

Abstract
Seabuckthorn ( Hippophae rhamnoides L.) is known for its medicinal, nutritional and environmental importance since ancient times. However, very limited efforts have been made to characterize the genome and transcriptome of this wonder plant. Here, we report the use of next generation massive parallel sequencing technology (Illumina platform) and de novo assembly to gain a comprehensive view of the seabuckthorn transcriptome. We assembled 86,253,874 high quality short reads using six assembly tools. At our hand, assembly of non-redundant short reads following a two-step procedure was found to be the best considering various assembly quality parameters. Initially, ABySS tool was used following an additive k-mer approach. The assembled transcripts were subsequently subjected to TGICL suite. Finally, de novo short read assembly yielded 88,297 transcripts (> 100 bp), representing about 53 Mb of seabuckthorn transcriptome. The average length of transcripts was 610 bp, N50 length 1198 BP and 91% of the short reads uniquely mapped back to seabuckthorn transcriptome. A total of 41,340 (46.8%) transcripts showed significant similarity with sequences present in nr protein databases of NCBI (E-value < 1E-06). We also screened the assembled transcripts for the presence of transcription factors and simple sequence repeats. Our strategy involving the use of short read assembler (ABySS) followed by TGICL will be useful for the researchers working with a non-model organism’s transcriptome in terms of saving time and reducing complexity in data management. The seabuckthorn transcriptome data generated here provide a valuable resource for gene discovery and development of functional molecular markers.
Introduction
Seabuckthorn ( Hippophae rhamnoides L.) is a hardy, deciduous shrub of family Elaeagnaceae. The plant is a wind pollinated dioecious with a diploid chromosome number of 24 [1]. Its natural habitat extends widely across Europe and Asia in countries like China, Mongolia, India, Russia, Sweden, Finland and Norway [2,3]. In India, seabuckthorn is generally found growing in hilly tracks of Ladakh in Jammu and Kashmir, Lahaul-Spiti in Himachal Pradesh and parts of Uttarakhand, Arunachal Pradesh and Sikkim [4]. All parts of the seabuckthorn plant are known for their medicinal value. Seabuckthorn has been used in traditional medicinal systems in Tibet, Mongolia, Uzbekistan, Pakistan, Turkey, China, Russia and India to treat bowel irregularities, gastric ulcers, skin infection/wounds, influenza infections, cough and cold [5–8]. Apart from medicinal importance, seabuckthorn is also popular for its ornamental and environmental importance. It has long been recognized as an important plant for stabilization of mobile sand dunes [9]. The plant is known to harbor Frankia , a nitrogen fixing actinomycetes, in its roots and therefore has been used in land reclamation [10]. Due to its high nutritional and medicinal value, seabuckthorn has attracted focus of agricultural scientists for domestication and breeding programmes. Surprisingly, limited information about seabuckthorn genome and transcriptome is available in public domain. This is reflected by repository of a small number of seabuckthorn ESTs (3412) in the dbEST of NCBI [11] and a solitary recent report of 454 sequencing based transcriptome profiling of seabuckthorn berries [12].
During past three decades, Expressed Sequence Tags (ESTs) have played a significant role in gene discovery and gene function analysis, particularly for non-model organisms. ESTs generated from Sanger’s sequencing approach have longer read lengths facilitating easy assembly into longer consensus sequences for further downstream analysis. However, over the last few years, new high-throughput and cost effective sequencing technologies have become available which greatly outperform the standard Sanger technology in terms of massive data generation at a much reduced cost and labor. Ultra high-throughput RNA sequencing has allowed transcriptome analysis in several species and offered an attractive approach for qualitative and quantitative analysis of the whole transcriptome. Next Generation Sequencing (NGS) technologies are now increasingly been considered to be an alternative to microarrays [13] for analyzing differential gene expression also. Among NGS technologies, Roche-454 platform has been widely used in transcriptome sequencing of various non-model organisms [14,15] due to its ability to generate relatively long reads which greatly facilitates de novo assembly as compared to Illumina platform and ABI SOLiD system. Though advancement with regard to increase of read length in Illumina platform reads is underway, several new de novo assembly tools are being developed to assemble short sequence reads generated by these NGS platforms. However, performance of these assembly tools across various data sets has been investigated in a relatively few studies only [16–18].
Prior knowledge of the efficiency of different de novo short read assembly tools is useful while undertaking a project on transcriptome analysis in an unexplored species. In the present study, we have optimized de novo transcriptome assembly of short reads generated from Illumina HiSeq 2000 platform using six frequently used short read assemblers and following two different strategies to select the best transcriptome assembly obtained.
