology. Ann Med. 2004;36 (1):50–61. doi: 10.1080/07853890310017044. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Gold PW, Geracioti TD, Jr, Listwak SJ, Kling MA. Association of fluoxetine treatment with reductions in CSF concentrations of corticotropin-releasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry. 1993;150 (4):656–657. doi: 10.1176/ajp.150.4.656. [DOI] [PubMed] [Google Scholar]
- Dekeyne A, Denorme B, Monneyron S, Millan MJ. Citalopram reduces social interaction in rats by activation of serotonin (5-HT)(2C) receptors. Neuropharmacology. 2000;39 (6):1114–1117. doi: 10.1016/s0028-3908(99)00268-3. [DOI] [PubMed] [Google Scholar]
- Denninger JW, van Nieuwenhuizen AO, Wisniewski SR, Luther JF, Trivedi MH, Rush AJ, et al. Changes in depressive symptoms and social functioning in the sequenced treatment alternatives to relieve depression study. J Nerv Ment Dis. 2011;199 (10):807–810. doi: 10.1097/NMD.0b013e31822fcbe2. [DOI] [PubMed] [Google Scholar]
- Durand M, Berton O, Aguerre S, Edno L, Combourieu I, Mormede P, et al. Effects of repeated fluoxetine on anxiety-related behaviours, central serotonergic systems, and the corticotropic axis in SHR and WKY rats. Neuropharmacology. 1999;38(6):893–907. doi: 10.1016/s0028-3908(99)00009-x. [DOI] [PubMed] [Google Scholar]
- Flandreau EI, Ressler KJ, Owens MJ, Nemeroff CB. Chronic overexpression of corticotropin-releasing factor from the central amygdala produces HPA axis hyperactivity and behavioral anxiety associated with gene-expression changes in the hippocampus and paraventricular nucleus of the hypothalamus. Psychoneuroendocrinology. 2012;37(1):27–38. doi: 10.1016/j.psyneuen.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmor ML, Skelton KH, Nemeroff CB, Owens MJ. The effects of chronic treatment with the mood stabilizers valproic acid and lithium on corticotropin-releasing factor neuronal systems. J Pharmacol Exp Ther. 2003;305 (2):434–439. doi: 10.1124/jpet.102.045419. [DOI] [PubMed] [Google Scholar]
- Gold PW, Licinio J, Wong ML, Chrousos GP. Corticotropin releasing hormone in the pathophysiology of melancholic and atypical depression and in the mechanism of action of antidepressant drugs. Ann N Y Acad Sci. 1995;771:716–729. doi: 10.1111/j.1749-6632.1995.tb44723.x. [DOI] [PubMed] [Google Scholar]
- Grigoriadis DE, Pearsall D, De Souza EB. Effects of chronic antidepressant and benzodiazepine treatment on corticotropin-releasing-factor receptors in rat brain and pituitary. Neuropsychopharmacology. 1989;2 (1):53–60. doi: 10.1016/0893-133x(89)90007-9. [DOI] [PubMed] [Google Scholar]
- Hellerstein DJ, Batchelder ST, Hyler S, Arnaout B, Toba C, Benga I, et al. Escitalopram versus placebo in the treatment of dysthymic disorder. Int Clin Psychopharmacol. 2010;25 (3):143–148. doi: 10.1097/YIC.0b013e328333c35e. [DOI] [PubMed] [Google Scholar]
- Hesketh S, Jessop DS, Hogg S, Harbuz MS. Differential actions of acute and chronic citalopram on the rodent hypothalamic-pituitary-adrenal axis response to acute restraint stress. J Endocrinol. 2005;185 (3):373–382. doi: 10.1677/joe.1.06074. [DOI] [PubMed] [Google Scholar]
- Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res. 1994;28 (4):341–356. doi: 10.1016/0022-3956(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Hinkelmann K, Moritz S, Botzenhardt J, Muhtz C, Wiedemann K, Kellner M, et al. Changes in cortisol secretion during antidepressive treatment and cognitive improvement in patients with major depression: a longitudinal study. Psychoneuroendocrinology. 2012;37 (5):685–692. doi: 10.1016/j.psyneuen.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Ising M. Central CRH system in depression and anxiety—evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583 (2–3):350–357. doi: 10.1016/j.ejphar.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dube C, Maras PM, Grigoriadis DE, et al. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30 (39):13005–13015. doi: 10.1523/JNEUROSCI.1784-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayatissa MN, Bisgaard C, Tingstrom A, Papp M, Wiborg O. Hippocampal cytogenesis correlates to escitalopram-mediated recovery in a chronic mild stress rat model of depression. Neuropsychopharmacology. 2006;31 (11):2395–2404. doi: 10.1038/sj.npp.1301041. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Jessop DS, Harbuz MS, Mork A, Sanchez C, Mikkelsen JD. Acute and long-term treatments with the selective serotonin reuptake inhibitor citalopram modulate the HPA axis activity at different levels in male rats. J Neuroendocrinol. 1999;11 (6):465–471. doi: 10.1046/j.1365-2826.1999.00362.x. [DOI] [PubMed] [Google Scholar]
- Kasckow JW, Baker D, Geracioti TD., Jr Corticotropin-releasing hormone in depression and post-traumatic stress disorder. Peptides. 2001;22 (5):845–851. doi: 10.1016/s0196-9781(01)00399-0. [DOI] [PubMed] [Google Scholar]
