

Abstract
Previous studies have confirmed that natural bone apatite crystals are bound with citrate-rich molecules. Citrates on apatite crystals impact bone development and its load-bearing function. However, such understanding has never been translated into bone biomaterials design. Herein, a first citrate-based injectable composite material for orthopedic applications is developed based on our recently developed biodegradable poly(ethylene glycol) maleate citrate (PEGMC) and hydroxyapatite (HA). PEGMC contains rich carboxylic groups that could chelate with calcium-containing HA thus facilitating polymer/HA interactions, similar to natural citrate-bound apatite crystal. The crosslinking of poly(ethylene glycol) diacrylate (PEGDA) with PEGMC/HA composites allows an addition control over degradation and mechanical properties of the crosslinked PEGMC/HA (CPEGMC/HA) composites. CPEGMC/HA composite can serve as an ideal injectable cell carrier as confirmed by the enhanced DNA content, ALP activity, and calcium production through a human fetal osteoblast encapsulation study. Ex vivo study on porcine femoral head demonstrated that PEGMC/HA is a potentially promising injectable biodegradable bone material for the treatment of osteonecrosis of the femoral head. Development of biodegradable citrate-based injectable PEGMC/HA composite is an initial step for the development of the next generation of bone tissue engineering and orthopedic biomaterials.
Introduction
Mimicking nature with respect to the tissue molecular, biological, and dynamic environment, and the tissue compositional and morphological structure has been a widely adopted strategy and the ultimate goal in tissue engineering scaffolding design. Significant progress in bioconjugation,1–3 growth factor delivery,4 nanostructure fabrication,5 recombinant protein /matrix,6–8 self-assemble peptide,9, 10 and stem cell niche11, 12 has been made and are all within the scope of mimicking nature in tissue engineering. Bone scaffolding materials have been heavily studied in the past two decades, probably the most studied in tissue engineering. Examples include natural polymers such as polysaccharides (starch, alginate, chitosan, hyaluronic acid) and synthetic biodegradable polymers such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA), and polycaprolacton (PCL). It has long been recognized that hydroxyapatite (HA, Ca10(PO4)6(OH)2) consists of about 60 wt% of bone13 in composite with collagen.14.Thus, mixing polymers with HA or other related calcium phosphate (e.g. α-TCP and β-TCP) to form micro/nano-composites has been a conventional strategy to improve mechanical property and bioactivity of scaffolds with considerable success both in vitro and in vivo for bone tissue engineering. Examples of degradable polymers that are composited with HA or related calcium phosphate include poly(L-lactic acid), poly(D,L-lactic acid), polycaprolactone, and poly(propylene fumarate) (PPF).15 However, seeking bioactive bone tissue engineering materials and scaffolds seems still to be an endless on-going effort and a challenge due to the unsatisfactory osteoinductivity and osteointegration and significant inflammatory responses of the existing bone biomaterials in vivo.
By carefully reviewing literatures on natural bone compositions, it is clear that one important fact has been inadvertently overlooked in the design of bone biomaterials and scaffolds in the past. Natural bone is an organic-inorganic nanocomposite where thin nanocrystals of apatite are embedded in collagen. The small nanocrystals (3 nm thick) control the mechanical properties of bone and prevent crack propagation. Early in 1960s, it was found that citrate molecules consist of about 5 wt% of the organic components in bone, which was thought to regulate the stabilization of thin nanocrystal apatite.16 More recently, a careful solid-state nuclear magnetic resonance (NMR) study revealed that the surface of apatite nanocrystals is studded with strongly bound citrate molecules.17 Citrate is not a dissolved calcium-solubilizing agent but a strongly bound, integral part of the bone nanocomposite. Surprisingly, citrate was not even mentioned in most of the literatures in the past 30 years, not even those on bone biomaterials and scaffolds.
The natural existence of citrate in bone hints that citrate should be considered in bone biomaterial and scaffold design although its function on bone development is still largely unknown. A recent study on poly(diol citrate)/HA composites further support this strategy. Poly(diol citrate) is the first citrate-based biodegradable elastomer that holds promise for tissue engineering and other biomedical applications.18, 19 Use of poly (diol citrates) allows the formation of a degradable composite containing up to 60% of HA within it, while the conventional polylactide can only form a composite with up to 35% of HA. Poly(diol citrate)/HA demonstrated excellent osteoconductivity and induced rapid mineralization. Surprisingly, it elicited no chronic inflammatory response at the tissue-composite interface in vivo.19 It was inferred to that the rich –COOH from citrate units of poly(diol citrate) prompt calcium chelation thus facilitating poly(diol citrate)/HA interaction, enhancing mechanical properties, and promoting mineralization. Clear understanding on why poly(diol citrate)/HA exhibited excellent in vivo tissue/bone-compatibility is still unknown. Although poly(diol citrate)/HA was not developed under an hypothesis that the existence of citrate in bone may implicate the bone development, its excellent in vivo performance prompts further systematic study on the development of citrate-based orthopedic biomaterials.
With the goal of mimicking nature, it is our belief that citrate-based polymer/HA (or other related calcium phosphate) composite materials could constitute the next generation of bone tissue engineering and orthopedic biomaterials. Our lab has long been developing citrate-based biodegradable elastomers for versatile biomedical applications, including the recently developed crosslinked urethane-doped polyester (CUPE),20–22 poly(alkylene maleate citrate) (PAMC),23–25 and biodegradable photoluminescent polymer (BPLP).26, 27 With careful molecular design, these citrate-based polymers offer versatile processability and tunable mechanical, degradable, and optical properties. These versatile citrate-based biodegradable polymers form a platform for us to understand and custom-design biomaterials toward bone tissue engineering and orthopedic applications.
Recently, a significant attention has been placed on delivering various types of cells (autologous condrocytes, mesenchymal stem cells, and osteoblast) to treat irregularly shaped bone defect for bone regeneration. It has been recognized that in situ crosslinkable injectable hydrogels would be more beneficial for cell/drug deilvery in filling irregular defects as they can homogeneously fit into the void in situ prior to setting via a minimally invasive injection. However, most of the synthetic hydrogels lack of osteoconductivity. Therefore, injectable composites composed of injectable hydrogels with osteoconductive HA microparticles can be an alternative solutions. Very recently, few groups have started working on injectable hydrogel/HA composites for cell/drug delivery for orthopaedic applications using injectable polymers such as PPF15, Poly(ethylene glycol)-diacrylate28, Poly(2-hydroxyethyl methacrylate)29, poly(ethylene glycol)-poly(ε-caprolactone)-poly(ethylene glycol)30, and acrylamide-based polymers.31 It has been confirmed that HA microparticles within the hydrogels not only enhance the mechanical strengths but also serve as bioactive molecules to enhance cellular respones. However, none of these injectable composites considered the benefits of the aforementioned citrate effects in nature bone.