Materials and Methods
RNA Isolation, Illumina Sequencing and Quality Control
Seabuckthorn leaf and root tissues were harvested from the seedlings grown in a plant growth chamber as mentioned in Ghang et al. al. 2012 [11]. RNA was isolated as described previously by Ghang et al. al. 2009 [19]. RNA samples having more than 8.0 RIN (RNA Integrity Number) value were used for further processing. mRNA was purified from 3.0 µg of total RNA using oligo(dT) beads and fragmented to generate short mRNAs. Taking these short fragments as template, first strand cDNA was synthesized using random hexamer primers. Purified double stranded cDNAs containing sequencing adaptors were sequenced using Illumina HiSeq 2000. Construction of leaf and root cDNA libraries and their paired end sequencing was out sourced to a commercial service provider, Ocimum Biosolution Pvt. Ltd., Hyderabad. Various quality control checks on the short reads were performed using NGS QC Tool Kit [20].
Availability of Short Read Data
The Illumina short reads generated in this study have been submitted to NCBI’s Short Read Archive (SRA) with study accession number SRP011938, containing seabuckthorn leaf and root sample data under the accession numbers SRS304528 and SRS304529, respectively.
De novo Short Read Assembly
In this study, we systematically studied and compared the performance of six commonly used de novo short read assembly tools including Velvet [21], Oases [22], ABySS [23], SOAPdenovo [24], CLC Genomics Workbench (commercially available) and Trinity [25] following different approaches. Most of these assemblers are based on de Bruijn graphs wherein sequence reads are broken into smaller sequences of DNA, referred to as k-mers, where k denotes the length of these smaller sequences [26].
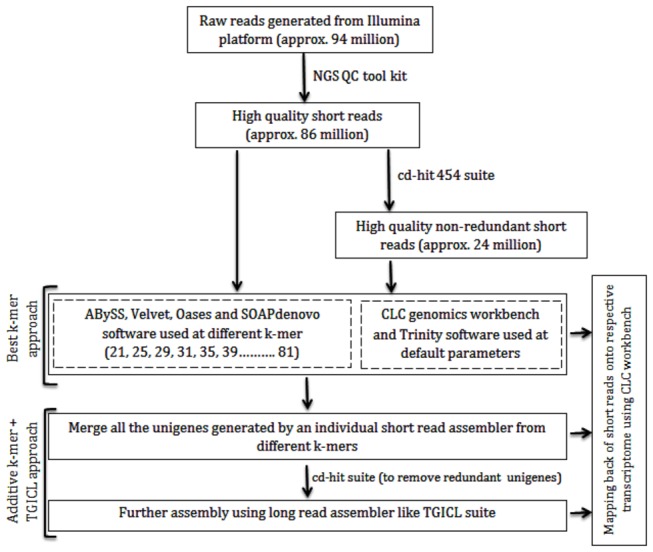
De novo transcriptome assembly was performed following two different approaches. The outline of the methodology employed in the present study has been shown in the flow chart (Figure 1). In the first approach (best k-mer strategy), redundant (approx. 86 million) and non-redundant (approx. 24 million) high quality short reads were assembled using short read assemblers mentioned above. The k-value ranged from 21 to 81 for Velvet, Oases, ABySS and SOAPdenovo, whereas CLC and Trinity softwares were run at default settings. The best k-mer assembly was identified on the basis of various assembly parameters. In the second approach (additive k-mer followed by TGICL), a two-step strategy was employed. Initially, contigs generated for all k-mer values by respective assembler were merged and redundant sequences were removed by cd-hit tool [27]. The non-redundant contigs thus generated were assembled again using TGICL suite [28], as TGICL suite is effective in assembly of long reads rather than short sequences generated by NGS platforms. To check the integrity of assembled transcriptome generated using various assembly tools and following different strategies, high quality short reads were mapped back onto respective assembled Unigenes using CLC workbench software and compared for total mapped back reads and unique mapped back reads. Total mapped back reads depict the number of reads involved in the formation of Unigenes , while uniquely mapped back reads signifies the number of short reads, each showing single target in the assembled transcriptome. Ideally, assembly finalized for further annotation should have high percentage of total as well as uniquely mapped back reads. Various other parameters like total number of contigs (> 100 bp), N50 length and average contig length were also taken into consideration as a function of k-mer length to select the best possible transcriptome assembly. N50 length is a weighted median statistics such that 50% of the entire assembly is contained in contigs equal to or larger than this value. Since higher N50 length indicates better performance of the assembly tool, more weightage was given to N50 value rather than average read length wherever a marginal difference was present in average read length of compared assemblies.
