

Abstract
Although protein kinases and phosphatases participate in integrin αIIbβ3 signaling, whether integrin functions are regulated by the catalytic subunit of protein phosphatase 1 (PP1c) isoforms are unclear. We show that siRNA mediated knockdown of all PP1c isoforms (α, β and γ1) in 293 αIIbβ3 cells decreased adhesion to immobilized fibrinogen and fibrin clot retraction. Selective knockdown of only PP1cγ1 did not alter adhesion or clot retraction, while depletion of PP1cβ decreased both functions. Unexpectedly, knockdown of PP1cα enhanced αIIbβ3 adhesion to fibrinogen and clot retraction. Protein interaction studies revealed that all PP1c isoforms can interact with the integrin αIIb subunit. Phosphoprofiling studies revealed an enhanced activation of mitogen-activated protein kinase (MAPK) p38 in the PP1cα depleted cells. Enhanced adhesive phenotype displayed by the PP1cα depleted 293 αIIbβ3 cells was blocked by pharmacological inhibition of p38. Conversely, the decreased adhesion of PP1cα overexpressing cells was rescued by the expression of constitutively active p38α or p38γ. Thus, PP1c isoforms have distinct contribution to the outside-in αIIbβ3 signaling-dependent functions in 293 αIIbβ3 cells. Moreover, PP1cα negatively regulates integrin function by suppressing the p38 pathway.
Keywords: Protein phosphatase 1, Adhesion, Clot retraction, Fibrinogen
INTRODUCTION
Hemostatic response at the site of vascular injury entails stable platelet-platelet and platelet-extracellular matrix adhesion that is mediated in part by platelet integrin αIIbβ3. Engagement of integrin αIIbβ3 generates outside-in signals, which facilitates the rearrangement of cytoskeletal compartment and contributes to processes like stable adhesion, spreading and fibrin clot retraction in platelets [1-2]. Outside-in αIIbβ3 signaling encompasses reversible phosphorylation of multiple effectors (cytoskeletal, signaling and adaptor proteins) in the pathways that are downstream of the integrin receptor. Interplay between the activities of protein kinases and phosphatases that associate with the cytoplasmic tails of the αIIbβ3 complex can initiate outside-in αIIbβ3 signaling [3]. We and others have reported that the catalytic subunit of protein phosphatase 1 (PP1c) associates with the integrin αIIbβ3 complex in platelets and in heterologous model cells overexpressing αIIbβ3 [4-5].
Protein phosphatase 1 is a major serine/threonine (Ser/Thr) phosphatase that regulates a variety of cellular functions ranging from protein synthesis, cell division, metabolism and apoptosis [6-7]. The catalytic subunit of PP1 (PP1c) associates with a wide variety of proteins called PP1c interacting proteins (PIPs). These proteins can either target PP1c to a subcellular location or act as substrates for PP1c or regulate the PP1c activity [6-7]. Three isoforms of PP1c that share ~90% amino acid sequence similarity have been identified and includes PP1cα, PP1cβ/δ and PP1cγ [6]. PP1cγ has two splice variants; PP1cγ1 (ubiquitous expression) and PP1cγ2 (testis specific expression).
Generic Ser/Thr phosphatase inhibitors like calyculin A and okadaic acid block PP1, PP2A and impair agonist-induced platelet aggregation, adhesion and spreading on immobilized fibrinogen [8-11]. In contrast to the studies using generic Ser/Thr phosphatase inhibitors, we noticed a moderate decrease in only low-dose thrombin-induced αIIbβ3 activation, fibrinogen binding and aggregation in PP1cγ−/− platelets. Importantly, outside-in αIIbβ3 signaling-dependent functions like adhesion, spreading and clot retraction were largely unaltered in PP1cγ−/− platelets [12]. A milder phenotype noted in the PP1cγ−/− platelets may reflect functional redundancies by other PP1c isoforms or represent a true PP1cγ isoform specific phenotype. Our ability to test whether platelet functions are regulated in a PP1c isoform specific manner is hampered by the lack of PP1cα−/− and PP1cβ−/− mice models and the non-availability of isoform specific pharmacological inhibitors. Here, we assessed the role of PP1c in regulating the outside-in αIIbβ3 functions by utilizing genetic approaches in αIIbβ3 overexpressing human kidney embryonal 293 cell model system. We report that the depletion of all PP1c isoforms in the 293 αIIbβ3 cells moderately decreased cell adhesion and clot retraction. Unexpectedly, analysis of the individual PP1c isoforms revealed opposing roles for the PP1cα and PP1cβ isoforms in outside-in αIIbβ3 signaling-dependent functions. Moreover, PP1cα negatively regulated αIIbβ3 adhesion via the regulation of p38 mitogen-activated protein kinase (MAPK) pathway.