In the present study, a new bioactive citrate-based injectable biodegradable composite is developed for delivering cells to irregular bony defects. Given the aforementioned benefits of POC/HA used for orthopaedic applications, we have recently investigated and reported the synthesis and characterization of the first citrate-based water-soluble, in-situ crosslinkable, and biodegradable polymers, poly (ethylene glycol maleate citrate) (PEGMC) for cell delivery.24 These hydrogels were found to be elastic when compressed and demonstrated minimal cytotoxicity in vitro comparable to the FDA approved Poly (ethylene glycol) diacrylate (PEGDA) and completely degrade in vivo with minimal inflammatory responses.23, 32 Thus, an injectable composite derived by combining PEGMC and HA particles would have characteristics potentially ideal for injectable orthopaedic applications. The objective of this study is to evaluate the physicochemical properties, polymer and HA interaction, cytotoxicity, and injectability of CPEGMC/HA composites potentially serving as injectable materials for osteoblast delivery in orthopaedic applications.
Experimental Section
Materials
HA [Mw: 502.32, assay >90% (as Ca3(PO4)2); particle size: >75 μm (0.5%), 45–75 μm (1.4%), <45 μm (98.1%)] was purchased from Fluka (St Louis, MO, USA). All other chemicals were purchased from Sigma Aldrich (St Louis, MO), except where mentioned otherwise. All chemicals were used as received.
Synthesis and Preparation of CPEGMC/HA composite
PEGMC polymer was synthesized by a simple and controlled polycondensation reaction of poly(ethylene glycol) (PEG) (Mw=200), citric acid (CA) and maleic anhydride (MA) as reported previously23, 24. In a typical procedure, PEG, CA and MA were added to a 250 ml round-bottom flask fitted with an inlet and an outlet adapter. The flask was immersed in a silicon oil bath at 160°C at constant stirring under continuous flow of nitrogen. Following the melting of the monomers, the temperature of the oil bath was reduced to 140°C until the desired PEGMC polymer viscosity was achieved. To remove any unreacted monomers and small molecular weight oligomers, PEGMC was dissolved in deionized (DI) water and dialyzed (Spectrum, 500 Dalton cut-off) for 2 days with frequent changes of DI water, followed by freeze-drying.
The HA content of the PEGMC/HA composite was defined by the weight percentage of HA over the entire composite. Purified PEGMC polymer was dissolved in DI water to make a 30%(w/v) solution in a container (centrifuge tube or culture dish). The pH of the mixture was adjusted to 5.5 by adding sodium bicarbonate under continuous monitoring of pH. HA powder was then added to the polymer solution under thoroughly mixing. In the next step, 15%(w/v) of poly(ethylene glycol diacrylate) (PEGDA) (Mw=3.4K) was added as a crosslinker followed by the addition of an aqueous radical initiator ammonium persulfate (APS) and N,N,N',N'- tetramethylenediamine (TEMED) at 25mM concentration. The entire container was tightly sealed and then placed in an incubator at 37°C for ~15 minutes to obtain crosslinked poly(ethylene glycol maleate citrate)/HA (CPEGMC/HA) composites. The schematic of PEGMC synthesis and CPEGMC/HA crosslinking is shown in Scheme 1. Three different compositions of CPEGMC/HA were fabricated where the weight ratios of PEGMC:HA was varied from 70:30, 55:45 and 40:60 to yield CPEGMC/HA (30), CPEGMC/HA (45) and CPEGMC/HA (60) respectively.
Scheme.

A) Schematic representation of PEGMC synthesis and crosslinking; B) schematic representation of CPEGMC/HA cmposites. PEGMC is crosslinked using redox initiators APS/TEMED in the presence of HA and PEGDA to produce crosslinked PEGMC/HA network.
Characterization of CPEGMC/HA composites
The sol content, swelling ratio, and degradation of the CPEGMC/HA composites were investigated by a mass differential method. CPEGMC/HA composites with different HA composition were made as previously described. The CPEGMC/HA samples were cut into circular discs of dimensions 10 mm diameter by 2 mm thickness (n = 8). Samples were lyophilized for 48 hours to remove all water and weighed (Mi).
For the sol-gel fraction study, samples were immersed in 10 ml of 1,4-dioxane for 24 hours, freeze-dried and weighed (Md). Sol fraction (SF) of the hydrogel composites was determined by using following equation (1):
| (1) |
For the swelling study, the crosslinked composites were swollen in DI water to equilibrium swelling, blotted with Kimwipe paper, and weighed (Mw) and then lyophilized and weighed again (Md). The swelling ratio (SR) was calculated using the formula in equation (2)
| (2) |
For degradation study, CPEGMC/HA disks were incubated in phosphate buffer saline (PBS) (pH = 7.4) at 37° C. The buffer was changed regularly to ensure that the pH of the buffer was maintained at 7.4 during degradation. The initial weight of CPEGMC/HA discs was recorded as Mi. At pre-determined time points, the samples were removed from PBS, washed in DI water thrice to remove any residual salts and lyophilized and weighed (Mt). Degradation was determined by weight loss ratio as shown in equation (3).
| (3) |
Microstructural characterization of CPEGMC/HA composites
The microstructures of the CPEGMC/HA composites were examined using a Leica DMLP microscope (Leica Microsystems Inc., Bannockburn, IL) fitted with a Nikon E500 camera (Nikon Corp., Japan). Hydrated CPEGMC/HA samples were first embedded in OCT freezing media (Polysciences Inc., Warrington, PA), cryosectioned into 10 μm thick sections, and then photomicrographed to characterize the porous morphology of the composites and the distribution of HA in the composites.
Mechanical Properties of CPEGMC/HA composites
Un-confined compressive mechanical tests of CPEGMC/HA composites were studied under a MTS Insight 2 mechanical tester equipped with a 10 N load cell (MTS, Eden Prairie, MN). Compressive tests of CPEGMC/HA at two different conditions were conducted, where CPEGMC/HA was compressed immediately after crosslinking (as prepared) and after equilibrium swelling (fully hydrated). Briefly, the cylindrical shaped specimens of dimensions 10 mm by 10 mm (diameter × height) were compressed at a rate of 1mm/min to 50% strain. The initial modulus was calculated by measuring the gradient at 10% of compression of the stress-strain curve. Fully hydrated samples were subjected to cyclic compression to 20% strain for ten cycles and the hysteresis curves of load vs strain were plotted. The results were presented as mean ± standard deviation (n = 5). Cancellous bone cylinders removed from a femoral head of a domestic pig was also decellularized and subjected to compression tests as a control.
In vitro mineralization on CPEGMC/HA composites
Simulated body fluids (SBF) were prepared as described previously33–35 to study in vitro mineralization on the CPEGMC/HA composites. The SBF with inorganic ion concentration of five times of those of human blood plasma was denoted as SBF (5×). Disk shaped polymer scaffolds were immersed in 6-well plates containing SBF (5×) up to 7 days. The SBF was replaced every other day. After incubation for various periods of times, the specimens were washed carefully with DI water to remove any soluble inorganic ions, and dried in air and coated with silver prior to analyze under Hitachi 3000N scanning electron microscope (SEM) (Hitachi, Pleasanton, CA).