Figure 1. Flow chart showing the strategy used for generation of de novo short read transcriptome assembly.
Similarity Search and Functional Annotation of Unigenes
Similarity search is one of the very first and easiest computational approaches for annotation of any transcriptome dataset. BLASTX module of the BLAST tool pack was employed for functional annotation because BLAST [29] is the most commonly used tool considering the statistical model applied for measuring the significance of local sequence similarities and computational speed. Only the best hit results were extracted and hits with an E-value <1E-06 were considered to be significant. Gene Ontology (GO) annotations for molecular function, biological process, and cellular component categories were based on the BLASTX hit from BLAST2GO suite [30].
In silico Mining for Simple Sequence Repeats and Transcription Factors
Seabuckthorn transcriptome was screened for the presence of microsatellites or simple sequence repeats (SSRs) using MISA (MIcroSAtellite) [31], a perl script, using default parameters. The number of repeating units taken in the present study was atleast ten for mono-nucleotides, six for di-nucleotides, five for tri-nucleotides, five each for tetra-, penta- and hexa-nucleotides. For the identification of transcription factors represented in seabuckthorn transcriptome, transcripts were searched against all the transcription factor protein sequences available at Plant Transcription Factor Database [32].
Results and Discussion
Next Generation (Illumina) Sequencing and Quality Control
Recent advances in NGS technologies have markedly increased the amount of data generated and significantly reduced the sequencing cost. Although continuous strive is on to achieve long read lengths, it is still shorter when compared to that of Sanger sequencing technology. However, the field is undergoing revolutionary changes providing gradual improvement and advancements towards development of a perfect transcriptome assembly. In this study, a total of 94,013,936 (approx. 94 million) raw short reads (91 bp) were obtained from seabuckthorn leaf and root tissue libraries using Illumina HiSeq 2000 platform. The short reads were first filtered using NGS QC toolkit to remove low-quality reads and those having primer/adaptor contaminations. This exercise yielded a total of 86,253,874 high quality paired end reads. Although generation of single end reads is much cheaper than paired end reads in NGS techniques, paired end reads are important for de novo assembly due to their linked nature [33]. A summary of short read data (raw and filtered) generated for seabuckthorn transcriptome in this study has been presented in Table 1. As evident from this tabulated summary, collectively for both the tissues, more than 90% of the actual short reads were retained as high quality reads signifying the use of good quality RNA for next generation sequencing.
Table 1. Summary of Illumina HiSeq 2000 short read data used for seabuckthorn transcriptome assembly.
| Data item | Leaf | Root |
|---|---|---|
| Total number of reads | 44791536 | 49222400 |
| Read length | 91 | 91 |
| Total number of HQ reads | 41067522 | 45186352 |
| Percentage of HQ reads | 91.69% | 91.80% |
| Total number of bases | 4031238240 (4.0 Gb) | 4430016000 (4.4 Gb) |
| Total number of bases in HQ reads | 3696076980 (3.7 Gb) | 4066771680 (4.0 Gb) |
| Total number of HQ bases in HQ reads | 3619124516 | 3985259341 |
| Percentage of HQ bases in HQ reads | 97.92% | 98% |
HQ stands for high quality obtained after running NGS QC toolkit on raw data
For short read assemblers that perform assembly as a function of k-mer, lower the k-value, higher is the computer’s RAM requirement. Thus for RAM intensive assembly tools, high quality reads obtained from Illumina platform (approx. 86 million) were fed to cdhit-454 suite to remove redundancy resulting in the generation of 24,405,576 (approx. 24 million) non-redundant short reads. The assembly obtained from this non-redundant dataset has been mentioned as NR in the figures and following text.