MATERIALS AND METHODS
Reagents
SiImporter, PP2Ac and PP1cβ antibodies were from Millipore (Millipore, Billerica, MA). Antibodies to PP1cα, PP1cγ1 and HA were from Santacruz Biotechnology (Santacruz, CA). Antibodies to integrin αIIb (SEW-8 and 132.1) were kindly provided by Dr. P. Newman, Blood Research Institute (Milwaukee, WI). Antibody 10E5 and integrilin were gifts from Drs. B. Coller, Rockefeller University (New York, NY) and D. Phillips (Portola Pharmaceuticals Inc., San Francisco, CA). Human Phospho-Kinase Array kit was from BD BioScience (San Jose, CA). Fibrinogen was from Enzyme Research Laboratories Inc. (South Bend, IN). Fibronectin and lipofectamine were from Invitrogen (Carlsbad, CA). ECL system was from GE Healthcare Life Science (Piscataway, NJ). Phospho-p38 (Thr180, Tyr182) and p38 antibodies were from Cell Signaling Technologies (Beverly, MA). SB203580 hydrochloride was from Tocris Bioscience (Ellisville, MO).
Adhesion and clot retraction in 293 αIIbβ3 cells
Human embryonal kidney 293 cells overexpressing αIIbβ3 (293 αIIbβ3) were generated as we have previously described [13]. 293 αIIbβ3 cells were transfected with 100 nM siRNA using siImporter. A (smart pool) of four independent siRNAs targeting PP1cα, PP1cβ, PP1cγ1 along with a non-specific control siRNA (Dharmacon, Thermo Fisher Scientific, Lafayette, CO) were used in this study. For overexpression studies, 293 αIIbβ3 cells were transiently transfected with cDNAs for haemagglutinin (HA)-tagged PP1cα or the control vector (gift from Dr. M. Eto, Thomas Jefferson University, Philadelphia, PA) using lipofectamine [14]. In certain experiments, cells were also transfected with empty vector or HA tagged constitutively active p38α (MKK6–p38α) or p38γ (MKK6-p38γ) fusion constructs (gift from Dr. G. Chen, Medical College of Wisconsin, Milwaukee, WI) [15]. After 48-72 hours, the cells were used for either Western blotting or cell adhesion experiments. For adhesion experiments, 1 × 105 cells in Tyrode's buffer were incubated for 15 or 30 minutes on a ninety six well plate coated with 12.5 μg/ml fibrinogen or 12.5 μg/ml fibronectin and blocked with 5% BSA. Control wells were coated with 5% BSA. Wells were washed three times and adhered cells were calorimetrically assayed for acid phosphatase at 405 nm as we have described [16]. The absorbance value obtained for BSA coated wells was subtracted from that of fibrinogen and fibronectin coated wells. A standard curve for absorbance versus cell numbers was used to obtain the number of adhered cells. Percent adhesion was calculated as the number of fibrinogen/fibronectin bound cells divided by the total cells added per well and multiplied by 100. In certain experiments, cells were pretreated with 10 μM water soluble p38 inhibitor (SB203580) for 30 minutes prior to adhesion studies. Integrin expression levels in PP1c depleted/overexpressing cells were monitored by flow cytometric studies using anti-αIIb-PE and anti-αIIbβ3 (P2) antibodies as described before [13].
Fibrin clot retraction assays were performed as in our previous study [13]. 293 αIIbβ3 cells (4 × 106) treated with either control, PP1cα, PP1cβ, PP1cγ1 or PP1c (α, β, γ1) siRNAs were suspended in 300 μl DMEM containing 28 mM CaCl2 and 25 mM HEPES. Clot formation was initiated by the addition of thrombin (2.5 U). After incubation at 37°C for varying time periods, clot retraction was quantified by measuring the volume of liquid not incorporated into the clot. The percentage of clot retraction was calculated as the (amount of liquid collected)/ (the amount of liquid before the clot) × 100. Control siRNA treated cells pretreated with 5 μM Integrilin (αIIbβ3 blocker) were also used. In certain experiments, cells were treated with 10 μM SB203580 prior to clot retraction assays.
GST pulldown, co-immunoprecipitation and immunoblotting
The cDNAs for PP1cα, PP1cβ and PP1cγ1 in pcMV vector was amplified by PCR and subcloned into a glutathione S-transferase (GST) vector pGEX 4T-1. GST or GST tagged PP1cα, PP1cβ, PP1cγ1 proteins were expressed in Escherichia coli following induction with isopropyl –β-D-thiogalactopyranoside and purified using glutathione beads. Purified GST proteins were characterized by Commassie staining. Purified GST or GST-PP1c α, β or γ1 were pre-coupled with glutathione beads and mixed with lysate from 293 αIIbβ3 cells (300 μg) and platelets (150 μg) for 3 hours at 4°C. Beads were washed and the PP1cα, PP1cβ and PP1cγ1 interacting proteins separated by SDS-PAGE and immunoblotted with anti-αIIb antibody. For co-immunoprecipitation assays, lysate (~300 μg) from 293 αIIbβ3 cells was immunoprecipitated with rabbit IgG or αIIb (Sew-8) antibodies using protein A sepharose beads, and immunoblotted with antibodies to all PP1c isoforms and αIIb. In some experiments, forty μg of lysate from 293 cells were separated by SDS-PAGE and immunoblotted with either anti-PP1cα, PP1cβ, PP1cγ1, PP2Ac, phospho-p38, p38, HA or actin antibodies using ECL. The signals on the films were scanned and the densitometric quantification performed using Image J software from NIH.