In vitro cell culture on CPEGMC/HA composites
CPEGMC and CPEGMC/HA films were cut into cylindrical discs (10 mm in diameter and 1mm in thickness). The samples were sterilized by soaking in 70% ethanol for 30 minutes, then exposing to UV light for 1 hour, and then washed with PBS for 3×5 minutes. Human fetal osteoblasts (hFOBs) (ATCC) were cultured according to ATCC protocol in phenol free Dulbecco's modified Eagle's medium (DMEM)- Ham's F12 1:1 medium supplemented with 10% fetal bovine serum (FBS) (HyClone) and geneticin (300 μg/ml; Sigma-Aldrich, St.Louise). The culture flasks were kept in an incubator maintained at 37°C, 5% CO2, and 95% relative humidity. The cells were trypsinized, centrifuged, and suspended into media to obtain a seeding density of 2×105 cells/ml on CPEGMC/HA films. After 48 hours of culture, the cells were fixed in 3%(v/v) of gluteraldehyde in PBS and sequentially dehydrated with a graded series of ethanol, lyophilized, and sputter coated with silver. The samples were then observed under SEM to view the morphology of the attached cells. In addition to SEM images, films seeded with hFOBs were stained with carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) (Invitrogen, Carlsbad, CA) green fluorescent cell tracer according to the manufacturer's protocol and viewed under a Zeiss Auxiovert inverted microscope (Carl Zeiss MicroImaging, Thornwood, NY).
Cytotoxicity of the composites was further evaluated by encapsulating hFOB cells within the networks of the CPEGMC/HA composites. Briefly, PEGMC solution was formulated as described in section 2.2. The solution was then sterilized via filtering through 0.22μm filter. hFOBs were added to polymer solution to achieve a final cell density of 15×106 cells/ml. After being held at 37°C for approximately 15min in an incubator, the cells/composite constructs were transferred to a 6 well plate and incubated in osteoblast media, During the 21-day culture period, the culture media was changed every 48 hr and the cell-encapsulated composites were taken out at week 1, 2 and 3 and stained using LIVE/DEAD assay (LIVE/DEAD Viability/Cytotoxicity Kit, Invitrogen, Carlsbad, CA). Cell viability and distribution were observed under a Zeiss Auxiovert inverted microscope (Carl Zeiss MicroImaging, Thornwood, NY).
For biochemical analysis, at various time points, the cell-cultured CPEGMC/HA constructs were removed from media and homogenized in PBS. 500 μl of 0.2% Triton-×100 solution was added to the constructs. The samples were subjected to two freeze-thaw cycles. The homogenates were then sonicated for 30 sec. The homogenates were centrifuged for 10 minutes at 4000 rpm. The supernatant after centrifuging was used to measure the DNA content of each construct using PicoGreen assay according to the manufacturer protocol. Measurements were performed in triplicate.36, 37
To characterize the alkaline phosphatase (ALP) activity within the composite, alkaline buffer solution was added to the constructs and sonicated to ensure that the constructs were completely homogenized. The lysate was centrifuged at 4000 rpm for 10 min at 4 °C. 100 μl of supernatant was incubated with 100 μl of p-nitrophenyl phosphate solution in a 96 well plate at 37°C for 1 hour. The reaction was stopped by adding 0.2M NaOH solution to each well. The production of p-nitrophenyl in the presence of ALP was measured by monitoring the light absorbance at 405 nm using a microplate reader (Infinite 200, Tecon Group Ltd, Switzerland).38
To quantify the calcium content within the composite, supernatant was collected and 50μl of the supernatant was added to a 96 well plate. The calcium concentration of the cell lysate was analyzed using cresolphthalein complexone (Sigma). After 10 minutes, the absorbance was read at 575 nm using the Tecan microplate reader (Infinite 200).38
Ex vivo injectability of CPEGMC/HA composite
Ex vivo study was conducted on a porcine femoral head. Femoral head was cored to create a cylindrical cavity. PEGMC/HA solution was injected into the cavity and incubated at 37°C for in-situ crosslinking. To visualize the in-situ formed composites, the femur head was further sectioned and photographed using a digital camera.
Statistical Analysis
Data was expressed as mean ± standard deviation. The statistical significance between two sets of data was calculated using oneway ANOVA. Data were taken to be significant, when p < 0.05 was obtained.
Results
Polymer synthesis, crosslinking and characterization of PEGMC/HA composite
PEGMC polymer was synthesized by a simple, one pot, and controlled polycondensation reaction from maleic anhydride, PEG, and citric acid. The polymer chains consist of degradable ester bonds, crosslinkable carbon-carbon double bonds, and pendant carboxyl and hydroxyl functional group as confirmed by previous FTIR and 1H-NMR.23. Due to these pendent carboxylic groups present throughout the polymeric network, it provide strong interaction between calcium ions of HA molecules. This was confirmed in figure 1,as preak maxima due to bond viberation of hydroxyl groups at 3570 cm−1 of the polymer shift to 3590 cm−1 as in the case of PEGMC/HA composite where as characteristic HA peaks (1049 cm−1 for PO−4) and degradable ester bonds (C=O at 1690–1750 cm−1 and C-O at 1000–1260 cm−1) are still evident at their respective wave number. PEGMC is completely soluble in water and can be homogeneously mixed with inorganic minerals such as hydroxyapatite without observable sedimentation before, during, and after crosslinking process. In our previous study, detail rheology study of PEGMC/HA composite demonstrated that the hydrogel composite precursor is highly injectable (via 27-gauge needle) with low viscosity even in higher (50%) HA content and can be crosslinked through radical polymerization aided by PEGDA crosslinkers within 3 to 15 minutes at 37° C to create mechanically stable hydrogel composite.32
Fig. 1.
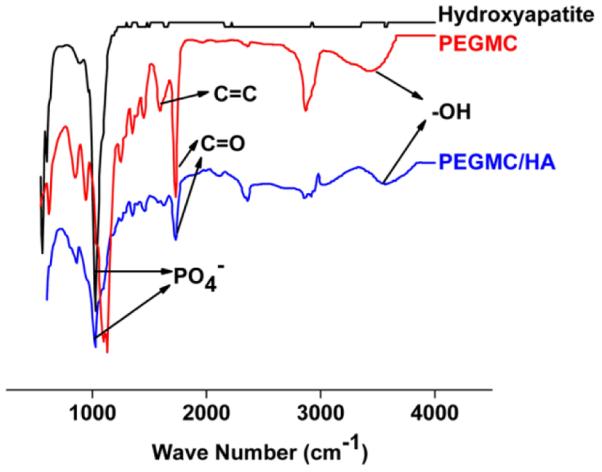
FTIR Spectra of HA powder, PEGMC hydrogel and CPEGMC/HA composite.
