

Abstract
Campylobacter jejuni is a major human pathogen and a leading cause of bacterial derived gastroenteritis worldwide. C. jejuni regulates gene expression under various environmental conditions and stresses, indicative of its ability to survive in diverse niches. Despite this ability to highly regulate gene transcription, C. jejuni encodes few transcription factors and its genome lacks many canonical transcriptional regulators. High throughput deep sequencing of mRNA transcripts (termed RNAseq) has been used to study the transcriptome of many different organisms, including C. jejuni; however, this technology has yet to be applied to defining the transcriptome of C. jejuni during in vivo colonization of its natural host, the chicken. In addition to its use in profiling the abundance of annotated genes, RNAseq is a powerful tool for identifying and quantifying, as-of-yet, unknown transcripts including non-coding regulatory RNAs, 5’ untranslated regulatory elements, and anti-sense transcripts. Here we report the complete transcriptome of C. jejuni during colonization of the chicken cecum and in two different in vitro growth phases using strand-specific RNAseq. Through this study, we identified over 250 genes differentially expressed in vivo in addition to numerous putative regulatory RNAs, including trans-acting non-coding RNAs and anti-sense transcripts. These latter potential regulatory elements were not identified in two prior studies using ORF-based microarrays, highlighting the power and value of the RNAseq approach. Our results provide new insights into how C. jejuni responds and adapts to the cecal environment and reveals new functions involved in colonization of its natural host.
Introduction
Campylobacter jejuni is an important human pathogen and a leading cause of bacterial derived gastroenteritis worldwide [1,2]. Human Campylobacter infections are sporadic foodborne diseases commonly associated with foods of animal origin, with C. jejuni being the predominant species associated with human illness [3]. Preventative measures to limit human exposure have focused on identifying genes and loci required for colonization [4–8] as these represent targets for anti- Campylobacter strategies. Although much research has been carried out characterizing regulatory mechanisms in this human pathogen, most of this work has focused on in vitro studies with little knowledge generated towards understanding the mechanisms of global gene expression during colonization.
The five C. jejuni strains sequenced to date contain approximately 1,650 to 1,800 protein-encoding genes [9–11]. C. jejuni highly regulates gene expression under various stresses and environmental conditions [4,12–18]. Despite its ability to rapidly alter gene expression, C. jejuni encodes only three sigma factors and a total of 34 other identified transcriptional regulators [9,10]. Furthermore, C. jejuni lacks homologues of many canonical transcriptional regulators such as FNR, CRP, OxyR, RpoS, SoxRS and ArcA [9,10]. Although there appears to be a paucity of encoded transcription factors, other mechanisms of gene regulation have been identified in C. jejuni. Slip-strand mispairing, leading to phase variation gene expression, has been demonstrated to be a regulatory mechanism of flagella biosynthesis [19,20], lipooligosaccharide glycosylation [21], and capsule production [22]. Furthermore, genome rearrangements [23], interspecies genetic exchange [24] and modification of RNA polymerase [25,26] are alternative mechanisms of genetic regulation employed by C. jejuni.
Regulatory networks in C. jejuni have been characterized principally through in vitro methods including quantitative reverse transcriptase PCR (qRT-PCR), transcriptional reporter strains and microarray studies. These have yielded tremendous amounts of information as to how this organism regulates gene expression; however, many of these studies focused on regulatory mechanisms of specific genes and loci in response to specific substrates and culture conditions in vitro. Only two studies have evaluated the transcriptome of C. jejuni during in vivo colonization of an animal model, including the day-of-hatch chicken model [18] and the rabbit ileal loop model [27]. These studies, utilizing microarray approaches, determined that there are marked differences in gene expression profiles between in vivo and in vitro samples, illustrating the importance for continued research into how C. jejuni regulates gene expression during colonization.
High throughput sequencing of cDNA libraries (RNAseq) has emerged as a powerful approach for mapping transcriptomes and profiling gene expression in diverse bacteria [28–32]. RNAseq has several key advantages over microarray analysis, including 1) the ability to detect and quantify transcripts derived from all regions of the genome, 2) a large dynamic range that affords high sensitivity for low-abundance transcripts, and 3) single nucleotide resolution [33].
Regulatory mechanisms involving small, non-coding RNAs (ncRNAs), or riboswitches have not been well-characterized in C. jejuni. Recently, two in vitro RNAseq studies mapped the transcriptome of C. jejuni and other Campylobacter species [29,30]. One study characterized the regulon of the sigma factor RpoN and correlated transcriptome expression data with protein expression profiles [29]. In the other study, RNAseq was used to map and compare transcriptional start sites, characterize promoter structures and analyze CRISPR RNAs in multiple Campylobacter strains and species [30]. Moreover, both studies identified a wide repertoire of potential non-coding RNA species. These in vitro RNAseq studies have yielded much information into regulatory mechanisms encoded by Campylobacter species; however, this powerful tool has yet to be applied to characterizing the transcriptome of C. jejuni during colonization of its natural host. Although, C. jejuni encodes non-coding RNA species, it does not encode the conserved RNA binding protein Hfq [9,10], a key component of sRNA regulatory mechanisms in other organisms [34–37]. C. jejuni does, however, encode another conserved RNA binding protein CsrA [9,10] which, in other organisms, plays a role in regulating many processes including motility [38], virulence [39], biofilm formation and central carbon metabolism [40]. The C. jejuni CsrA homologue is involved in the oxidative stress response, biofilm formation, and host cell invasion [41]. Unlike other microbes that use CsrA, C. jejuni does not encode any obvious homologue of the CsrA-antagonizing small RNA csrB; thus the mechanism of CsrA regulation in this pathogen is unknown. Although the regulatory mechanisms of non-coding RNAs have yet to be characterized in C. jejuni, the genome of the closely related organism Helicobacter pylori (which like Campylobacter encodes CsrA but not Hfq), encodes non-coding RNAs as well as riboswitches and antisense transcripts involved in regulatory processes [32,42], suggesting C. jejuni may also regulate gene expression with non-coding RNAs.