Phosphoprofiling studies
Lysate (~500 μg in 150 μl) from control and PP1cα depleted cells were diluted with the buffer in the Phospho-kinase antibody array kit and incubated with the nitrocellulose membrane spotted with various phospho kinase antibodies. The membrane was washed and incubated with the biotinylated detection antibody cocktail, followed by streptavidin-HRP and detected by ECL as per the manufacturer's instructions.
Statistics
Statistical significance of the data was analyzed by using a paired Student's t test. Data is expressed as mean ± SEM. P<0.05 was considered as significant.
3. RESULTS
PP1c depletion moderately decreased adhesion of 293 αIIbβ3 cells to fibrinogen
Since the pharmacological inhibitors that block PP1 also exert some inhibition on PP2A, one approach to study the role of PP1c in αIIbβ3 signaling is to utilize PP1c knockdown or overexpression strategies. However, such genetic approaches are challenging in anucleate platelets. Cell model system overexpressing integrin αIIbβ3 are amenable to genetic manipulations and have immensely contributed to the understanding of integrin αIIbβ3 outside-in signaling and function [17-19]. Therefore, in this study, we utilized the previously characterized 293 αIIbβ3 cell model [13].
PP1c isoforms in 293 αIIbβ3 cells were depleted by siRNA and evaluated for the adhesion to immobilized fibrinogen. PP1c knockdown assessed by an antibody that recognizes all PP1c isoforms was specific, since the levels of PP2Ac and actin were comparable between the control and PP1c depleted cells (Figure 1A). Compared to the control siRNA treated cells, PP1c depleted cells exhibited a moderate but significant decrease in adhesion to fibrinogen (Figure 1B). Antibody 10E5 inhibited the adhesion of 293 αIIbβ3 cells to fibrinogen, suggesting that the adhesion assays were integrin αIIbβ3 specific (Figure 1B). In contrast, adhesion of control and PP1c depleted cells to fibronectin, a ligand for the endogenous α5β1 on 293 cells was not significantly different (Figure 1C). This suggests that the differential adhesion due to PP1c depletion is integrin (αIIbβ3) and ligand (fibrinogen) specific. Mean fluorescence intensity (MFI) for the αIIbβ3 expression was 26.9 ± 6.7 (control) and 29.35 ± 4.8 (PP1c depleted) and could not account for the differences in adhesion. These studies suggest that PP1c in part stabilized αIIbβ3 mediated adhesive interactions to fibrinogen.
Figure 1. Effect of depleting the PP1c isoforms on 293 αIIbβ3 cell adhesion.

(A) Lysate from the control siRNA and PP1c depleted cells were immunoblotted with anti-PP1c pan antibody (recognizes all PP1c isoforms), anti-PP2Ac and anti-actin (loading). (B) Adhesion of 293 αIIbβ3 cells treated with either control or PP1c α, β or γ1 siRNAs to fibrinogen. Compared to control siRNA, the decreased adhesion of all PP1c isoform depleted cells were significant at ◆p<0.001, *p=0.002. n=4-7. (C) Adhesion of 293 αIIbβ3 cells treated with control or PP1c α, β or γ1 siRNAs to fibronectin. The adhesion difference between control and PP1c depleted cells from 3 experiments were not significant (P>0.05).