Fig. 2A showed the overall percentage of leachable (sol) content of the crosslinked composites. The sol content did not vary significantly with increasing HA concentration. For example, The sol content for CPEGMC/HA(30), CPEGMC/HA(45), and CPEGMC/HA(60) were 6.90 ± 0.74 %, 7.06 ± .28 %, and 5.66 ± 1.13 %, respectively. Fig. 2B showed the swelling kinetics of CPEGMC/HA containing various concentration of HA particles. These hydrogel composites started uptaking water and reached equilibrium swelling as early as 10 min and did not significantly increase swelling till 12 h. Inreasing the amount of HA in composites decreased swelling ratios. For instance, the swelling ratio reduced from 9.56 ± 0.56 (CPEGMC/HA(30)) to 7.89 ± 0.96 (CPEGMC/HA(45)) and 3.42 ± 0.66 (CPEGMC/HA(60)).
Fig. 2.
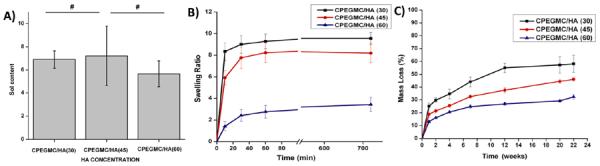
Physiochemical characterization of CPEGMC/HA. A) Degree of sol content as a function of HA concentration; B) Degree of swelling as a function of HA concentrations; C) In vitro mass loss of CPEGMC/HA in PBS (pH 7.4; 37°C) over time up to 22 weeks.
Degradation profile of CPEGMC/HA in PBS showed increasing mass losses with lowering concentrations of HA. As shown in Fig. 2C, CPEGMC/HA (30) showed a mass loss of 58.18 ± 6.60% at the end of 22 weeks. The high HA concentration CPEGMC/HA(60) showed a mass loss of 32.24 ± 1.79 % at the end of 22 weeks. This result indicated that the mass loss of the composites was primarily due to the degradation of the polymers and that HA particles did not leach out from the network in noticeable amounts during degradation. The composites maintained their integrity throughout the 22 weeks of degradation study.
Microstructure Characterization of CPEGMC/HA composites
The pore morphology of the fully hydrated CPEGMC/HA composite sections (5 μm) was observed under microscope. It was observed that the pure PEGMC gel network was highly porous with pore size of 200–400 μm (Fig. 3A). Multiple sections of the fully hydrated cylindrical hydrogels cut at various locations and angles were analyzed and observed with similar morphologies. This technique allowed us to visualized the interaction of the polymer and HA particles under fully hydrated conditions which is otherwise impossible to observed by SEM or cryo-SEM.28 The pore structure of the composites was not altered with the addition of HA particles. In fact while under a fully hydrated state, these mineral particles were distributed within the polymer matrix rather than simply encapsulated in the pores. As shown in Fig. 3B–D, the more HA was incorporated in the composite, the more HA could be observed within the polymer matrix.
Fig. 3.

Representative light microphotographs of macroporous CPEGMC hydrogel and CPEGMC/HA composite thin sections (10 μm). A) CPEGMC; B) CPEGMC/HA(30) composite; C) CPEGMC/HA(45) composite; and D) CPEGMC/HA(60) composite. The black dots denote the HA micro-particles within the polymer matrix. The white spaces represent void pore spaces.
Mechanical Properties of CPEGMC/HA composite
CPEGMC/HA composite with varying HA content (30, 45, 60)% were prepared and subjected to unconfined compressive tests as soon as it is freshly prepared (as-prepared) and after equilibrium swelling (fully hydrated). As shown in Fig. 4A–B, the modulus of as-prepared CPEGMC/HA(30)(10 ± 5 KPa) did not change significantly as compared to CPEGMC/HA(45)(26 ± 12 KPa). The modulus of CPEGMC/HA(30)(12.5 ± 5 KPa) and CPEGMC/HA(45)(30 ± 7 KPa) also showed no significant difference when tested after fully hydration. However, the modulus of CPEGMC/HA(60) (205 ± 26 KPa) was significantly increased compared to CPEGMC/HA(30) and CPEGMC/HA(45) and it also significantly decreased under fully hydrated state (100 ± 20 KPa). Good overlaps of stress–strain curves were observed when 10 consecutive compressive loading and unloading cycles with 20% strain on fully hydrated CPEGMC/HA samples (Fig. 4C). These mechanical results demonstrated that the CPEGMC/HA confers the constructs with excellent elastic property and the HA contributed to the increased strength. As shown in Fig. 4D, increasing crosslinking times also increases composite mechanical strength. The dried PEGMC/HA (60) demonstrated a close ultimate compressive strength to that of decellularized cancellous bone of femur head of porcine model.
Fig. 4.
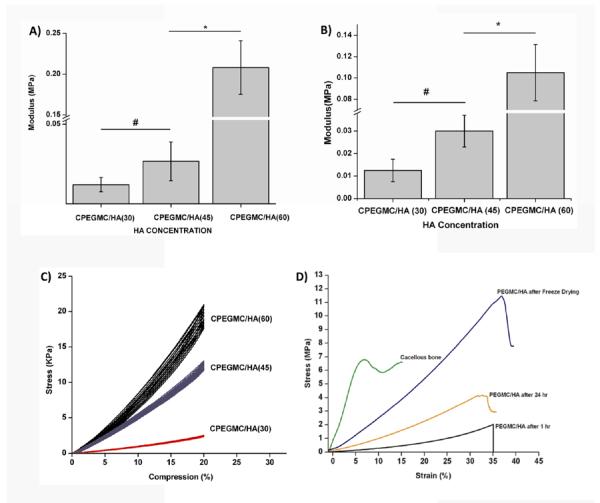
Mechanical tests of as-prepared and fully hydrated CPEGMC/HA composites A) Compressive modulus of as-prepared CPEGMC/HA composites; B) Compressive modulus of fully hydrated CPEGMC/HA composites; C) Cyclic compression tests of fully hydrated CPEGMC/HA composites. Ten consecutive cycles of controlled loading and unloading was applied to fully hydrated CPEGMC/HA specimens using MTS Insight II mechanical tester using a 10N load cell. D) Compression test of decellularized cancellous bone and synthetic hydrogel composite at different conditions.
In vitro mineralization
When incubated in simulated body fluid, by day 1, homogenoeus nucleation and 2-dimention growth of calcium phosphate was evident as mineral nodules appeared on the surface of CPEGMC/HA composite (Fig. 5A). By allowing mineralization to proceed for longer period of time (day 7), more distinct mineral nodules continuously covered the entire surface (several micron) of the CPEGMC/HA composite (Fig. 5B). Upon evaluation by the calibrated energy dispersive spectroscopy (EDS), the apatite nodules on the surface of the composite revealed a Ca/P ratio (1.6 ± 0.1) similar to that of synthetic HA.