De novo Short Read Assembly
De novo assembly was performed following two strategies. In the first approach (best k-mer strategy), high quality reads (all approx. 86 million and non-redundant approx. 24 million) were assembled separately using Velvet, Oases, ABySS and SOAPdenovo programs at different k-mer lengths ranging from 21 to 81, whereas CLC Genomics Workbench and Trinity were used at default parameters (Table S1). A comparative analysis of assemblies generated by different assembly tools used in the present study revealed that best k-mer value varied for different tools. For example, in the case of Oases, decrease in k-mer value (at k=29-35) showed improved assembly statistics. On the other hand, in case of Velvet and SOAPde novo, increase in k-mer value (at k=69-79) led to the improvement in the generated assembly. Moreover, in case of ABySS the mid k-mer values were found to be the best as a gradual increase in assembly statistics up to k=55-59 (redundant sequences) and k=45-49 (non-redundant sequences) was observed that gradually decreased till end point k-mer (Table S1). However, on the basis of various parameters as a function of k-mer length as mentioned in material and methods, assembly generated by Oases at k=31 taking non-redundant short read dataset (NR-Oases) was considered to be the best among assemblies generated by first strategy (best k-mer strategy) with average read length of 794 bp and N50 read length of 1378 bp (Table 2; Table S1). Moreover, among all the short read assembler used in our study, Oases was found to be the most RAM intensive as about 512 GB RAM was required to perform assembly at k=21 using 86 million redundant high quality reads as an initial input.
Table 2. Comparison of different short read assemblers and strategies employed on the basis of various assembly parameters.
| Intial input reads |
Redundant short reads (86 million)
|
Non-redundant short reads (24 million)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Short Read Assembler | Velvet | Oases | ABySS | SOAP | CLC | Trinity | Velvet | Oases | ABySS | SOAP | CLC | Trinity | |
| Assembly Parameters | Best k-mer | (K=71) | (K=31) | (K=55) | (K=71) | Default | Default | (K=71) | (K=31) | (K=49) | (K=71) | Default | Default |
| Total number of Unigenes | 77022 | 62804 | 102826 | 97306 | 279673 | 240723 | 72302 | 55238 | 139923 | 83813 | 283777 | 234329 | |
| Maximum read length | 3293 | 8567 | 6650 | 3548 | 7070 | 14182 | 4182 | 8849 | 5001 | 3793 | 6257 | 9473 | |
| Average read length | 378 | 733 | 698 | 330 | 310 | 548 | 403 | 794 | 335 | 368 | 300 | 460 | |
| N50 read length | 454 | 1314 | 1144 | 429 | 486 | 1284 | 485 | 1378 | 482 | 474 | 434 | 791 | |
| Total map back reads (%) | 73.79 | 68.28 | 89.21 | 76.65 | 85.77 | 90.8 | 77.69 | 72.99 | 69.42 | 81.55 | 83.41 | 90.24 | |
| Unique map back reads (%) | 71.15 | 66.95 | 41.93 | 73.2 | 84.74 | 52.27 | 75.5 | 72.16 | 68.91 | 79.01 | 82.64 | 55.95 | |
| Short Read Assembler | Additive k-mer | Velvet | Oases | ABySS | SOAP | CLC | Trinity | Velvet | Oases | ABySS | SOAP | CLC | Trinity |
| Total number of Unigenes | 347958 | 115006 | 111040 | 314576 | 279673 | 240723 | 332830 | 97691 | 167892 | 295569 | 283777 | 234329 | |
| Maximum read length | 4668 | 10607 | 7659 | 4654 | 7070 | 14182 | 4931 | 10387 | 6552 | 5384 | 6257 | 9473 | |
| Average read length | 321 | 798 | 804 | 339 | 310 | 548 | 330 | 856 | 421 | 352 | 300 | 460 | |
| N50 read length | 419 | 1504 | 1317 | 458 | 486 | 1284 | 435 | 1533 | 647 | 484 | 434 | 791 | |
| Total map back reads (%) | 93.73 | 94.89 | 94.14 | 94.07 | 85.77 | 90.8 | 94.53 | 94.77 | 94.05 | 95.05 | 83.41 | 90.24 | |
| Unique map back reads (%) | 66.1 | 46.97 | 45.27 | 69.28 | 84.74 | 52.27 | 66.17 | 48.9 | 71.54 | 70.28 | 82.64 | 55.95 | |
| Short Read Assembler | Additive + TGICL | Velvet | Oases | ABySS | SOAP | CLC | Trinity | Velvet | Oases | ABySS | SOAP | CLC | Trinity |
| Total number of Unigenes | 228750 | 101055 | 85902 | 211863 | 270245 | 212093 | 219896 | 86124 | 88297 | 202713 | 274248 | 202089 | |
| Maximum read length | 7608 | 16199 | 14243 | 11102 | 9026 | 14182 | 9030 | 17565 | 10252 | 9015 | 6257 | 9474 | |
| Average read length | 374 | 871 | 930 | 396 | 317 | 475 | 385 | 932 | 610 | 407 | 307 | 444 | |
| N50 read length | 631 | 1606 | 1539 | 690 | 513 | 1035 | 658 | 1629 | 1198 | 725 | 456 | 766 | |
| Total map back reads (%) | 93.43 | 94.79 | 94.08 | 93.69 | 85.83 | 91.66 | 94.22 | 94.76 | 93.21 | 94.77 | 83.46 | 91.03 | |