Here we report the first complete transcriptome of C. jejuni during in vivo colonization of the chicken cecum using RNAseq. We used a strand-specific, library construction protocol to identify both sense and antisense transcripts. Through this study we identified 272 genes that are differentially expressed in vivo compared to in vitro mid-log and stationary phase grown cultures and identified 51 potential RNA regulators including small, non-coding RNAs (ncRNAs), anti-sense transcripts and probable riboswitches. We also identified the structure of the chick cecal microbiome and identified differences in microbiome structure in C. jejuni colonized chicks compared to mock infected controls. Overall, we identified in vivo expression patterns and possible uncharacterized regulatory mechanisms that provide insight into how C. jejuni is able to adapt to diverse environments.
Results and Discussion
Mapping the in vivo and in vitro transcriptome of C. jejuni using Illumina-based RNAseq
C. jejuni gene expression is highly regulated; however, the mechanisms of this regulation are still largely uncharacterized. To profile the C. jejuni transcriptome in vivo, RNA was isolated from the cecal contents of chicks seven days post colonization with an average of ~7.5 x 108 C. jejuni CFU/g cecal contents. To generate enough starting RNA for library construction (5 µg of DNA-free RNA), RNA isolated from five separate chicks housed in the same brooder, was pooled together. In total, three pools of RNA were isolated from three separate sets of chicks. Campylobacter strand specific, bar-coded cDNA libraries were generated using methods described by Mandlik et al. [31]. Bar-coded cDNA libraries were also generated from three independent cultures of C. jejuni grown in Mueller Hinton Broth (MHB) to mid-log and stationary phase, respectively. The six in vitro libraries were sequenced on a total of two lanes of the Illumina HiSeq2000 platform; each of the three in vivo samples was sequenced in its own lane. The average number of reads obtained for in vitro samples was ~125.4 million reads per sample, with ~87.2% of reads mapping to the C. jejuni 81-176 chromosome and two plasmids, pVir and pTet (Table 1). Sequencing of the in vivo samples resulted in ~378.8 million reads per sample, in which ~14.6% of reads mapped to the C. jejuni chromosome and plasmids (Table 1). Most reads mapped to open reading frames; however, many reads mapped to intergenic non-coding regions on the chromosome as well as locations anti-sense to open reading frames (ORFs), suggesting that C. jejuni encodes ncRNAs that could be involved in uncharacterized regulatory mechanisms (Figure 1A).
Table 1. Summary of sequencing results of in vitro and in vivo Illumina based cDNA libraries.
| Growth Condition | Sample # | Number of Reads (in millions) | % Reads Aligned to Genome | % Reads Aligned in Pairs |
|---|---|---|---|---|
| 1 | 111.72 | 90.21 | 80.24 | |
| Mid-Log | 2 | 149.09 | 90.87 | 82.90 |
| 3 | 106.84 | 89.61 | 80.52 | |
| 1 | 114.91 | 84.14 | 76.22 | |
| Stationary | 2 | 112.21 | 83.53 | 74.01 |
| 3 | 157.25 | 84.33 | 75.51 | |
| 1 | 389.47 | 15.34 | 89.58 | |
| Chick | 2 | 379.03 | 11.17 | 87.63 |
| 3 | 367.79 | 17.29 | 85.56 |
Figure 1. Mapping and expression profiles of in vitro and in vivo sequence reads to the C. jejuni 81-176 genome.

A. Distribution of reads (as percent of total reads) that mapped back to specific locations of the C. jejuni 81-176 genome. B. Venn diagram of genes with increased and decreased abundance in vivo compared to in vitro samples. ORF: open reading frame, IGR: intergenic region, rRNA: ribosomal RNA, tRNA: transfer RNA, ncRNA: non-coding RNA, Anti-sense: anti-sense RNA.
Structural differences in the chicken microbiome do not correlate to changes in C. jejuni global gene expression during colonization
To determine the relative abundance of C. jejuni in the chick ceca during colonization, we performed 454-pyrosequencing of the 16S rRNA gene isolated from the same C. jejuni colonized chicks used in RNAseq experiments, as well as PBS (mock) infected chicks. DNA based 454-pyrosequencing analysis of the V3-V5 region of the 16S rRNA gene yielded a total of 87,663 sequences, which after trimming and chimera removal by the analysis program mothur [43], yielded 48,571 sequences with minimum read length of 250 base pairs. On average, Campylobacters represented between 5–10% of the total number of sequences identified in the ceca of brooder 1 (10%), 2 (5%) and 3 (5%), (Figure 2A). We identified differences in the structure of the cecal microbiota at the phylum, family and OTU (Operational Taxonomic Unit) level between brooder sets and PBS control chicks (Figures 2 and S1). Previous studies investigating the cecal microbiota of pathogen free and C. jejuni colonized chicks showed a difference in phylotype distribution; however, no changes in the microbiota functional gene content was identified [44]. In this study, chicks colonized with C. jejuni had microbiome structures distinct from the PBS control group (Figure 2B). The structure of the cecal microbiota of chicks colonized with C. jejuni housed in brooder 1 was statistically different from chicks housed in brooders 2 and 3 (Figure 2B). Moreover, at the family level, chicks in brooder 1 showed an increase (compared to the other brooders) in the relative abundance of Ruminococcaceae, Leuconostocaceae and Enterococcaceae families and a decrease in members of the Lachnospiraceae family (Figure 2A). Brooder 1 chicks also had an increase in the relative abundance of Campylobacteraceae (~10%) compared to brooders 2 and 3 (~5%). This poses questions as to whether the indigenous cecal microbiota dictates the level of Campylobacter colonization, or if the level of Campylobacter colonization results in distinct shifts in the microbiota. Although there was a significant difference in the bacterial community structure between brooders, there was no significant difference (avg. R-value of 0.979) in the global transcriptome profiles (Figure S2, G-I), indicating that changes in the cecal microbiota, observed in this study, did not result in changes in the global gene expression of C. jejuni. This is in agreement with the result that changes in the microbiota of chicks colonized with C. jejuni did not alter functional gene content of the microbiome within the chick cecum [44]. In this study, we observed no changes in transcriptome profiles between animal replicates (Figure S2 G–I); in contrast, a previous transcriptome study of C. jejuni, using the rabbit illeal loop model (RIL), showed wide variances in transcript abundance between animals (R value of 0.49) [27]. The difference between the two sets of findings could be due to differences in animal models (RIL v. 1-day-old chicks), including host immune response and the host microbiota structure and function, and/or the result of differences in transcriptomic technologies (microarray v. RNAseq).