- Keen-Rhinehart E, Michopoulos V, Toufexis DJ, Martin EI, Nair H, Ressler KJ, et al. Continuous expression of corticotropin-releasing factor in the central nucleus of the amygdala emulates the dysregulation of the stress and reproductive axes. Mol Psychiatry. 2009;14 (1):37–50. doi: 10.1038/mp.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005a;62 (6):593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005b;62 (6):617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, McCormick JA, Chapman KE, Kelly PA, Seckl JR, Yau JL. Differential regulation of corticosteroid receptors by monoamine neurotransmitters and antidepressant drugs in primary hippocampal culture. Neuroscience. 2003;118 (4):975–984. doi: 10.1016/s0306-4522(03)00038-1. [DOI] [PubMed] [Google Scholar]
- Lenze EJ, Mantella RC, Shi P, Goate AM, Nowotny P, Butters MA, et al. Elevated cortisol in older adults with generalized anxiety disorder is reduced by treatment: a placebo-controlled evaluation of escitalopram. Am J Geriatr Psychiatry. 2011;19 (5):482–490. doi: 10.1097/JGP.0b013e3181ec806c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson D, Lakoma MD, Petukhova M, Schoenbaum M, Zaslavsky AM, Angermeyer M, et al. Associations of serious mental illness with earnings: results from the WHO World Mental Health surveys. Br J Psychiatry. 2010;197 (2):114–121. doi: 10.1192/bjp.bp.109.073635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RB, Nemeroff CB. The role of corticotropin-releasing hormone in the pathophysiology of depression: therapeutic implications. Curr Top Med Chem. 2011;11 (6):609–617. doi: 10.2174/1568026611109060609. [DOI] [PubMed] [Google Scholar]
- Lockhart P, Guthrie B. Trends in primary care antidepressant prescribing 1995–2007: a longitudinal population database analysis. Br J Gen Pract. 2011;61 (590):e565–e572. doi: 10.3399/bjgp11X593848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey L, Leeds C, Giltay EJ, van Veen T, Vreeburg SA, Penninx BW, et al. Antidepressant use and salivary cortisol in depressive and anxiety disorders. Eur Neuropsychopharmacol. 2011;21 (9):691–699. doi: 10.1016/j.euroneuro.2011.03.002. [DOI] [PubMed] [Google Scholar]
- McConathy J, Capello C, Jarkas N, Stowe ZN, Owens MJ. Preparation of antidepressants for use in preclinical research. Int J Neuropsychopharmacol. 2007;10 (6):759–763. doi: 10.1017/S1461145706007474. [DOI] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R. The use of behavioral test batteries: effects of training history. Physiol Behav. 2001;73 (5):705–717. doi: 10.1016/s0031-9384(01)00528-5. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72 (10):8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272 (5259):263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nikisch G, Agren H, Eap CB, Czernik A, Baumann P, Mathe AA. Neuropeptide Yand corticotropin-releasing hormone in CSF mark response to antidepressive treatment with citalopram. Int J Neuropsychopharmacol. 2005a;8 (3):403–410. doi: 10.1017/S1461145705005158. [DOI] [PubMed] [Google Scholar]
- Nikisch G, Mathe AA, Czernik A, Thiele J, Bohner J, Eap CB, et al. Long-term citalopram administration reduces responsiveness of HPA axis in patients with major depression: relationship with S-citalopram concentrations in plasma and cerebrospinal fluid (CSF) and clinical response. Psychopharmacology (Berl) 2005b;181 (4):751–760. doi: 10.1007/s00213-005-0034-3. [DOI] [PubMed] [Google Scholar]
- Okugawa G, Omori K, Suzukawa J, Fujiseki Y, Kinoshita T, Inagaki C. Long-term treatment with antidepressants increases glucocorticoid receptor binding and gene expression in cultured rat hippocampal neurones. J Neuroendocrinol. 1999;11 (11):887–895. doi: 10.1046/j.1365-2826.1999.00405.x. [DOI] [PubMed] [Google Scholar]
- Overstreet DH, Keeney A, Hogg S. Antidepressant effects of citalopram and CRF receptor antagonist CP-154,526 in a rat model of depression. Eur J Pharmacol. 2004;492 (2–3):195–201. doi: 10.1016/j.ejphar.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43 (4):425–473. [PubMed] [Google Scholar]
- Paslakis G, Heuser I, Schweiger U, Deuschle M. A single DEX/CRH test in male drug-free depressed patients is associated with the clinical response to treatment with fluoxetine. J Psychiatr Res. 2010;44 (16):1154–1157. doi: 10.1016/j.jpsychires.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. Vol. 1. Elsevier Academic Press; Amsterdam, Boston: 2005. (unpaged)pp. [Google Scholar]