3.2 Isoform specific roles for PP1c in αIIbβ3 outside-in signaling-dependent functions
To clarify the contribution of PP1c isoforms in integrin function, we selectively depleted each of the PP1c isoforms and evaluated αIIbβ3 outside-in signaling dependent functions in 293 αIIbβ3 cells. PP1cγ1 siRNA depleted PP1cγ1 but not PP1cα and PP1cβ isoforms in 293 cells (Figure 2A). Functional studies revealed a comparable adhesion between the control and PP1cγ1 depleted cells (Figure 3A). PP1cβ depleted cells (Figure 2B) showed a modest but significant reduction in the adhesion to fibrinogen (Figure 3B). MFI for the αIIbβ3 expression was comparable between the control (28.4 ± 8.1) and PP1cβ depleted cells (28.9 ± 10.8). Unexpectedly, loss of PP1cα (Figure 2C) moderately enhanced the adhesion of 293 αIIbβ3 cells to fibrinogen (Figure 3C). Integrin αIIbβ3 expression was not significantly (p>0.05) altered in the PP1cα depleted cells (MFI of 23.3 ± 0.8 and 21.1 ± 1.4 for control and PP1cβ depleted cells). Compared to the PP1cα depleted cells, an opposing adhesive phenotype displayed by the PP1cα depleted cells prompted us to further validate the PP1cα knockdown data. In a complementary approach, we overexpressed a HA-tagged PP1cα in 293 αIIbβ3 cells and evaluated adhesion to fibrinogen. Immunoblotting with anti-HA and anti-PP1cα antibodies confirmed the overexpression of PP1cα (Figure 2D). Consistent with the PP1cα knockdown studies, overexpression of PP1cα in 293 αIIbβ3 cells moderately decreased the adhesion to fibrinogen (Figure 3D). Integrin expression levels were unaltered in these cells (MFI of 28.1 ± 4.3 and 29.3 ± 5.4 for control and PP1cα HA cells).
Figure 2. Knockdown of PP1c isoforms and overexpression of PP1cα in 293 αIIbβ3 cells.
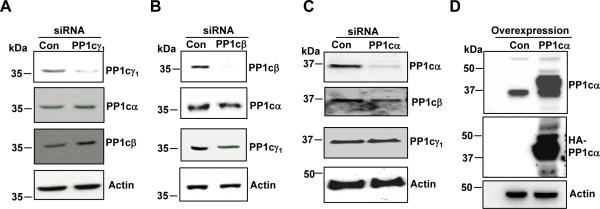
(A) Lysate from the control siRNA and PP1cγ1 siRNA-treated cells were immunoblotted with PP1cγ1 PP1cα, PP1cβ and actin antibodies. (B) Lysate from control and PP1cβ siRNA-treated cells were immunoblotted with anti-PP1cβ, anti-PP1cα, anti-PP1cγ1 and actin antibodies. (C) PP1cα, PP1cβ, PP1cγ1 and actin expression in 293 αIIbβ3 cells treated with control and PP1cα siRNAs. (D) Immunoblotting with anti-PP1cα and HA antibodies in the control vector and HA-tagged PP1cα overexpressing cells. Knockdown and overexpression blots are representative of 2-3 experiments.
Figure 3. Effect of depleting the PP1c isoforms and overexpression of PP1cα on cell adhesion.

(A) Depletion of PP1cγ1 did not affect fibrinogen adhesion. n=4. (B) Compared to control siRNA, the decreased adhesion of PP1cβ depleted cells were significant at ◆p=0.005, *p=0.001. n=5-6. (C). Compared to control siRNA, the increased adhesion of PP1cα depleted cells were significant at ◆p=0.01, *p<0.0001. n=8-12. (D) Compared to control HA, the decreased adhesion of PP1cα HA overexpressing cells were significant at ◆p<0.001, *p=0.006. n=5-9.
To further validate the PP1c isoform specific phenotypes, we examined fibrin clot retraction, an independent αIIbβ3 outside-in signaling function that involves the rearrangement of actin cytoskeleton. Compared to the control siRNA treated cells, depletion of all PP1c isoforms led to a decreased fibrin clot retraction at all time points (Figure 4A). Loss of PP1cα moderately increased clot retraction compared to the control siRNA at 30 and 60 minutes. In contrast, depletion of PP1cβ decreased clot retraction. Loss of PP1cγ1 did not significantly alter clot retraction process (Figure 4A). Pretreatment of control siRNA cells with integrilin blocked fibrin clot retraction, indicating that clot retraction by 293 αIIbβ3 cells was αIIbβ3 dependent. Taken together, these studies indicate that the PP1c isoforms distinctly contribute to the αIIbβ3 outside-in signaling functions in 293 αIIbβ3 cells.
Figure 4. Effect of depleting the PP1c isoforms on fibrin clot retraction.

(A) The decreased fibrin clot retraction of PP1c (α, β, γ1) depleted cells compared to the control siRNA treated cells was significant at *p=0.03, *p=0.01, *p=0.001 for 30, 60 and 120 minutes, respectively. The increased fibrin clot retraction of PP1cα depleted cells compared to the control siRNA treated cells was significant at ◆p=0.05, ◆p=0.04, p=0.07 for 30, 60 and 120 minutes, respectively. The decreased fibrin clot retraction of PP1cβ depleted cells compared to the control siRNA treated cells was significant at †p=04, †p=.0.03, †p=0.005 for 30, 60 and 120 minutes, respectively. Integrilin (5 μM) blocked the clot retraction of control siRNA cells at 120 minutes *p=0.003. n=3. (B) PP1c α, β and γ1 GST proteins separated on a SDS-PAGE gel were detected by Coomassie Blue staining. M is Molecular weight marker. (C). αIIb in the lysate from the 293 αIIbβ3 cells and platelets interact with PP1cα–GST, PP1cβ-GST, PP1cγ1–GST but not GST. Comparable levels of αIIb in lysate (input) was used for pull down studies. n=2-3. (D) 293 αIIbβ3 cell lysate was immunoprecipitated (IP) with IgG (control) or αIIb and immunoblotted (IB) with anti- PP1cα, PP1cβ, PP1cγ1 and αIIb. Lys is cell lysate. n=2.