Figure 2. The cecal microbiota of chickens colonized with C. jejuni.

A. Defining members of the cecal microbiota by relative rank abundance plots (>1%) at the bacterial family level. The average relative abundance for each treatment group (PBS control n=5; brooder 1 n=5; brooder 2 n=5; brooder 3 n=5) is represented in the bar plot. B. Principle coordinates analysis (PCoA) illustrating the community structure relationship between chicks from different treatment groups. This ordination was generated using a Yue and Clayton-based distance matrix representing the relative abundance of OTUs in each community at a 3% OTU definition level. The community of each chick is indicated by a colored symbol (PBS control = blue; brooder 1 = black; brooder 2 = green; brooder 3 = red). All brooder bacterial communities were significantly different from PBS controls (p=0.008 for brooder1 and p=0.007 for brooder 2 and 3; AMOVA). Brooder 1 was significantly different from Brooder 2 and 3 (p=0.003 and p=0.015, respectively; AMOVA).
Differential gene expression during colonization of the chicken cecum and validation of RNAseq
To analyze differential expression of transcripts between in vivo and in vitro samples we utilized the variance analysis package DESeq [45]. A substantial number of genes were significantly differentially expressed (>4-fold, padj <0.05) in vivo compared to both in vitro conditions. Differential expression profiles of chick samples compared to mid-log samples identified 149 genes that were differentially expressed; 135 transcripts showed increased abundance in vivo while 14 genes had decreased abundance (Figure 1B, Tables S1 and S2). Comparing DESeq of chick samples to stationary samples, 152 genes were significantly differentially expressed (>4-fold, padj <0.05); 95 transcripts with increased abundance compared to 57 with decreased abundance (Figure 1B and Tables S3 and S4). Of the genes with increased abundance in vivo, 29 were increased in comparison to both in vitro conditions (Figure 1B, Table 2). No transcripts were found to be decreased in abundance in both conditions with a >4-fold cutoff. All genes significantly differentially expressed (>4-fold, padj <0.05) during in vivo colonization compared to in vitro growth, and those differentially expressed between in vitro growth conditions, are listed in Tables S1-S6. Expression profiles of every gene, under all growth conditions, are listed in Table S9.
Table 2. Genes increased in abundance in vivo compared to both in vitro mid-log and stationary phase cultures.
| CJJ Locus Number | Gene Name / Function | Fold Change (ML)* | Fold Change (Stat)* | |
|---|---|---|---|---|
| Energy and Metabolism | CJJ81176_0044 | dsbB | 4.47 | 8.04 |
| CJJ81176_0064 | Cytochrome c Fmaily | 10.46 | 20.99 | |
| CJJ81176_0403 | Sulphite Oxidase | 4.52 | 14.00 | |
| CJJ81176_0880 | dsbA | 5.30 | 5.10 | |
| Stress Response | CJJ81176_1387 | katA | 320.82 | 46.11 |
| CJJ81176_1574 | cgb | 12.29 | 4.72 | |
| Transport | CJJ81176_0642 | pstS | 8.00 | 44.54 |
| CJJ81176_0643 | pstA | 6.75 | 31.31 | |
| CJJ81176_0644 | pstC | 7.92 | 5.74 | |
| CJJ81176_0750 | ABC transporter periplasmic substrate binding protein | 5.36 | 24.95 | |
| CJJ81176_0753 | ABC transporter periplasmic substrate binding protein | 6.39 | 11.85 | |
| CJJ81176_0754 | ABC transporter periplasmic substrate binding protein | 8.69 | 10.82 | |
| CJJ81176_1601 | chuA | 8.75 | 10.54 | |
| CJJ81176_1602 | chuB | 6.73 | 5.20 | |
| CJJ81176_1603 | chuC | 6.61 | 6.79 | |
| CJJ81176_1604 | chuD | 7.91 | 8.19 | |
| CJJ81176_1619 | exbB-2 | 16.40 | 43.35 | |
| CJJ81176_1620 | exbD | 8.59 | 49.24 | |
| Other | CJJ81176_1388 | Ankyrin repeat protein | 87.98 | 18.96 |
| Hypothetical | CJJ81176_0045 | Hypothetical | 4.72 | 15.76 |
| CJJ81176_0063 | Hypothetical | 27.27 | 20.74 | |
| CJJ81176_0065 | Hypothetical | 6.53 | 31.66 | |
| CJJ81176_0522 | Hypothetical | 19.65 | 5.19 | |
| CJJ81176_0751 | Hypothetical | 6.42 | 16.33 | |
| CJJ81176_0752 | Hypothetical | 7.05 | 21.44 | |
| CJJ81176_0954 | Hypothetical | 24.06 | 9.70 | |
| CJJ81176_1062 | Hypothetical | 11.11 | 11.19 | |
| CJJ81176_1386 | Hypothetical | 23.72 | 30.08 | |
| CJJ81176_1746 | Hypothetical | 21.00 | 7.06 |
padj < 0.05, a corrected p-value analogous to a false detection rate of < 5%.
To validate expression patterns identified by RNAseq, we performed qRT-PCR on RNA isolated from in vitro and in vivo grown cultures. RNA used in qRT-PCR experiments was isolated independent from samples used in RNAseq library construction. Primers were designed to amplify genes with increased, decreased or unchanged levels of transcript abundance in vivo compared to in vitro samples (Table S8). RNAseq identified katA as one of the highest abundant transcripts during colonization compared to both in vitro growth conditions, and this increase in abundance was confirmed by qRT-PCR (Figure 3). Furthermore, genes identified as having decreased transcript abundance (Cjj0315 and Cjj0067) or no changes in transcript abundance (cjaC and ccoN) during colonization, compared to in vitro grown cultures, were also confirmed by qRT-PCR, validating the RNAseq expression profiles (Figure 3). Because differential expression analysis (DESeq) combined and averaged sequencing results from the three biological replicates, we wanted to confirm that transcript abundance between replicates was similar. Expression profiles between each biological replicate were compared by plotting the abundance (log2) of each ORF between all replicates of each growth condition. Transcript abundance between replicates, of all three growth conditions, showed no statistical differences (Figure S2).