Fig. 5.

Scanning electron microscope images of in vitro mineralization of CPEGMC/HA composite. A) CPEGMC/HA (45) composite when incubated in 5× SBF for 1 day; B) CPEGMC/HA(45), C) CPEGMC/HA(30), and D) CPEGMC/HA(60) composites when incubated in 5× SBF for 7 day.
In vitro cell culture
hFOB seeded on the surface of the PEGMC/HA composites support cell adhesion and promote cell spreading throughout the surface over the experimental period of 48 hrs when observed under fluorescent microscope and SEM (Fig. 6A and B).
Fig. 6.
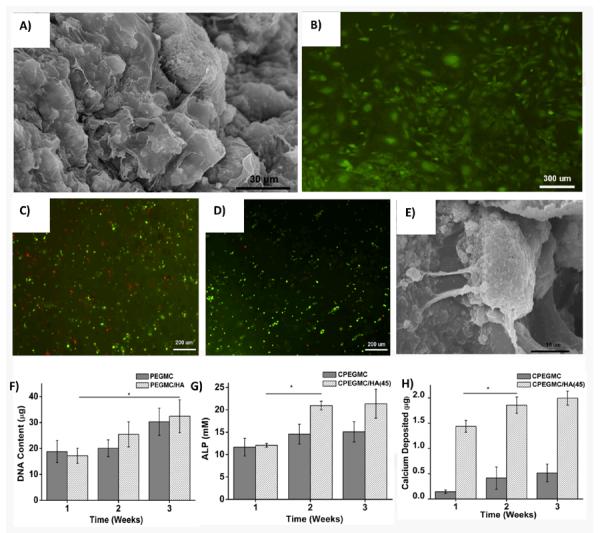
hFOBs attachment on the surface of CPEGMC/HA (45) films 48 hours post-seeding. A) SEM image of hFOB-seeded PEGMC/HA(45) composite and B) Microphotograph of hFOBs on CPEGMC/HA(45) composite stained with CFDA-SE flourecent dye. hFOBs encapsulated in CPEGMC/HA(45) composites. C) Live/Dead stain of hFOBs encapsulated in CPEGMC/HA(45) after 1 day culture; D)) Live/Dead stain of hFOBs encapsulated in CPEGMC/HA(45) after 21 day culture; Live cells are green fluorescent and dead cells are red fluorescent; E) Crossectional SEM image of hFOBs encapsulated within the matrix; F) DNA content assay; G) alkaline phosphatase (ALP) assay; and H) Calcium assay. In F-H, hFOBs were encapsulated in CPEGMC and CPEGMC/HA(45).
hFOB cells were further encapsulated within the CPEGMC/HA hydrogel composites to test their potential for cell encapsulation/delivery. It was found that the cells survived the encapsulation procedure and proliferated during the 3 weeks of subculture. Fluorescent images of the encapsulated osteoblasts stained by LIVE/DEAD assay at day 1 and day 21 of encapsulation are shown in Fig. 6C and D, where red fluorescence denotes dead cells and green fluorescence indicates live cells. In Fig. 6D, majority of hFOB cells were green indicating that CPEGMC/HA polymer network provided a cytocompatible environment for hFOB delivery. Only few cells died upon crosslinking conditions. Furthermore, the images also indicated a uniform cell distribution in the composites. hFOBs could adhere and spread on CPEGMC/HA composites, which could be clearly seen in Fig. 6E.
Deoxyribonucleic acid (DNA) content of hydrogels containing osteoblasts was assessed using the PicoGreen assay and the results were shown in Fig. 6F. DNA content increased from week 1 to week 3 demonstrating cells were proliferating. No significant difference was found from both pure PEGMC group and CPEGMC/HA group at each time points. As shown in Fig. 6G, the ALP production of the encapsulated hFOBs in CPEGMC/HA showed significant increased after two weeks. In contrast, the ALP production on pure CPEGMC did not show significant changes within 3 week of culture. The calcium content deposited by the hFOBs in CPEGMC/HA was significantly higher as compared to that of pure CPEGMC and CPEGMC/HA composite without cells. The significant increase was observed at 2nd week (Fig. 6H).
Ex vivo study
An ex vivo in-situ delivery study was conducted on a porcine femoral head (Fig. 7A–D). PEGMC/HA could be easily injected in the defect in the central region of the femur head using syringe fitted with biopsy cannula. No leakage was observed prior to crosslinking. Visual examination of the polymerization site after sectioning the femoral head suggested that the in-situ crosslinked CPEGMC/HA completely filled up the irregular implantation site and reinforced the femoral head.
Fig. 7.
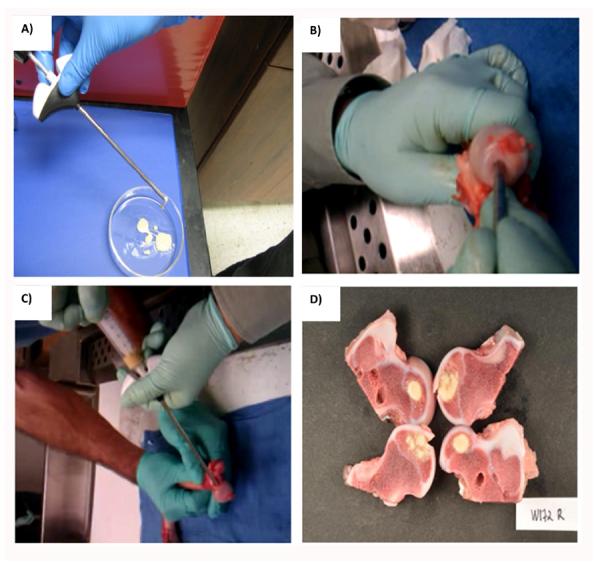
Ex vivo study demonstrates that biodegradable injectable PEGMC/HA(45) can be injected into collapsed femoral head for reinforcement. A) PEGMC/HA(45) solution loaded in cannula injection tool; B) The cored femoral head with a cylindrical cavity; C) PEGMC/HA(45) being injected into collapsed femoral head; and D) The sectional view of cemented femoral head with CPEGMC/HA(45) composite. Crosslinking was achieved within 5 minutes of injection.