Interaction of all PP1c isoforms with the αIIb integrin subunit
We previously showed that αIIb integrin subunit co-immunoprecipitates with PP1c using an antibody that recognizes all isoforms [4]. Because of the distinct role for each PP1c isoforms in the αIIbβ3 outside-in signaling, we examined if specific isoforms of PP1c associates with αIIb. PP1cα, PP1cβ and PP1cγ were expressed as a GST fusion protein in E.coli (Figure 4B). Integrin αIIb from the lysate of 293 αIIbβ3 cells and platelets interacted with the GST-PP1cα, GST-PP1cβ and GST-PP1cγ1 but not with GST alone in a GST pull down assays (Figure 4C). Moreover, co-immunoprecipitation assays revealed that all PP1c isoforms can associate with αIIb in 293 αIIbβ3 cells (Figure 4D). These studies indicate that all the PP1c isoforms can interact with the αIIbβ3 complex.
PP1cα negatively regulates αIIbβ3 functions by suppressing the p38 signaling
It is possible that PP1c isoforms distinctly modulate integrin function by engaging discrete effectors downstream of the integrin pathway. Therefore, we sought to identify potential PP1cα specific effectors that participate in negatively regulating integrin function. PP1cα depleted cell lysate was subjected to phosphoprofiling studies using a phospho-kinase antibody array kit. Compared to the control siRNA cells, PP1cα depleted cells showed altered phosphorylation response to several signaling proteins, including a robust enhancement in the phosphorylation of residues Thr180 and Tyr182 of the MAPK p38, which is a surrogate marker for p38 activation (Supplementary data, Table 1). A ~1.5 fold increased p38 activation in the PP1cα depleted cells by immunoblotting studies validated the phosphoprofiling studies (Figure 5A). Interestingly, depletion of PP1cβ or PP1cγ did not overtly impact p38 activation (Figure 5A), suggesting that p38 may be a PP1c specific effector.
Figure 5. PP1cα negatively regulates αIIbβ3 adhesion by suppressing the p38 signaling pathway.

(A) Lysate from the 293 cells treated with either control, PP1c (α, β, γ1) siRNAs were immunoblotted with anti-phospho-Thr180, Tyr182 p38 (pp38) and p38 antibodies. Densitometric quantification of the activation of p38 was expressed as pp38/total p38 from 2-3 experiments in arbitrary units. The increased P38 activation in PP1cα depleted cells compared to the control siRNA treated cells was significant at ◆p=0.04. Effect of the water soluble p38 inhibitor on the increased fibrinogen adhesion (B) and clot retraction (C) displayed by the PP1cα depleted cells. Compared to control siRNA, the increased adhesion of PP1cα depleted cells at 30 minutes and the increased clot retraction at 60 minutes were significant at ◆p=0.001 and p=0.03. Treatment with SB203580 abolished the increased adhesion and clot retraction differences p=0.618 and p=.138 n=4-5. (D) Expression of PP1cα with either MKK6-P38α or MKK6-p38γ fusion proteins detected by immunoblotting with anti-HA antibody in 293 αIIbβ3 cells overexpressing the appropriate constructs. The blot is a representative of 2-3 experiments. (E) Effect of MKK6-p38α and MKK6-p38γ on the decreased adhesion of PP1cα overexpressing cells to fibrinogen at 15 minutes. Compared to the control HA, the decreased adhesion of PP1cα HA overexpressing cells were significant at ◆p<0.001. These differences were lost in cells expressing MKK6-p38α (p>0.05) or MKK6-p38γ (p>0.0.1) *p=0.006. n=4.