| Unique map back reads (%) | 90.07 | 50.25 | 54.11 | 90.67 | 85.15 | 67.2 | 90.7 | 52.15 | 91.03 | 91.16 | 83.07 | 70.15 | |
In the second approach (additive k-mer followed by long read assembler TGICL), a two-step strategy was employed. In the first step, contigs obtained from all the k-mer of respective assembler were merged and redundant sequences removed. In the second step, non-redundant contigs thus obtained in the above step were then again assembled using long read assembler TGICL suite. Groba and Burgos [34] suggested additive multiple-k method to be a better approach than single best k-mer as it improves transcript diversity and increases contiguity. We also observed that additive k-mer approach increased the average and N50 read length to a large extent thereby improving the transcriptome assembly significantly (Table 2). Moreover, except for CLC, assemblies obtained from all other short read assemblers showed more than 90% of the short reads mapping back onto their respective transcriptome assembly (Table 2). In contrast, employing best k-mer approach, only Trinity assembly had more than 90% mapped back reads (Table 2). This observation further supports the use of additive k-mer approach over best k-mer to get a better assembly. However, when percentage of uniquely mapped reads was compared, it showed a completely different picture. Although the average read length, N50 length and percentage of total mapped back reads increased, there was a sharp decrease in the percentage of unique mapped back reads (Table 2) thereby suggesting redundant use of short reads in final assembly and formation of chimeric Unigenes as one short read must map back to a single locus and take part in the formation of a single transcript. To overcome this problem and further improve assembly statistics, transcriptome assembly obtained from additive k-mer approach was subjected to long read TGICL assembler. This step further increased the average read length and N50 length of all the assemblies. Although there was a marginal increase in the percentage of total mapped reads, a marked increase in the percentage of unique mapped back reads was observed in some of the assemblies (Table 2). A preliminary examination suggested the assembly generated by Oases taking non-redundant short read dataset to be the best (NR-Oases) as it had highest average read length, N50 length and total mapped back reads (Table 2). Nevertheless, when uniquely mapped reads were taken into consideration, only 52% (44,978,322) of the total reads uniquely mapped back onto NR-Oases assembly (Table 2), thus indicating the possibility of generation of chimeric Unigenes . Therefore, this assembly was not used for further analysis. On the basis of mapping results, NR-SOAPdenovo and NR-ABySS assemblies were found to be most impressive as more than 90% of the short reads uniquely mapped back onto the transcriptome (Table 2). However, when other parameters like total number of contigs (88,297), N50 read length (1198 bp) and average contig length (610 bp) were considered, assembly generated by NR-ABySS was found to be the best (Table 2; File S1). Although different strategies employed in this study gradually increased the N50 and average read length, use of only additive k-mer approach decreased the proportion of unique mapped back reads with respect to total mapped back reads. However, the use of TGICL on additive k-mer assemblies increased the map back proportion to a large extent suggesting the use of long read assembly tools on data initially generated by preliminary short read assemblers.
Similarity Search and Functional Annotation of Unigenes
Keeping a significant E-value cutoff of < 1E-06, we used BLASTX algorithm to annotate 88,297 seabuckthorn transcripts (obtained from NR-ABySS following second strategy) against non-redundant (nr) protein database of NCBI. A total of 41,340 (46.8%) seabuckthorn transcripts were found to have a significant hit with protein entries present in the NCBI database (Table S2). This figure is comparable with that reported by Fatima et al. 2012 [12] where 46% of seabuckthorn transcripts obtained from 454 sequencing showed similarity to existing proteome database. However, in our previous pilot study based on a small seabuckthorn dataset comprising of 1,665 Unigenes , we were able to annotate nearly 77% of the Unigenes [11].