Discussion
Hydroxyapatite (HA, Ca10(PO4)6(OH)2), consisting of around 60 wt% of bone, has long been recognized as a crucial biomaterial to design a tissue-engineered bone substitutes.13 It has been confirmed that HA and related calcium phosphate (e.g. α-TCP and β-TCP) not only enhances mechanical properties but also plays a critical role in the osteoconduction and osseointegration of the implanted bone graft.39, 40 Thus, developing composite materials based on biodegradable polymers and HA became an intense focus in bone tissue engineering. Such composites take advantage of the formability of polymers and the bioactivity of HA to enhance the mechanical properties of the fabricated scaffolds. In the meantime, the poor bioactivity of most of synthetic polymers can be improved.41 Examples of degradable polymers compositing with HA or related calcium phosphate include poly(L-lactic acid), poly(D,L-lactic acid), polycaprolactone, and poly(propylene fumarate).15
These prefabricated polymer composites may be excellent choice for critical bone defect, however, may not be applicable in the case of irregular bone defects, as in the case of femoral head osteonecrosis where complete evacuation of the necrotic tissue is performed by core decompression technique. It is desirable to use an injectable scaffolding system that can perfectly fill the void, solidifies within the site of injection, and induce/promote bone tissue regeneration. In fact, hydrogels based upon materials such as gelatin,42 poly(propylene fumarate),15 polymethylmethacrylate,43 and hydroxypropylmethyl cellulose44 have been combined with HA particles as a non-invasive injectable composite system for bone tissue replacement.
Herein, we reported a new elastomeric injectable PEGMC/HA composite for bone cell delivery. The water soluble PEGMCs would be a good choice of biomaterial for injectable application, as it has demonstrated excellent injectability, in situ crosslinkability, presence of pendant functional groups, elastic mechanical properties, controlled degradability, and excellent cytocompatibility both in vitro and in vivo.
In a previous study, we have systematically studied the viscoelastic properties of newly developed citric acid-based injectable biodegradable PEGMC/HA composites via rheology study. It was demonstrated that the intrinsic affinity of the pendent carboxyl group in the bulk of crosslinked PEGMC (CPEGMC) matrix to the HA particles provide a strong interfaces between the organic and inorganic components, a mimic to the natural citrate-bound apatite crystals in bone. The dispersion of HA particles within the matrix of PEGMC have very little effects on the gelation kinetics of chemically crosslinked gels but help to improve the elastic moduli due to ionic interactions between HA molecules and pendant –COOH groups from the citrate units of the polymer.32 Consequently, this interaction provides even dispersion of HA particle throughout the polymer matrix and futher avoid rapid material degradation.
One of the prime concern regarding injectables is the leachable (sol content) component following the solidification. Especially on the hydrogel/particles composites, tendency of leaching of particles in a fully hydrated condition is high. As demonstrated earlier, PEGMCs were found to have lower sol content after crosslinking (<15 w/w%).23 Similar results were found with our CPEGMC/HA composites. In fact, increase in HA component did not show any significant increase in leachable components from the system. HA particles were also found to be homogeneously dispersed within the matrix without any settling by gravity during solidification. Excellent structural integration of the organic and inorganic components was evident FTIR spectroscopy (Fig. 1) and morphology analysis (Fig. 2). Further, no evident amount of HA particles were leached out from the composite for 22 week of incubation in PBS during degradation study. Another crucial parameter to be address while designing bone scaffold is its porosity.45 Cancellous bone, where osteonecrosis occurs, is a highly porous environment with 50–90% porosity with average pore size of 10–400 um to facilitate nutrient exchange and osteoblast cellular infusion.46 Interestingly, CPEGMC/HA composites were found to be highly porous with open and interconnected pores within the range of 200 to 400 um as similar to other hydrogel systems. Increase in HA content did not influence pore formation within the formulations were investigated. Rather, the particles distributed evenly within the polymer matrices.
In our previous study, redox crosslinked PEGMC hydrogel degraded almost 70% of its mass by the end of one month in PBS at 37 °C.23 Interestingly, even at the end of 22 weeks of in vitro degradation study, the CPEGMC/HA composites were found to maintain their structural integrity without any noticeable amount of HA release from the system. It was inferred that the residual polymer network could still well chelate with HA particles to maintain the integrity of the composites. Morphology change of the composites at longer degradation times need to be studied in the future. Nonetheless, CPEGMC/HA maintained excellent integrity during degradation, a favourable factor when used for tissue engineering applications. The degradation of CPEGMC/HA networks in vivo will be reported in our future studies as others have suggested that local enzymes and biological environments may impact materials degradation in vivo.47–52
In general, polymer themselves have poor osteoconductivity. Therefore, incorporation of HA into polymers to induce mineralization has been a common way in designing bone tissue engineering composite biomaterials.53, 54 It was reported that the apatite formed as a result of biomimetic process could have biological properties that are more similar to natural bone mineral than that of synthetic calcium phosphate bioceramics and provide a more conducive environment for bone formation38, 55, 56 and differentiation of mesenchymal cells to osteoblasts. Studies have shown that osteoblasts seeded on PLGA/HA scaffolds covered with a bone like apatite coating boost cell growth, ALP activity, and mineralization. Thus, bone like apatite may provide a favourable environment for osteoblast attachment and growth.57 Inducing mineralization is one of the important property that should be inherit by bone scaffolds. Various strategies have been employed in biomaterials to demonstrate template-driven biomineralization of the system. For example, urea mediated surface hydrolysis was performed on Poly(2-hydroxyethyl methacrylate) (pHEMA) to create pandent carboxylic group with an aim to create high mineral content hydrogel14. Our CPEGMC/HA is saturated with carboxylic acid groups from citrate molecules of CEPGMC that not only provide strong HA cleation within the network but also promote high affinity nucleation and growth of calcium phosphate in simulated body fluid. After 7 days of incubation, SEM micrographs indicated a robust surface mineral layer due to apatite deposition covering entire surface of the composite. The induced apatite crystals on the composite hydrogel demonstrated typical `cauliflower'. These data clearly demonstrated that PEGMC/HA composite is a promising candidate for bone tissue engineering with high affinity for calcium ions.
Material stiffness has been reported to play an important role in adhesion, proliferation, and differentiation of cells seeded/encapsulated on/in biomaterials. Studies showed that matrix with stiffness of 20–110 kPa could promote differentiation of mesenchymal stem cells (MSCs) into osteoblasts.58 Thus, we formulated hydrogel composites mimicking similar stiffness. The as-prepared CPEGMC/HA composites exhibited stiffness within 110KPa. Given the results, our composites injected into bone defects should exhibit favourable mechanical stiffness conducive for bone differentiation. It is also noteworthy that the complete hydration did not sacrifice the ability of CPEGMC/HA composites to withstand cyclic compression with 20% compressive strain. Furthermore, the mechanical properties of CPEGMC/HA composite can be further tuned by adjusting the polymer concentrations and crosslinking times.
In addition to surface mineralization, PEGMC/HA could also serve as a template for surface cell adhesion and proliferation. Upon implantation, it is extremely important for scaffolds to encourage cell adhesion and tissue infiltration within the scaffold that helps to improve tissue-material integration. Further, we encapsulated osteoblast within the matrix of PEGMC/HA composite and evaluated their functionalities for a 3-week period. Although minimal cytotoxicity was expected during the process of encapsulation, viable cells within CEPGMC/HA proliferated and displayed characteristic osteoblastic functionality in bone formation. ALP, an ectoenzyme usually produced in the early stages of osteoblastic differentiation,59 was found to be significantly increased by week 2, in agreement with the increase of calcium deposition at week 2 and week 3 of the subculture. In review of literatures, it was rarely reported that cells could be encapsulated in injectable polymer/HA composites. Most of the cell encapsulation carriers are pure hydrogels with or without bioconjugation of biological molecules.60, 61 The presence of HA within injectable PEGMC/HA composites favoured ALP production and calcium deposition as compared to CPEGMC gels alone.