Figure 3. Validation of RNAseq expression analysis by qRT-PCR.

Relative expression levels of genes determined by RNAseq to be differentially expressed in vivo compared to A. in vitro mid-log cultures and B. in vitro stationary phase cultures. Relative expression was assessed using the 2(-Delta Delta C(T)) method, error bars represent standard deviation of three independent biological replicates.
Insights into the intestinal environment during C. jejuni colonization derived from global transcript regulatory patterns
To identify global regulation patterns occurring in vivo, we graphed DESeq analyses on MA plots (Figure 4). MA plots represent the log2 of the ratio of abundances of each ORF between the indicated conditions [M], plotted against the average log2 abundance of that ORF in all conditions [A]. Three functional gene groups were identified as increased in abundance during colonization including iron transport, phosphate transport and oxidative and nitrosative stress responses. Iron has been consistently identified as a critical micronutrient for C. jejuni growth and colonization [46–50]. Increased expression of the heme transport genes chuA and chuB were previously identified in an C. jejuni in vivo microarray transcriptome study [18]. Utilizing RNAseq, we confirmed the increased abundance of chuA and chuB transcripts along with chuC and chuD of the heme transport operon (chuABCD). Additionally, we identified two other iron transporters with increased expression in vivo, including the outer membrane TonB-dependent energy transduction system exbB-exbD-tonB and the ferric transport system FTR1-p19. The increased abundance of three iron transport systems suggests that the chicken cecum is an extremely iron-limiting environment.
Figure 4. Differential gene expression (DESeq) of C. jejuni during in vivo colonization and in vitro growth.
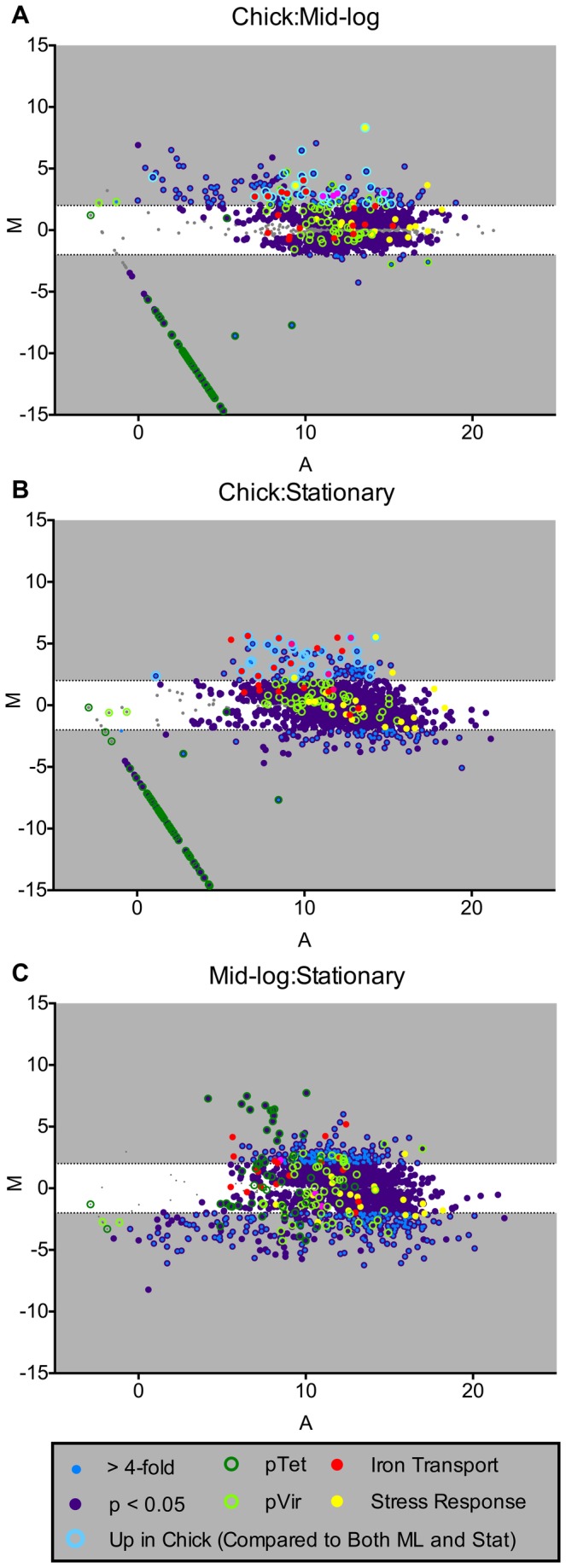
(A–C) The log2 of the ratio of abundances of each gene between the indicated conditions [M] plotted against the average log2 of abundance of that gene in all conditions [A]. For each plot, [M] and [A] values were generated with DESeq [45] using data from three biological replicates of each in vivo and in vitro growth condition. Genes significantly differentially expressed (>4-fold padj<0.05) as well genes of specific functional groups are highlighted (see legend for functional group highlight annotation). Grey regions highlight expression differences greater-than and less-than 4-fold.
Transcripts from the phosphate transport system (pstSCAB) were increased in abundance in vivo compared to both in vitro conditions (Figure 5B, Tables S1 and S3); however, in contrast to the iron transport systems noted above, increased expression of this locus was not observed in either previous in vivo microarray transcriptome study [18,27]. In addition to this ABC-type transport system, C. jejuni encodes a low affinity non-specific phosphate transport system pitAB (Cjj1208, Cjj1209) [51], which was not differentially expressed to any significant extent in our studies. In response to phosphate limitation, the pst transport system is activated by the two-component system PhoSR; pst is required for growth in phosphate-limited media [52] while the pitAB locus has yet to be characterized. A role for pitAB and pstSCAB in vivo has yet to be determined in C. jejuni, but increased abundance of the pst system in vivo suggests that the chick cecum is limited in phosphate and that this system could be required for colonization based on its importance in vitro [52].
Figure 5. Gene expression profiles of C. jejuni during in vitro growth and in vivo colonization.