In accordance with our previous study [11] and findings of Fatima et al. 2012 [12], we also observed significant sequence homology between Vitis vinifera (grapes) and seabuckthorn. According to amino acid homology, species showing top-hits at NCBI database included V. vinifera, Ricinus communis, Populus trichocarpa and Glycine max (Figure 2), in that order. We used BLAST2GO tool to assign gene ontology (GO) terms associated with top 20 BLAST hits for each transcript. Of the 41,340 sequences having BLAST hit (E-value cutoff of < 1E-06), 38,830 had at least one GO term associated with each of them representing 43.9% of the seabuckthorn transcriptome. Seabuckthorn transcripts could be grouped in different categories viz. biological process (Figure S1), cellular component (Figure S2) and molecular function (Figure S3) depicting a broad spectrum of the analyzed transcriptome. In the category of biological process, the largest groups were “primary metabolic process”, “cellular metabolic process” and “biosynthetic process”. However, presence of groups like “cellular response to stimulus”, “response to external stimulus”, “response to biotic stimulus”, response to endogenous stimulus”, “response to abiotic stimulus” and “response to stress” in our dataset indicated that a large number of transcripts are expressed in response to environmental stresses (Figure S1). In the category of molecular function, Unigenes with “catalytic activity”, “protein binding” and “nucleotide binding” formed the largest groups (Figure S3).
Figure 2. Top hit species representation of seabuckthorn transcriptome based on sequence similarity search (BLASTX).

In silico SSR Screening and Detection of Transcription Factors
EST based markers, owing to high polymorphism, abundance and ease in development are important resource for determining functional genetic variation. Mining of SSRs from seabuckthorn transcriptome was performed using MISA search tool at default parameters. A total of 13,299 SSRs were identified in 10,980 (12.4%) seabuckthorn transcripts (Table 3). Though mononucleotide SSRs represented the largest fraction (56.4%) but due to the presence of polyA tail in eukaryotic mRNA which confounds with microsatellite repeat, these in silico mined mono-nucleotide SSRs should be considered with care for further analysis. Among the rest, dinucleotide repeats (21.5%) were found to be most common EST-SSR in seabuckthorn transcriptome followed by trinucleotide repeats (18.9%) which is in accordance with our previous report [11] on a limited number of 1,665 Unigenes . Among dinucleotide repeats, AG/CT accounted for the highest occurrence (67%) in seabuckthorn transcriptome followed by AT/TA (23%), AC/GT (9.5%) and marginal CG/CG (0.2%). While in case of trinucleotide repeats, occurrence of various constituting repeats was uniform except for AAG/CTT showing higher frequency (38%) and CCG/CGG showing least abundance (0.4%).
Table 3. Data on distribution of Simple Sequence Repeats (SSRs) in seabuckthorn transcriptome.
| Data item | Number (%) |
|---|---|
| Total number of sequences screened | 88297 |
| Total number of identified SSRs | 13299 |
| Number of sequences containing SSRs | 10980 (12.4) |
| Number of sequences containing more than one SSR | 1850 (16.8) |
| Number of SSRs present in compound formation | 1099 (8.2) |
| Relative distribution of different repeat types | |
| Mono-nucleotides | 7502 (56.4) |
| Di-nucleotides | 2860 (21.5) |
| Tri-nucleotides | 2520 (18.9) |
| Tetra-nucleotides | 213 (1.6) |
| Penta-nucleotides | 62 (0.46) |
| Hexa-nucleotides | 142 (1.06) |
The seabuckthorn transcriptome generated in this study was also screened for the presence of transcription factors by sequence comparison to known transcription factor gene families. In total, 7,421 putative seabuckthorn transcription factor genes, distributed in at least 80 different families, were identified representing 8.4% of seabuckthorn transcripts. The most frequent transcription factors belonged to C3H, MADS, bHLH, NAC, and FAR1 families (Figure 3). Transcription factors involved in abiotic stress tolerance have also been identified in our dataset. Earlier we reported expression analysis of HMG transcription factor in response to freeze tolerance in seabuckthorn [11].
Figure 3. Distribution of seabuckthorn transcripts in different transcription factor families.

Conclusions
Next generation sequencing technologies are becoming increasingly affordable, accessible and robust for non-model organisms also enabling faster and cheaper data generation. However, task of extracting meaningful information from such a huge amount of data has become more challenging. Here, with the objective to generate transcriptome of a non-model organism, seabuckthorn, using Illumina short reads, we compared the utility of six de novo short read assembly tools following different approaches. The study suggested removal of redundancy in short read dataset prior to assembly by short read assembler ABySS following additive k-mer approach. Subsequent assembly by long read assembler TGICL yielded the optimum assembly. Looking at the prospects of using TGICL after initial short read assemblers, as observed in the present study, it is recommended to use long read assemblers after de novo short read assemblers in future transcriptome assembly projects. Functional annotation of seabuckthorn transcriptome revealed conservation of genes involved in various biological processes. Our study demonstrates that NGS technique is a powerful tool for gene function discovery even in organisms with no annotated genomes.