- Paylor R, Spencer CM, Yuva-Paylor LA, Pieke-Dahl FS. The use of behavioral test batteries. II: Effect of test interval. Physiol Behav. 2006;87 (1):95–102. doi: 10.1016/j.physbeh.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci USA. 2001;98 (20):11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24 (20):4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed AL, Anderson JC, Bylund DB, Petty F, El Refaey H, Happe HK. Treatment with escitalopram but not desipramine decreases escape latency times in a learned helplessness model using juvenile rats. Psychopharmacology (Berl) 2009;205 (2):249–259. doi: 10.1007/s00213-009-1535-2. [DOI] [PubMed] [Google Scholar]
- Regev L, Neufeld-Cohen A, Tsoory M, Kuperman Y, Getselter D, Gil S, et al. Prolonged and site-specific over-expression of corticotropin-releasing factor reveals differential roles for extended amygdala nuclei in emotional regulation. Mol Psychiatry. 2011;16 (7):714–728. doi: 10.1038/mp.2010.64. [DOI] [PubMed] [Google Scholar]
- Research IoLA, Sciences CoL, Council NR. Guide for the Care and Use of Laboratory Animals. The National Academies Press; 1996. [Google Scholar]
- Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol. 2002;2 (1):23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Stein MB. Role of corticotropin releasing factor in anxiety disorders: a translational research perspective. Horm Behav. 2006;50 (4):550–561. doi: 10.1016/j.yhbeh.2006.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JC, Belkin BM, Krishnan KR, Nemeroff CB, Carroll BJ. Plasma dexamethasone concentrations and the dexamethasone suppression test. Biol Psychiatry. 1990;27 (2):159–173. doi: 10.1016/0006-3223(90)90646-j. [DOI] [PubMed] [Google Scholar]
- Sink KS, Walker DL, Freeman SM, Flandreau EI, Ressler KJ, Davis M. Effects of continuously enhanced corticotropin releasing factor expression within the bed nucleus of the stria terminalis on conditioned and unconditioned anxiety. Mol Psychiatry. 2012 doi: 10.1038/mp.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton KH, Nemeroff CB, Knight DL, Owens MJ. Chronic administration of the triazolobenzodiazepine alprazolam produces opposite effects on corticotropin-releasing factor and urocortin neuronal systems. J Neurosci. 2000;20 (3):1240–1248. doi: 10.1523/JNEUROSCI.20-03-01240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout SC, Owens MJ, Lindsey KP, Knight DL, Nemeroff CB. Effects of sodium valproate on corticotropin-releasing factor systems in rat brain. Neuropsychopharmacology. 2001;24 (6):624–631. doi: 10.1016/S0893-133X(00)00243-8. [DOI] [PubMed] [Google Scholar]
- Strekalova T, Gorenkova N, Schunk E, Dolgov O, Bartsch D. Selective effects of citalopram in a mouse model of stress-induced anhedonia with a control for chronic stress. Behav Pharmacol. 2006;17 (3):271–287. doi: 10.1097/00008877-200605000-00008. [DOI] [PubMed] [Google Scholar]
- Tonissaar M, Mallo T, Eller M, Haidkind R, Koiv K, Harro J. Rat behavior after chronic variable stress and partial lesioning of 5-HT-ergic neurotransmission: effects of citalopram. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32 (1):164–177. doi: 10.1016/j.pnpbp.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Uys JD, Muller CJ, Marais L, Harvey BH, Stein DJ, Daniels WM. Early life trauma decreases glucocorticoid receptors in rat dentate gyrus upon adult re-stress: reversal by escitalopram. Neuroscience. 2006;137 (2):619–625. doi: 10.1016/j.neuroscience.2005.08.089. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213 (4514):1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Curtis AL. Antidepressant interactions with corticotropin-releasing factor in the noradrenergic nucleus locus coeruleus. Psychopharmacol Bull. 1991;27 (3):263–269. [PubMed] [Google Scholar]
- von Bardeleben U, Holsboer F. Human corticotropin releasing hormone: clinical studies in patients with affective disorders, alcoholism, panic disorder and in normal controls. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12(Suppl):S165–S187. doi: 10.1016/0278-5846(88)90079-6. [DOI] [PubMed] [Google Scholar]
- Welberg LA, Kinkead B, Thrivikraman K, Huerkamp MJ, Nemeroff CB, Plotsky PM. Ketamine-xylazine-acepromazine anesthesia and postoperative recovery in rats. J Am Assoc Lab Anim Sci. 2006;45 (2):13–20. [PubMed] [Google Scholar]
- Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8 (3):231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau JL, Noble J, Chapman KE, Seckl JR. Differential regulation of variant glucocorticoid receptor mRNAs in the rat hippocampus by the antidepressant fluoxetine. Brain Res Mol Brain Res. 2004;129 (1–2):189–192. doi: 10.1016/j.molbrainres.2004.06.033. [DOI] [PubMed] [Google Scholar]
- Yau JL, Noble J, Hibberd C, Seckl JR. Short-term administration of fluoxetine and venlafaxine decreases corticosteroid receptor mRNA expression in the rat hippocampus. Neurosci Lett. 2001;306 (3):161–164. doi: 10.1016/s0304-3940(01)01890-0. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72 (12):9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.