Histograms illustrating strand-specific coverage per nucleotide across multiple loci on the C. jejuni chromosome. Red lines represent reads sequenced and mapped from in vivo chick libraries. Blue (mid-log) and green (stationary) lines represent reads sequenced and mapped from in vitro libraries. ORF’s are labeled below each histogram in blue. Specific loci A) katA, B) pstSCAB, C) cgb, which were increased in abundance during in vivo colonization, are highlighted in red.
Furthermore, increased in vivo transcript abundance (compared to in vitro growth) was observed from genes involved in oxidative and nitrosative stress responses (Figure 4, Figure 5, Tables S1 and S3). The second highest differentially expressed gene in vivo, katA (encoding catalase), is critical for the C. jejuni response to H2O2 and is required for colonization of the chicken cecum (Figure 5A) [53–55]. Additionally, the truncated flavohemoprotein (cgb), involved in the nitrosative stress response [56], was highly expressed in vivo (Figure 5C). The high level of expression of these two important stress response genes during in vivo colonization corresponds with studies demonstrating that C. jejuni induces a host immune response in the chicken [57–59]. Specifically, RNAseq analysis of chicken cecal tissue during colonization with C. jejuni showed increased expression of NOXO1, (NADPH oxidase organizer 1) which regulates respiratory burst [60]. Moreover, infection of human HCT-8 intestinal epithelial cells with C. jejuni activated host Nox1 and Duox2, resulting in increased H2O2 production and consequent inactivation of CjtK. CjtK, a tyrosine kinase encoded by C. jejuni, activates GalE, required for glycosylation and capsule formation, important factors in adherence and invasion [61]. The high level of catalase gene expression during in vivo growth suggests that C. jejuni is responding to host H2O2 production, which might be a mechanism to avoid inactivation of CjtK allowing for adherence and invasion. Furthermore, C. jejuni elicits a TLR response in chicks, specifically activating TLR-2, TLR-4 and TLR-21 [62]. Most importantly, TLR signaling was shown to be induced when using C. jejuni cell lysates compared to whole cells, proposing that bacterial lysis (evoking a hostile host environment) is required for full TLR activation [62]. Although C. jejuni is considered a commensal that does not induce human-like disease symptoms in chicks, it does elicit an immune response; therefore, increasing expression of stress response genes could possibly result in a mechanism for C. jejuni to postpone lysis, preventing a robust immune response, resulting in persistent colonization.
Overall, we identified 272 transcripts that were differentially expressed in vivo (Tables S1-S4). Many of the genes whose abundance increased in vivo contribute to cellular processes including central carbon metabolism, electron transport, biosynthetic processes and transport systems, indicative of a nutrient-restricted environment in the chick cecum compared to the rich in vitro growth medium MHB. We observed that in vivo abundance of RNA transcripts from the pTet plasmid were extremely low, with some transcripts never detected (Figure 4). This suggests either that C. jejuni strongly down-regulates expression of this plasmid in vivo, or that the plasmid is lost at a high rate during colonization. Both pTet and pVir are conjugative plasmids in C. jejuni 81-176 and both contain type-4 secretion system (T4SS) genes, with only genes encoded on pVir functionally characterized [63–65]. Involvement of pVir in attachment, invasion and disease during colonization of ferrets has been demonstrated [65]; however, no mechanism by which pTet might contribute to colonization has been uncovered. Genes involved in histidine biosynthesis and nitrogen metabolism (hisH and hisF) [66] were decreased in vivo (Table S2), suggesting that the chick cecum could be replete with histidine and/or glutamate. In C. jejuni, hisH and hisF have been implicated in the biosynthesis of intermediates of the flagellar glyscosylation pathway [67]; however, no role in chick colonization has been determined. Similarly, the major antigenic peptide (PEB3) was also determined to be decreased in abundance in vivo (Table S2). This glycoprotein is immunoreactive [68,69] suggesting that decreased expression could be a mechanism to evade host recognition leading to persistent colonization.
Comparison of RNAseq expression profiles to those of a previous in vivo transcriptome study using microarrays revealed many differences in expression patterns along with some similarities [18]. Most evident are the total number of differentially regulated genes between the two experiments, with microarray identifying only 68 differentially expressed genes (>2-fold), while RNAseq identified 272 differentially expressed genes, even using a higher stringency cutoff (>4-fold). Functional groups identified as differentially expressed in both experiments included components of electron transport, central carbon metabolism and transport systems. C. jejuni encodes a highly branched electron transport chain allowing for adaptation to specific environments [7,8,70]. C. jejuni is considered a microaerophile but has been shown to grow anaerobically in the presence of alternative electron acceptors including DMSO and nitrate [8]. RNAseq identified increased abundance of many transcripts associated with electron transport, including DMSO reductase, sulphite oxidase, the low-affinity oxidase (cioAB) and cytochrome c biogenesis (Tables S1-S4). Alternatively, there was decreased in vivo abundance of NADH: ubiquinone oxidoreductase transcripts (~2-3-fold), which encode proteins responsible for using reduced flavodixin as an electron donor instead of NADH [71], and there was no change in the abundance of the high-affinity oxidase CcoN (Figure 3B). Taken together, and as previously hypothesized, C. jejuni most likely encounters a limited oxygen environment in the cecum, as CioAB is a low-affinity oxidase [72], and C. jejuni has been shown to be able to respire off substrates such as sulphite and DMSO [8,70,73]. We did not observe an increase in expression of the nitrate reductase genes (napAB) as identified in the previous chick in vivo microarray study [18]. This difference in expression of napAB could be a result of growth temperature variation of in vitro grown cultures. The nitrate reductase locus is up-regulated at 42°C, compared to 37°C [74]. In vitro cultures for RNAseq experiments, reported here, were grown at 42°C in an effort to identify genes that were differentially regulated based on host colonization factors and the host nutritional environment, not differences in temperature, as the internal temperature of the chicken is 42°C. Additionally, expression differences could be due to different colonization conditions (including strain differences, inoculum load, duration of colonization and in vitro growth conditions). Finally, these differences could also be attributed to differences in transcriptome mapping technologies, as RNAseq affords direct quantification of transcripts and high sensitivity for low-abundance transcripts. As transcriptome studies are snapshots of gene expression, it would be interesting to determine C. jejuni gene expression profiles over time during the course of colonization. This would allow for a more in-depth profile of the transcriptome during initial colonization through persistent colonization.