Supporting Information
Gene Ontology classification of seabuckthorn Unigenes on the basis of their role in biological processes.
(TIF)
Gene Ontology classification of seabuckthorn Unigenes on the basis of their occurrence in different cellular components.
(TIF)
Gene Ontology classification of seabuckthorn Unigenes on the basis of their molecular function.
(TIF)
Unigene sequences of de novo assembled seabuckthorn transcriptome.
(7Z)
Assembly statistics of various short read assemblers at different k-mers.
(XLS)
Functionally annotated seabuckthorn Unigenes on the basis of BLASTX results.
(XLSX)
Acknowledgments
Rajesh Ghangal is thankful to Ravi Kumar Patel for his support and guidance in handling of assembly tools.
Funding Statement
This work was financially supported by Department of Biotechnology, Government of India, New Delhi (BT/PR10798/NDB/51/170/2008); http://dbtindia.nic.in/index.asp. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Elena T, Capraru G, Rosu CM, Zamfirache MM, Olteanu Z et al. (2011) Morphometric pattern of somatic chromosomes in three Romanian seabuckthorn genotypes. Caryologia 64: 189-196. [Google Scholar]
- 2. Lu R (1997) Eco-geographical distribution of seabuckthorn and prospects of international cooperation. In: Lu S, Li M, Hu J, Liu S. Worldwide Research & Development of Seabuckthorn. Beijing, China: Science Publishing House & Technology Press; pp. 11-22. [Google Scholar]
- 3. Lian YS, Chen XL (2000) The regular patterns of distribution on the natural components in plants of the genus Hippophae L. J Northwest Normal University; (Natural Science Edition) 36: 113-128 [Google Scholar]
- 4. Stobdan T, Angchuk D, Singh SB (2008) Seabuckthorn: An emerging storehouse for researchers in India. Curr Sci 94: 1236-1237. [Google Scholar]
- 5. Sezik E, Yesilada E, Shadidoyatov H, Kulivey Z, Nigmatullaev AM et al. (2004) Folk medicine in UzbekistanI. Toshkent, Djizzax, and Samarqand provinces J Ethnopharmacol 92: 197-207. [DOI] [PubMed] [Google Scholar]
- 6. Shinwari ZK, Gilani SS (2003) Sustainable harvest of medicinal plants at Bulashbar Nullah, Astore (Northern Pakistan). J Ethnopharmacol 84: 289-298. doi:10.1016/S0378-8741(02)00333-1. PubMed: 12648828. [DOI] [PubMed] [Google Scholar]
- 7. Dhyani D, Maikhuri RK, Rao KS, Kumar L, Purohit VK et al. (2007) Basic nutritional attributes of Hippophae rhamnoides (Seabuckthorn) populations from Uttarakhand Himalaya, India. Curr Sci 92: 1148-1152. [Google Scholar]
- 8. Singh KN, Lal B (2008) Ethnomedicines used against four common ailments by the tribal communities of Lahaul-Spiti in western Himalaya. J Ethnopharmacol 115: 147-159. doi:10.1016/j.jep.2007.09.017. PubMed: 17980527. [DOI] [PubMed] [Google Scholar]
- 9. Ledwood JS, Shimwell DW (1971) Growth rates of Hippophae rhamnoides L. Ann Bot 35: 1053-1058. [Google Scholar]
- 10. Akkermans ADL, Roelofsen W, Blom J, Hussdanell K, Harkink R (1983) Utilization of carbon and nitrogen compounds by Frankia in synthetic media and in root nodules of Alnus glutinosa, Hippophae rhamnoides, and Datisca cannabina. Can J Bot 61: 2793-2800. doi:10.1139/b83-309. doi:10.1139/b83-309 [Google Scholar]
- 11. Ghangal R, Raghuvanshi S, Sharma PC (2012) Expressed sequence tag based identification and expression analysis of some cold inducible elements in seabuckthorn (Hippophae rhamnoides L.). Plant Physiol Biochem 51: 123-128. doi:10.1016/j.plaphy.2011.10.005. PubMed: 22153248. [DOI] [PubMed] [Google Scholar]
- 12. Fatima T, Snyder CL, Schroeder WR, Cram D, Datla R et al. (2012) Fatty acid composition of developing seabuckthorn (Hippophae rhamnoides L.) berry and the transcriptome of the mature seed. PLOS ONE 7(4): e34099. doi:10.1371/journal.pone.0034099. PubMed: 22558083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thakur V, Varshney R (2010) Challenges and Strategies for Next Generation Sequencing (NGS) Data Analysis. J Computer Sci Systems Biol 3: 040-042. doi:10.4172/jcsb.1000053. [Google Scholar]