To demonstrate the injectable feasibility of PEGMC/HA, an exvivo study was conducted on porcine femoral head after the removal of the cancellous bone from its central region. The experiment clearly showed that the composite precursor can be easily injected and crosslinked in situ after completely filing the entire void space of collapsed femoral head prior to crosslinking. PEGMC/HA with HA concentration up to 60% was still injectable and crosslinkable in situ. Once fully filled with PEGMC/HA, the pocketed femoral head could be reinforced without collapse.
Conclusion
In summary, an injectable citrate-based PEGMC/HA hydrogel composite was developed. PEGMC/HA demonstrated highly porous micro-architecture, tuneable mechanical and degradable properties, and exhibited excellent osteoconductivity supported by increased ALP production and calcium deposition by the cells seeded/encapsulated on/into the composites. PEGMC/HA with high concentration of HA (up to 60%) could be injected into porcine femoral head bone defect and reinforced it within a short crosslinking time. By mimicking the citrate-bound apatite crystals, PEGMC/HA was the first citrate-based injectable composite scaffold with great potential serving as a cell delivery carrier for bone tissue engineering.
The discovery of citrate-bound apatite crystal in natural bone left many unanswered questions.17 For examples, where is the citrate from in bone? When does the citrate come into play for bone development? How does the abundance of citrate on crystal surface influence on bone formation? These questions can be translated into questions in bone biomaterial design. How much citrate should be placed in bone biomaterials? How long and when should the citrate exist in biomaterials? Although our current study does not answer the above questions, it constitutes an initial step of developing citrate-based injectable biomaterial platform to study the above questions. The development of injectable PEGMC/HA also expands the repertoire of the existing orthopaedic biomaterials.
Acknowledgement
This work was supported in part by a R01 award EB012575 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB), a National Science Foundation (NSF) CAREER award 0954109, and small research funding by the Texas Scottish Rite Hospital for Children.
Notes and references
- 1.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 2.Elisseeff J, Yang F, Williams CG, Wang DA, Lee H, Manson PN. Biomaterials. 2005;26:5991–5998. doi: 10.1016/j.biomaterials.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Mikos AG, Shin H, Jo S. Biomaterials. 2003;24:4353–4364. doi: 10.1016/s0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 4.Mikos AG, Babensee JE, McIntire LV. Pharm Res. 2000;17:497–504. doi: 10.1023/a:1007502828372. [DOI] [PubMed] [Google Scholar]
- 5.Wei GB, Ma PX. Adv Funct Mater. 2008;18:3568–3582. doi: 10.1002/adfm.200800662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddi AH, Nakashima M. Nat Biotechnol. 2003;21:1025–1032. doi: 10.1038/nbt864. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan DL, Li CM, Vepari C, Jin HJ, Kim HJ. Biomaterials. 2006;27:3115–3124. doi: 10.1016/j.biomaterials.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Tirrell DA, Maskarinec SA. Curr Opin Biotech. 2005;16:422–426. doi: 10.1016/j.copbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Kisiday J, Jin M, Kurz B, Hung H, Semino C, Grodzinsky AJ. Proc Natl Acad Sci U S A. 2002;99:9996–10001. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stupp SI, Guler MO, Hsu L, Soukasene S, Harrington DA, Hulvat JF. Biomacromolecules. 2006;7:1855–1863. doi: 10.1021/bm060161g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vacanti JP, Khademhosseini A, Langer R, Borenstein J. Proc Natl Acad Sci U S A. 2006;103:2480–2487. doi: 10.1073/pnas.0507681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter JR, Ruckh TT, Popat KC. Biotechnol Prog. 2009;25:1539–1560. doi: 10.1002/btpr.246. [DOI] [PubMed] [Google Scholar]
- 14.Song J, Saiz E, Bertozzi CR. J Am Chem Soc. 2003;125:1236–1243. doi: 10.1021/ja028559h. [DOI] [PubMed] [Google Scholar]
- 15.Lee KW, Wang S, Yaszemski MJ, Lu L. Biomaterials. 2008;29:2839–2848. doi: 10.1016/j.biomaterials.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartles RL. Adv Oral Biol. 1964;1:225–253. doi: 10.1016/b978-1-4832-3117-4.50014-0. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt-Rohr K, Hu YY, Rawal A. Proc Natl Acad Sci U S A. 2010;107:22425–22429. doi: 10.1073/pnas.1009219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Webb AR, Pickerill SJ, Hageman G, Ameer GA. Biomaterials. 2006;27:1889–1898. doi: 10.1016/j.biomaterials.2005.05.106. [DOI] [PubMed] [Google Scholar]
- 19.Qiu H, Yang J, Kodali P, Koh J, Ameer GA. Biomaterials. 2006;27:5845–5854. doi: 10.1016/j.biomaterials.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 20.Dey J, Xu H, Shen J, Thevenot P, Gondi SR, Nguyen KT, Sumerlin BS, Tang L, Yang J. Biomaterials. 2008;29:4637–4649. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dey J, Xu H, Nguyen KT, Yang J. J Biomed Mater Res A. 2010;95:361–370. doi: 10.1002/jbm.a.32846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dey J, Tran RT, Shen J, Tang L, Yang J. Macromol Mater Eng. 2011;296 doi: 10.1002/mame.201100074. DOI:10.1002/mame.201100074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gyawali D, Nair P, Zhang Y, Tran RT, Zhang C, Samchukov M, Makarov M, Kim HKW, Yang J. Biomaterials. 2010;31:9092–9105. doi: 10.1016/j.biomaterials.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gyawali D, Tran RT, Guleserian KJ, Tang L, Yang J. J Biomater Sci Polym Ed. 2010;21:1761–1782. doi: 10.1163/092050609X12567178204169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran RT, thevenot P, Gyawali D, Zhang Y, Yang J. Soft Matter. 2010;6:2449–2461. doi: 10.1039/C001605E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Zhang Y, Gautam S, Liu L, Dey J, Chen W, Mason RP, Serrano CA, Schug KA, Tang L. Proc Natl Acad Sci U S A. 2009;106:10086–10091. doi: 10.1073/pnas.0900004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrano CA, Zhang Y, Yang J, Schug KA. Rapid Commun Mass Sp. 2011;25:1152–1158. doi: 10.1002/rcm.4974. [DOI] [PubMed] [Google Scholar]