Identification of non-coding and anti-sense RNA species
We next mined our RNAseq data for non-coding RNAs and identified 51 putative ncRNA candidates on the chromosome (Table 3) and on plasmid pTet (Table S7), including transcripts derived from intergenic regions and anti-sense to open reading frames (Figure 6). Our analysis identified nearly all the C. jejuni ncRNAs annotated in the Rfam database [75], including the thiamin pyrophosphate (TPP) riboswitch, the bacterial signal recognition particle (SRP), tmRNA and the RNA component of RNase P [9,76]. Similarly, we detected expression of 17 of the 25 ncRNAs identified in previous C. jejuni RNAseq studies [29,30]. In several cases, expression of ncRNAs previously identified in C. jejuni strain NCTC11168 were not detected in our analysis of strain 81-176, despite the conservation of these loci in both strains, an observation consistent with recent studies in Campylobacter sp. [30] and Listeria sp. [77] showing that expression of conserved ncRNAs may diverge significantly even among strains of the same species. Additionally, several ncRNAs previously identified in C. jejuni 81-176 [30] were not identified in our study, likely reflecting differences in growth temperatures, RNA library construction protocols, and/or ncRNA prediction algorithms.
Table 3. Non-coding RNAs identified by RNAseq and their differential expression under different growth conditions.
| ncRNA ID | Chromosome Location |
|
|
|
|---|---|---|---|---|
| nc 1 | 100050.100258 | 4.10 | 0.18 | 0.05 |
| nc 2 | 104909.105040 | 5.89 | 0.44 | 0.08 |
| nc 3 | 913140.913278 | 1.10 | 0.07 | 0.07 |
| nc 4 | 940044.940212 | 2.87 | 0.17 | 0.06 |
| nc 5 | 1032051.1032145 | 17.74 | 0.44 | 0.03 |
| nc 6 | 1190593.1190723 | 1.58 | 0.21 | 0.14 |
| nc 7 | 1278300.1278473 | 0.81 | 0.15 | 0.19 |
| nc 8 | 1536244.1536387 | 15.56 | 0.32 | 0.02 |
| nc 9 | 1566220.1566328 | 2.42 | 0.37 | 0.16 |
| nc 10 | 77586.77846 | 0.65 | 0.84 | 1.34 |
| nc 11 | 93760.93891 | 1.00 | 0.19 | 0.20 |
| nc 12 | 227132.227266 | 20.33 | 0.71 | 0.04 |
| nc 13 | 176680.176901 | 5.11 | 0.31 | 0.06 |
| nc 14 | 250947.251049 | 5.28 | 0.79 | 0.16 |
| nc 15 | 506475.506785 | 11.66 | 0.21 | 0.02 |
| nc 16 | 578796.579113 | 1.85 | 0.31 | 0.17 |
| nc 17 | 622442.622594 | 6.59 | 0.15 | 0.02 |
| nc 18 | 667317.667510 | 1.35 | 0.13 | 0.10 |
| nc 19 | 874073.874227 | 0.58 | 0.04 | 0.07 |
| nc 20 | 878326.878438 | 6.90 | 0.38 | 0.06 |
| nc 21 | 924301.924469 | 0.89 | 0.09 | 0.10 |
| nc 22 | 1036626.1036898 | 1.85 | 0.26 | 0.15 |
| nc 23 | 1075868.1076095 | 3.30 | 0.56 | 0.18 |
| nc 24 | 361854.361960 | 1.47 | 0.10 | 0.07 |
| nc 25 | 1248138.1248316 | 2.10 | 0.23 | 0.11 |
| nc 26 | 1502121.1502247 | 2.65 | 0.08 | 0.03 |
| nc 27 | 1551155.1551721 | 10.12 | 1.32 | 0.14 |
| nc 28 | 457707.457860 | 10.98 | 3.34 | 0.32 |
| nc 29 | 459841.460022 | 1.06 | 0.06 | 0.06 |
| nc 30 | 473652.473826 | 19.98 | 0.59 | 0.03 |
| nc 31 | 789226.789789 | 1.17 | 0.39 | 0.35 |
| nc 32 | 878386.878507 | 2.52 | 0.07 | 0.03 |
| nc 33 | 910988.911157 | 59.73 | 2.89 | 0.05 |
Figure 6. Expression profiles of non-coding RNA species during in vitro growth and in vivo colonization.

Histograms illustrating strand-specific coverage per nucleotide across multiple loci identifying non-coding RNA species. Red lines represent reads sequenced and mapped from in vivo chick libraries. Blue (mid-log) and green (stationary) lines represent reads sequenced and mapped from in vitro libraries. ORF’s are labeled below each histogram in blue. Specific ncRNAs, A. anti-sense nc4, B. intergenic nc18, C. anti-sense nc21, are highlighted by grey boxes.
The abundance of most of these candidate ncRNAs increased during stationary phase compared to mid-log phase (Figure 7A), suggesting they are likely induced in response to increased stress and/or nutrient limitation. The abundance of these putative ncRNAs was also induced in vivo compared to mid-log cultures, though not as much as in stationary phase (Figure 7B and 7C), again suggesting their expression is likely regulated by similar stimuli and conditions encountered by C. jejuni during its transition from log to stationary growth in culture.
Figure 7. Differential expression of putative non-coding RNA species identified by RNAseq and Rfam analyses.