- 14. Sun C, Li Y, Wu Q, Luo H, Sun Yet al (2010) De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics 11: 262. doi:10.1186/1471-2164-11-262. PubMed: 20416102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo S, Zheng Y, Joung JG, Liu S, Zhang Z et al. (2010) Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11: 384. doi:10.1186/1471-2164-11-384. PubMed: 20565788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin Y, Li J, Shen H, Zhang L, Papasian CJ et al. (2011) Comparative studies of de novo assembly tools for next generation sequencing technologies. Bioinformatics 27: 2031-2037. doi:10.1093/bioinformatics/btr319. PubMed: 21636596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garg R, Patel RK, Tyagi AK, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18: 53-63. doi:10.1093/dnares/dsq028. PubMed: 21217129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang W, Chen J, Yang Y, Tang Y, Shang J et al. (2011) A practical comparison of de novo genome assembly software tools for next-generation sequencing technologies. PLOS ONE 6: e17915. doi:10.1371/journal.pone.0017915. PubMed: 21423806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghangal R, Raghuvanshi S, Sharma PC (2009) Isolation of good quality RNA from a medicinal plant seabuckthorn, rich in secondary metabolites. Plant Physiol Biochem 47: 1113-1115. doi:10.1016/j.plaphy.2009.09.004. PubMed: 19804984. [DOI] [PubMed] [Google Scholar]
- 20. Patel RK, Jain M (2012) NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLOS ONE 7: e30619. doi:10.1371/journal.pone.0030619. PubMed: 22312429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821-829. doi:10.1101/gr.074492.107. PubMed: 18349386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28: 1086-1092. doi:10.1093/bioinformatics/bts094. PubMed: 22368243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117-1123. doi:10.1101/gr.089532.108. PubMed: 19251739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li R, Zhu H, Ruan J, Qian W, Fang X et al. (2009) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265-272. PubMed: 20019144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 26: 644-652. PubMed: 21572440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller JR, Koren S, Sutton G (2010) Assembly algorithms for next-generation sequencing data. Genomics 95: 315-327. doi:10.1016/j.ygeno.2010.03.001. PubMed: 20211242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658-1659. doi:10.1093/bioinformatics/btl158. PubMed: 16731699. [DOI] [PubMed] [Google Scholar]
- 28. Pertea G, Huang X, Liang F, Antonescu V, Sultana R et al. (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651-652. doi:10.1093/bioinformatics/btg034. PubMed: 12651724. [DOI] [PubMed] [Google Scholar]
- 29. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403-410. doi:10.1016/S0022-2836(05)80360-2. PubMed: 2231712. [DOI] [PubMed] [Google Scholar]
- 30. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674-3676. doi:10.1093/bioinformatics/bti610. PubMed: 16081474. [DOI] [PubMed] [Google Scholar]
- 31. Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106: 411-422. PubMed: 12589540. [DOI] [PubMed] [Google Scholar]
- 32. Riaño-Pachón DM, Ruzicic S, Dreyer I, Mueller-Roeber B (2007) PlnTFDB: an integrative plant transcription factor database. BMC Bioinformatics 8: 42. doi:10.1186/1471-2105-8-42. PubMed: 17286856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang Y, Lu J, Peatman E, Kucuktas H, Liu S et al. (2011) A pilot study for channel catfish whole genome sequencing and de novo assembly. BMC Genomics 12: 629. doi:10.1186/1471-2164-12-629. PubMed: 22192763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groba YS, Burgos JIM (2010) Optimization of de novo transcriptome assembly from next generation sequencing data. Genome Res 20: 1432-1440. doi:10.1101/gr.103846.109. PubMed: 20693479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene Ontology classification of seabuckthorn Unigenes on the basis of their role in biological processes.
(TIF)
Gene Ontology classification of seabuckthorn Unigenes on the basis of their occurrence in different cellular components.
(TIF)
Gene Ontology classification of seabuckthorn Unigenes on the basis of their molecular function.
(TIF)
Unigene sequences of de novo assembled seabuckthorn transcriptome.
(7Z)
Assembly statistics of various short read assemblers at different k-mers.
(XLS)
Functionally annotated seabuckthorn Unigenes on the basis of BLASTX results.
(XLSX)