To ascertain whether the increased p38 activation in PP1cα depleted cells contributed to their enhanced functional phenotype, adhesion and clot retraction assays were performed with a p38 pharmacological inhibitor. The enhanced adhesiveness and the clot retraction phenotype displayed by the PP1cα depleted cells were abrogated in the presence of p38 inhibitor SB203580 (Figure 5B and 5C). In a complementary approach, we transfected the constitutively active p38α (HA-tagged MKK6-p38α) or p38γ (HA-tagged MKK6-p38γ) fusion constructs [15] into 293 αIIbβ3 cells overexpressing HA-PP1cα and examined cell adhesion to immobilized fibrinogen. MAPK kinase 6 (MKK6) is an upstream activator of p38 and fusing the cDNA of p38 in frame with its activator MKK6 via a decapeptide linker (Gly-Glu)5 results in a constitutively active p38 form [15]. Such strategies have been used previously to generate constitutively active ERK [20] and constitutively active Jun [21]. Compared to the cells transfected with either control vectors, HA-MKK6-p38α or HA-PP1cα, dual transfection of HA-MKK6-p38α and HA-PP1cα showed expression of both MKK6-p38α fusion protein (~80 kDa) and the PP1cα (~38 kDa) (Figure 5D). Similar results were noticed for the combination of HA-PP1cα and HA-MKK6-p38γ (Figure 5D). MKK6-P38α is predominantly localized in the cytoplasm, while MKK6-p38γ tends to be localized to both the cytosol and the nucleus (15). Such distribution patterns may account for the lower intensity of MKK6-p38γ that we observed in our studies, compared to the MKK6-p38α. Compared to the cells expressing empty vectors (for HA-PP1cα and HA-MKK6-p38), overexpression of PP1cα decreased αIIbβ3 adhesion to fibrinogen (Figure 5E). More importantly, dual expression of PP1cα and MKK6-p38α or PP1cα and MKK6-p38γ rescued the decreased adhesiveness of the PP1cα overexpressing cells to fibrinogen (Figure 5E). Thus, PP1cα negatively regulates αIIbβ3–fibrinogen adhesion by suppressing the p38 signaling pathway.
DISSCUSSION
Previous studies have identified that calyculin A can inhibit platelet adhesion and spreading to fibrinogen [8]. However, since this compound can block Ser/Thr phosphatases like PP1, PP2A and PP4 [22], a specific role for PP1 and its isoforms in the functions of αIIbβ3 remain unclear. Although tautomycin or its close analogue tautomycetin was reported to be a PP1 preferential inhibitor [23-24], we noticed that tautomycin moderately inhibited platelet PP2A activity (not shown), thereby precluding the use of tautomycin in platelet studies. To overcome the challenges posed by the pharmacological agents and the inability to use platelets in studies to evaluate the role of PP1, we utilized genetic approach in heterologous cells expressing αIIbβ3. Depletion of all PP1c isoforms in 293 αIIbβ3 cells revealed a moderate decrease in adhesion to fibrinogen and clot retraction (Figures 1B and 4A). Furthermore, loss of PP1cγ1 did not alter adhesion or clot retraction (Figures 3A and 4A), while loss of PP1cβ decreased both functions (Figures 3B and 4A). Surprisingly, loss of PP1cα enhanced adhesion and clot retraction (Figures 3C and 4A). Thus, isoform specific roles for PP1c in outside-in αIIbβ3 signaling dependent functions were identified. Indeed, PP1c isoform specific roles have been described to modulate morphological and cell cycle changes in Hela cells [25] and sarcoplasmic reticulum mediated Ca+2 cycling in rat cardiomyocytes [26]. Interestingly, like PP1cγ depleted cells, PP1cγ−/− platelets also exhibited normal αIIbβ3 mediated adhesion and clot retraction [12]. Despite these observations, findings from the 293 αIIbβ3 cells should be cautiously interpreted in the context of platelets. Future studies with the platelets from PP1cα and PP1cβ null mice will be needed to validate the observed phenotypes.
It is not clear how PP1c isoforms selectively regulate αIIbβ3 functions. Studies in Figure 4C and 4D suggest that the distinct PP1c phenotypes are not due to isoform specific localization of PP1c with the cytoplasmic tail of the integrin αIIb subunit. However, specific or preferential interactions of PP1c isoforms with effectors exist. For example, PP1cα associates with a focal adhesion protein tensin [27]. PP1cβ interacts with focal adhesion protein (FAK) [28] and myosin phosphatase targeting subunit (MYPT) [29], while PP1cγ1 can interact with spinophilin, an actin binding scaffolding protein [30]. Isoform specific interactions of PP1c with distinct effectors in 293 αIIbβ3 cells could temporally place a particular PP1c isoform in a certain milieu within the focal adhesions. This in turn could facilitate a temporal dephosphorylation of Ser/Thr residues of specific substrates and thereby regulate the assembly and disassembly of cellular focal adhesions. Such mechanisms could support the biological relevance for an isoform specific PP1c role. It is not apparent how combined loss of all PP1c isoforms showed a moderate reduction in αIIbβ3 functions, despite the positive and negative contribution of PP1cβ and PP1cα isoforms, respectively. PP1c interacts with ~200 phosphatase interacting proteins, some of which are endogenous inhibitors that are regulated by phosphorylation. The complex nature of phosphatase interactome and their ability to integrate with other signaling networks, may have precluded the functional outcome of multiple PP1c knock down to be a mere additive of the outcomes displayed by single PP1c knockdown. Such observations were also noted with other signaling molecules. Broad spectrum non-isoform selective PKC inhibitors block platelet functions yet, several PKC isoforms have distinct and opposing roles in platelet activation events [31].