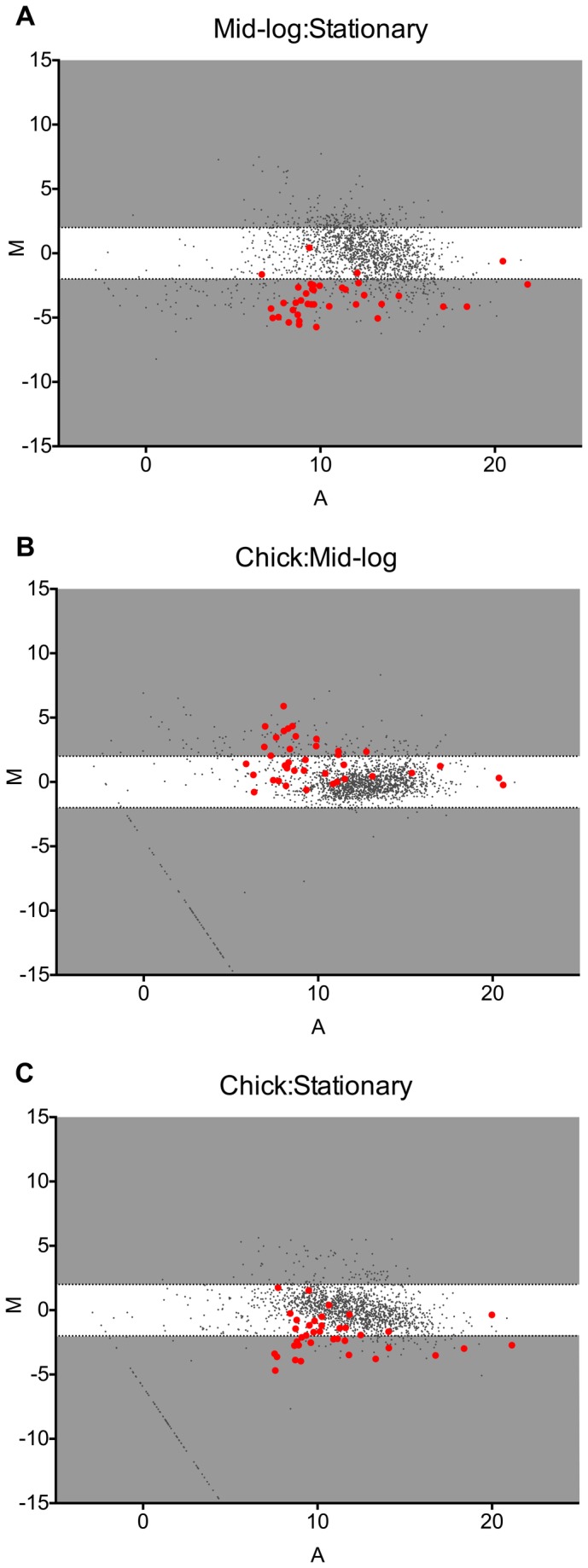
(A–C) The log2 of the ratio of abundances of each gene between the indicated conditions [M] plotted against the average log2 of abundance of that gene in all conditions [A]. For each plot, [M] and [A] values were based on data from three biological replicates from each in vivo and in vitro growth condition. ORFs are marked as dark grey circles. Non-coding RNAs are highlighted in red. Grey regions highlight expression differences greater-than and less-than 4-fold.
To investigate possible regulatory mechanisms of the newly identified ncRNAs we utilized the sRNA target prediction program TargetRNA [78]. Non-coding RNAs are a unique class of regulators that can regulate gene expression by different mechanisms, including altering mRNA interactions with the ribosome [79] and modulating mRNA stability [80–83]. Additionally, ncRNAs can act as cis-regulatory elements or as trans-regulatory elements [84]. We chose to focus on the intergenic ncRNAs for TargetRNA analysis, as these are more likely to act as trans-regulatory elements and potentially have multiple targets. TargetRNA identified at least 4 targets (p-value < 0.01) for eight intergenic encoded ncRNAs (Table 4). The pattern of abundance of many of these targets was the same or opposite of their cognate ncRNAs among the conditions tested (Table 4), suggesting their stability may be regulated by the ncRNA and lending further credence to the TargetRNA predictions. Those targets whose expression patterns were not well correlated with their cognate ncRNA may represent false predictions or correspond to ncRNA: mRNA interactions that do not significantly alter message stability. Further characterization of these putative ncRNAs and target mRNAs will likely yield key insights into C. jejuni regulatory mechanisms and pathways.
Table 4. mRNA targets of ncRNAs analyzed by TargetRNA and their differential expression in different growth conditions.
| ncRNA ID | Gene Name / locus number | TargetRNA Score | DESeq of mRNA Target |
||||
|---|---|---|---|---|---|---|---|
| Chick:ML | Chick:Stat | ML:Stat | |||||
| nc3 | CJJ81176_0481 | -84 | 3.01 | 1.02 | -2.82 | ||
| mviN | -80 | 1.04 | -1.91 | -1.89 | |||
| CJJ81176_0193 | -77 | -2.08 | 1.81 | 3.96 | |||
| CJJ81176_0024 | -77 | 2.11 | -6.92 | -13.98 | |||
| nc5 | CJJ81176_1304 | -96 | 1.16 | -1.62 | -1.79 | ||
| CJJ81176_0289 | -93 | 1.40 | 1.01 | 1.08 | |||
| CJJ81176_1389 | -81 | 1.37 | 1.35 | 1.04 | |||
| porA | -79 | -1.02 | -1.55 | -1.44 | |||
| nc6 | CJJ81176_0565 | -112 | 1.40 | -1.33 | -1.77 | ||
| tuf | -107 | 2.31 | -1.46 | -3.22 | |||
| rpsG | -105 | 1.61 | 1.64 | 1.06 | |||
| flgB | -102 | 2.63 | -4.63 | -11.63 | |||
| nc9 | chuA | -91 | 8.75 | 10.54 | 1.26 | ||
| pgk | -86 | -1.15 | -1.19 | 1.01 | |||
| neuB1 | -84 | -1.31 | 1.68 | 2.31 | |||
| fliN | -79 | 1.09 | -2.57 | -2.67 | |||
| nc14 | argC | -79 | -1.65 | -1.22 | 1.42 | ||
| mutY | -75 | -1.75 | 2.02 | 3.71 | |||
| CJJ81176_0579 | -74 | -1.78 | -2.41 | -1.29 | |||
| CJJ81176_0213 | -68 | -2.21 | 1.63 | 3.78 | |||
| nc18 | CJJ81176_1193 | -93 | 1.23 | -1.84 | -2.17 | ||
| CJJ81176_0481 | -84 | 3.01 | 1.02 | -2.82 | |||
| secF | -71 | -1.71 | 1.42 | 2.53 | |||
| CJJ81176_0973 | -70 | -1.79 | -1.53 | 1.22 | |||
| nc29 | CJJ81176_0193 | -96 | 1.16 | 1.81 | 3.96 | ||
| nuoM | -87 | -2.07 | 2.45 | 5.31 | |||
| CJJ81176_0481 | -84 | 3.01 | 1.01 | -2.82 | |||
| mviN | -80 | 1.04 | -1.90 | -1.90 | |||
| nc32 | CJJ81176_1746 | -85 | 21.01 | 7.06 | -2.85 | ||
| mobB | -84 | 1.64 | -2.03 | -3.19 | |||
| CJJ81176_1370 | -83 | -1.06 | -1.32 | -1.19 | |||
| nspC | -80 | -1.97 | -1.74 | 1.19 | |||
| nc33 | cutE | -93 | 2.38 | 4.59 | 2.02 | ||
| tlyA | -84 | -1.57 | 1.89 | 3.11 | |||
| CJJ81176_0076 | -79 | 1.27 | 2.42 | 1.98 | |||
| napL | -79 | -2.08 | -1.72 | 1.27 | |||
The small size of its genome, its lack of an Hfq homologue, and the relatively limited success of genomics-based approaches for identifying ncRNAs in C. jejuni [85] have led to speculations that C. jejuni does not encode the sizeable arsenal of ncRNAs found in many other bacteria. However, the discovery of numerous putative C. jejuni ncRNAs in this study, and in other recent RNAseq-based studies, suggests that C. jejuni does indeed possess a robust RNA-mediated regulation system and provides insights into the mechanisms by which this organism, which encodes so few canonical transcription factors, is able to effectively adapt to rapidly changing environmental conditions and niches.