- 28.Gaharwar AK, Dammu SA, Canter JM, Wu CJ, Schmidt G. Biomacromolecules. 2011;12:1641–1650. doi: 10.1021/bm200027z. [DOI] [PubMed] [Google Scholar]
- 29.Song J, Xu JW, Filion T, Saiz E, Tomsia AP, Lian JB, Stein GS, Ayers DC, Bertozzi CR. J Biomed Mater Res A. 2009;89A:1098–1107. doi: 10.1002/jbm.a.32110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu SZ, Gun G, Gong CY, Zeng S, Liang H, Luo F, Zhang XN, Zhao X, Wei YQ, Qian ZY. J Phys Chem B. 2009;113:16518–16525. doi: 10.1021/jp907974d. [DOI] [PubMed] [Google Scholar]
- 31.Demirtas TT, Karakecili AG, Gumusderelioglu M. J Mater Sci Mater Med. 2008;19:729–735. doi: 10.1007/s10856-007-3008-7. [DOI] [PubMed] [Google Scholar]
- 32.Jiao Y, Gyawali D, Stark JM, Akcora P, Nair P, Tran RT, Yang J. Soft Matter. 2012;8:1499–1507. doi: 10.1039/C1SM05786C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tas C. Biomaterials. 2000;21:1429–1438. doi: 10.1016/s0142-9612(00)00019-3. [DOI] [PubMed] [Google Scholar]
- 34.Oyane A, Kim H, Furuya T, Kokubo T, Miyazaki T, Nakamura T. J Biomed Mater Res A. 2003;65:188–195. doi: 10.1002/jbm.a.10482. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, Sun Park M, Jeon O, Yong Choi C, Kim B. Biomaterials. 2006;27:1399–1409. doi: 10.1016/j.biomaterials.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Wang D, Williams C, Yang F, Cher N, Lee H, Elisseeff J. Tissue Eng. 2005;11:201–213. doi: 10.1089/ten.2005.11.201. [DOI] [PubMed] [Google Scholar]
- 37.Burdick J, Anseth K. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Park M, Gwak S, Choi C, Kim B. Tissue Eng. 2006;12:2997–3006. doi: 10.1089/ten.2006.12.2997. [DOI] [PubMed] [Google Scholar]
- 39.Khanna R, Katti KS, Katti DR. Acta Biomater. 7:1173–1183. doi: 10.1016/j.actbio.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 40.Yoshikawa T, Ohgushi H, Tamai S. J Biomed Mater Res. 1996;32:481–492. doi: 10.1002/(SICI)1097-4636(199611)32:3<481::AID-JBM23>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 41.Weiner S, Wagner H. Annu Rev Mat Sci. 1998;28:271–298. [Google Scholar]
- 42.Hillig WB, Choi Y, Murthy S, Natravali N, Ajayan P. J Mater Sci Mater Med. 2008;19:11–17. doi: 10.1007/s10856-007-0154-x. [DOI] [PubMed] [Google Scholar]
- 43.Moursi AM, Winnard AV, Winnard PL, Lannutti JJ, Seghi RR. Biomaterials. 2002;23:133–144. doi: 10.1016/s0142-9612(01)00088-6. [DOI] [PubMed] [Google Scholar]
- 44.Weiss P, Bohic S, Lapkowski M, Daculsi G. J Biomed Mater Res. 1998;41:167–170. doi: 10.1002/(sici)1097-4636(199807)41:1<167::aid-jbm20>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 45.Karageorgiou V, Kaplan D. Biomaterials. 2005;26:5474–5491. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 46.Dadsetan M, Hefferan T, Szatkowski J, Mishra P, Macura S, Lu L, Yaszemski M. Biomaterials. 2008;29:2193–2202. doi: 10.1016/j.biomaterials.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peter SJ, Miller ST, Zhu G, Yasko AW, Mikos AG. J Biomed Mater Res A. 1998;41:1–7. doi: 10.1002/(sici)1097-4636(199807)41:1<1::aid-jbm1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 48.Tracy M, Ward K, Firouzabadian L, Wang Y, Dong N, Qian R, Zhang Y. Biomaterials. 1999;20:1057–1062. doi: 10.1016/s0142-9612(99)00002-2. [DOI] [PubMed] [Google Scholar]
- 49.Suuronen R, Pohjonen T, Hietanen J, Lindqvist C. J Oral Maxil Surg. 1998;56:604–614. doi: 10.1016/s0278-2391(98)90461-x. [DOI] [PubMed] [Google Scholar]
- 50.Smith R, Oliver C, Williams D. J Biomed Mater Res. 1987;21:991–1003. doi: 10.1002/jbm.820210805. [DOI] [PubMed] [Google Scholar]
- 51.Williams D. Clinical materials. 1992;10:9–12. doi: 10.1016/0267-6605(92)90078-8. [DOI] [PubMed] [Google Scholar]
- 52.Timmer M, Shin H, Horch R, Ambrose C, Mikos A. Biomacromolecules. 2003;4:1026–1033. doi: 10.1021/bm0300150. [DOI] [PubMed] [Google Scholar]
- 53.Kikuchi M, Itoh S, Ichinose S, Shinomiya K, Tanaka J. Biomaterials. 2001;22:1705–1711. doi: 10.1016/s0142-9612(00)00305-7. [DOI] [PubMed] [Google Scholar]
- 54.Chou YF, Chiou WA, Xu Y, Dunn JCY, Wu BM. Biomaterials. 2004;25:5323–5331. doi: 10.1016/j.biomaterials.2003.12.037. [DOI] [PubMed] [Google Scholar]
- 55.Kalita S, Rokusek D, Bose S, Hosick H, Bandyopadhyay A. J Biomed Mater Res A. 2004;71:35–44. doi: 10.1002/jbm.a.30118. [DOI] [PubMed] [Google Scholar]
- 56.Lickorish D, Ramshaw JAM, Werkmeister JA, Glattauer V, Howlett CR. J Biomed Mater Res A. 2004;68:19–27. doi: 10.1002/jbm.a.20031. [DOI] [PubMed] [Google Scholar]
- 57.Loty C, Sautier JM, Boulekbache H, Kokubo T, Kim HM, Forest N. J Biomed Mater Res A. 2000;49:423–434. doi: 10.1002/(sici)1097-4636(20000315)49:4<423::aid-jbm1>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 58.Kong HJ, Polte TR, Alsberg E, Mooney DJ. Proc Natl Acad Sci U S A. 2005;102:4300–4305. doi: 10.1073/pnas.0405873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Temenoff J, Park H, Jabbari E, Sheffield T, LeBaron R, Ambrose C, Mikos A. J Biomed Mater Res. 2004;70:235–244. doi: 10.1002/jbm.a.30064. [DOI] [PubMed] [Google Scholar]
- 60.Nicodemus G, Bryant S. Tissue Eng Part B Rev. 2008;14:149–165. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benoit D, Durney A, Anseth K. Tissue Eng. 2006;12:1663–1673. doi: 10.1089/ten.2006.12.1663. [DOI] [PubMed] [Google Scholar]