How could loss of PP1cα negatively regulate outside-in αIIbβ3 functions? Phosphoprofiling and immunoblot analysis revealed increased p38 activation in PP1cα depleted cells (Figure 5A). p38 is required for αIIbβ3 functions such as clot retraction [32] and spreading of platelets pretreated with PAR4-AP to fibrinogen and collagen [33]. Thus, enhanced p38 activation could be predicted to potentiate outside-in αIIbβ3 signaling function. Indeed, p38 activation in PP1cα depleted cells was responsible for potentiating adhesive and clot retraction functions in the PP1cα depleted cells. p38 inhibitor blocked the increased adhesion and clot retraction displayed by the PP1cα depleted cells (Figures 5B and 5C). Conversely, the constitutively active p38α or p38γ rescued the decreased adhesive phenotype showed by PP1cα overexpressing cells (Figure 5E). Interestingly, p38 phosphorylates and regulates the activity of PP1cα interacting protein tensin [34].
In summary, a genetic approach revealed for the first time that outside-in αIIbβ3 signaling-dependent functions are regulated in part by PP1c signaling, with an opposing role for PP1cα and PP1cβ. Mechanistically, regulation of the p38 pathway underpins the PP1cα mediated negative regulation of αIIbβ3 adhesion.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by in whole or in part by grants HL081613 (National Institutes of Health), 09GRNT2260224 (American Heart Association) and the Alkek Foundation. K.V.V was supported by the Mary R. Gibson Foundation. S.P was supported by grant T32 HL072754 from the NIH.
REFERENCES
- 1.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–15. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 2.Coller BS, Shattil SJ. The GPIIb/IIIa (integrin αIIbβ3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112:3011–25. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shattil SJ. The β3 integrin cytoplasmic tail: protein scaffold and control freak. J.Thromb.Haemost. 2009;7:210–13. doi: 10.1111/j.1538-7836.2009.03397.x. [DOI] [PubMed] [Google Scholar]
- 4.Vijayan KV, Liu Y, Li TT, et al. Protein phosphatase 1 associates with the integrin αIIb subunit and regulates signaling. J.Biol.Chem. 2004;279:33039–42. doi: 10.1074/jbc.C400239200. [DOI] [PubMed] [Google Scholar]
- 5.Raab M, Daxecker H, Edwards RJ, et al. Protein interactions with the platelet integrin αIIb regulatory motif. Proteomics. 2010;10:2790–2800. doi: 10.1002/pmic.200900621. [DOI] [PubMed] [Google Scholar]
- 6.Cohen PT. Protein phosphatase 1--targeted in many directions. J.Cell Sci. 2002;115:241–56. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 7.Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 8.Lerea KM, Cordero KP, Sakariassen KS, et al. Phosphorylation sites in the integrin β3 cytoplasmic domain in intact platelets. J.Biol.Chem. 1999;274:1914–19. doi: 10.1074/jbc.274.4.1914. [DOI] [PubMed] [Google Scholar]
- 9.Nishikawa M, Toyoda H, Saito M, et al. Calyculin A and okadiac acid inhibit human platelet aggregation by blocking protein phosphatases types 1 and 2A. Cell Signal. 1994;6:59–71. doi: 10.1016/0898-6568(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 10.Mauco G, Artcanuthurry V, Pidard D, et al. Total inhibition of phospholipase C and phosphatidylinositol 3-kinase by okadaic acid in thrombin-stimulated platelets. Cell Signal. 1997;9:117–24. doi: 10.1016/s0898-6568(96)00119-2. [DOI] [PubMed] [Google Scholar]
- 11.Hoyt CH, Lerea KM. Aggregation-dependent signaling in human platelets is sensitive to protein serine/threonine phosphatase inhibitors. Biochemistry. 1995;34:9565–70. doi: 10.1021/bi00029a033. [DOI] [PubMed] [Google Scholar]
- 12.Gushiken FC, Hyojeong H, Pradhan S, et al. The catalytic subunit of protein phosphatase 1 gamma regulates thrombin-induced murine platelet αIIbβ3 function. PLoS.One. 2009;4:e8304. doi: 10.1371/journal.pone.0008304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vijayan KV, Goldschmidt-Clermont PJ, Roos C, et al. The PlA2 polymorphism of integrin β3 enhances outside-in signaling and adhesive functions. J.Clin.Invest. 2000;105:793–802. doi: 10.1172/JCI6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eto M, Kirkbride JA, Brautigan DL. Assembly of MYPT1 with protein phosphatase-1 in fibroblasts redirects localization and reorganizes the actin cytoskeleton. Cell Motil.Cytoskeleton. 2005;62:100–9. doi: 10.1002/cm.20088. [DOI] [PubMed] [Google Scholar]
- 15.Qi X, Pohl NM, Loesch M, et al. p38αantagonizes p38γactivity through c-Jun-dependent ubiquitin proteasome pathways in regulating Ras transformation and stress response. J.Biol.Chem. 2007;282:31398–408. doi: 10.1074/jbc.M703857200. [DOI] [PubMed] [Google Scholar]
- 16.Gushiken FC, Patel V, Liu Y, et al. Protein phosphatase 2A negatively regulates integrin αIIbβ3 signaling. J.Biol.Chem. 2008;283:12862–69. doi: 10.1074/jbc.M708804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Toole TE, Loftus JC, Du XP, et al. Affinity modulation of the αIIbβ3 integrin (platelet GPIIb- IIIa) is an intrinsic property of the receptor. Cell Regul. 1990;1:883–93. doi: 10.1091/mbc.1.12.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han J, Lim CJ, Watanabe N, et al. Reconstructing and deconstructing agonist-induced activation of integrin αIIbβ3. Curr.Biol. 2006;16:1796–1806. doi: 10.1016/j.cub.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Pradhan S, Alrehani N, Patel V, et al. Cross-talk between serine/threonine protein phosphatase 2A and protein tyrosine phosphatase 1B regulates Src activation and adhesion of integrin αIIbβ3 to fibrinogen. J.Biol.Chem. 2010;285:29059–68. doi: 10.1074/jbc.M109.085167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson MJ, Stippec SA, Goldsmith E, et al. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr.Biol. 1998;8:1141–50. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- 21.Zheng C, Xiang J, Hunter T, et al. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J.Biol.Chem. 1999;274:28966–71. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- 22.McCluskey A, Sim AT, Sakoff JA. Serine-threonine protein phosphatase inhibitors: development of potential therapeutic strategies. J.Med.Chem. 2002;45:1151–75. doi: 10.1021/jm010066k. [DOI] [PubMed] [Google Scholar]
- 23.Mitsuhashi S, Matsuura N, Ubukata M, et al. Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1. Biochem.Biophys.Res.Commun. 2001;287:328–31. doi: 10.1006/bbrc.2001.5596. [DOI] [PubMed] [Google Scholar]
- 24.Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol.Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada T, Fujii T, Tanuma N, et al. Analysis of isoform specific function of PP1 catalytic subunits in mammalian cells using siRNA. Int.J.Oncol. 2004;25:1383–88. [PubMed] [Google Scholar]
- 26.Aoyama H, Ikeda Y, Miyazaki Y, et al. Isoform-specific roles of protein phosphatase 1 catalytic subunits in sarcoplasmic reticulum-mediated Ca2+ cycling. Cardiovasc.Res. 2011;89:79–88. doi: 10.1093/cvr/cvq252. [DOI] [PubMed] [Google Scholar]
- 27.Eto M, Kirkbride J, Elliott E, et al. Association of the tensin N-terminal protein-tyrosine phosphatase domain with the α isoform of protein phosphatase-1 in focal adhesions. J.Biol.Chem. 2007;282:17806–15. doi: 10.1074/jbc.M700944200. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi M, De LS, Vietri M, et al. Reciprocally interacting domains of protein phosphatase 1 and focal adhesion kinase. Mol.Cell Biochem. 2005;272:85–90. doi: 10.1007/s11010-005-7639-z. [DOI] [PubMed] [Google Scholar]
- 29.Moorhead G, Johnson D, Morrice N, et al. The major myosin phosphatase in skeletal muscle is a complex between the β isoform of protein phosphatase 1 and the MYPT2 gene product. FEBS Lett. 1998;438:141–44. doi: 10.1016/s0014-5793(98)01276-9. [DOI] [PubMed] [Google Scholar]
- 30.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc.Natl.Acad.Sci.U.S.A. 1997;94:9956–61. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harper MT, Poole AW. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J.Thromb.Haemost. 2010;8:454–62. doi: 10.1111/j.1538-7836.2009.03722.x. [DOI] [PubMed] [Google Scholar]
- 32.Flevaris P, Li Z, Zhang G, et al. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113:893–901. doi: 10.1182/blood-2008-05-155978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazharian A, Roger S, Berrou E, et al. Protease-activating receptor-4 induces full platelet spreading on a fibrinogen matrix: involvement of ERK2 and p38 and Ca2+ mobilization. J.Biol.Chem. 2007;282:5478–87. doi: 10.1074/jbc.M609881200. [DOI] [PubMed] [Google Scholar]
- 34.Hall EH, Balsbaugh JL, Rose K, et al. Comprehensive Analysis of Phosphorylation Sites in Tensin1 Reveals Regulation by p38MAPK. Mol.CellProteomics. 2010;12:2853–63. doi: 10.1074/mcp.M110.003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.