The Campylobacter jejuni RNAseq transcriptome browser
The RNAseq data generated during the course of this study contains a wealth of information that will be a valuable resource to the numerous researchers studying C. jejuni. To make these data readily accessible to this community, we have generated coverage plots for each RNAseq dataset that can be visualized in the GenomeView browser [86] using the following link:
http://www.broadinstitute.org/software/genomeview/supplemental/CjVJD13/. This browser allows visualization of expression profiles of all annotated ORFs (under all growth conditions), as well as non-coding RNA species; including 5’-UTRs, anti-sense transcripts and intergenic ncRNAs.
Summary
This is the first report of the complete transcriptome of C. jejuni during colonization of the chicken cecum using RNAseq. Through our strand specific library preparation, we identified 51 probable non-coding RNA species, 29 of which were not previously annotated. We also determined the expression profile of C. jejuni during colonization, identifying 272 genes that are significantly differentially expressed in vivo. Additionally, we characterized the microbiota of the chick cecum during C. jejuni colonization. It was shown that C. jejuni represents ~5-10% of the bacterial population and that structural differences in microbiota profiles do not result in changes in the global C. jejuni transcriptome. C. jejuni must rapidly regulate gene expression in response to diverse environmental conditions; however, the regulatory mechanisms underpinning these responses are poorly understood. This work provides key insights into the genes and functions involved in the transition of C. jejuni from logarithmic to stationary growth and during its adaption to the chicken cecal environment, as well as revealing a number of putative regulatory ncRNAs that may play key roles in these adaptations.
Materials and Methods
Bacterial strains and growth conditions
C. jejuni strain DRH212 (an 81-176 streptomycin resistant derivative [87]) was grown on Mueller Hinton agar (BD, Sparks, MD) supplemented with 10% sheep blood (BA) or in Mueller Hinton Broth (MHB) (BD, Sparks, MD). C. jejuni was cultured at 42°C microaerobically in a tri-gas incubator, constantly maintained at 5% O2, 10% CO2, balanced with N2. Liquid cultures of C. jejuni were grown in MHB to mid-log or stationary phase (as monitored by optical density Ab600).
Chicken Colonization
Animal work carried out in this study followed the recommendations of the National Institutes of Health in the Guide for the Care and Use of Laboratory Animals. This protocol was approved by the University Committee on Use and Care of Animals at the University of Michigan (Protocol 10462-1). All efforts were made to minimize suffering of animals throughout the course of the experiment.
One-day-old chicks were inoculated orally with 1x103 CFU of C. jejuni in PBS. Cells used in the inoculum were grown on BA for ~16 h, washed and resuspended in PBS to 104 CFU/mL. Chicks were inoculated with 100 µl of cell suspension via oral gavage. Seven days post inoculation, chicks were sacrificed and cecal contents were removed. For RNA isolation, cecal contents were immediately suspended in 1 mL of RNAlater (Qiagen, Valencia CA) and frozen at -80°C. For colonization load and DNA extraction for 454-pyrosequencing, a subsample of cecal contents was removed and diluted in PBS. Colonization load was determined by serially diluting cecal contents and plating on Campylobacter selective media (BA supplemented with Vancomycin [40 µg/ml], Cefoparazone [40 µg/ml], Trimethoprim [10 µg/ml] and Cycloheximide [100 µg/ml]). Plates were incubated at 42°C under microaerobic conditions until countable colonies appeared (~2-3 days). CFU counts were standardized to gram wet weight of cecal content.
RNA isolation
In vitro RNA was isolated from C. jejuni grown in MHB under microaerobic conditions to mid-log and stationary phase (as monitored by optical density Ab600). An equal volume of RNA stop solution (95% EtOH, 5% phenol) was added to the cultures immediately after removal from microaerobic conditions and cells were harvested by centrifugation at 4°C. RNA was extracted with TRIzol® (Invitrogen, Grand Island, NY) as per manufacturer’s instructions. Contaminating DNA was removed with two treatments of TURBO™ DNase (Invitrogen) per manufacturer’s instructions. DNase treated RNA was cleaned with RNA clean and concentrate 25 columns after each DNase treatment step (Zymo Research, Irvine, CA). Conformation of DNA removal was assessed by PCR with three sets of primers ranging in amplification products of 100, 150, 250 bp and the Qubit dsDNA high sensitivity assay kit (Life Technologies) (data not shown).
For in vivo RNA isolation, cecal contents were harvested from individual birds and immediately submerged in 1 ml of RNAlater/cecum. C. jejuni was enriched from cecal contents as performed by Jerome et al. in an effort to deplete contaminating eukaryotic cells and cecal debris [88]. After C. jejuni enrichment, RNA was extracted with TRIzol® and contaminating DNA was removed with TURBO™ DNase as described previously. Purified DNA-free RNA from 5 birds (housed in the same brooder) was pooled to create enough starting DNA-free RNA for Illumina cDNA library construction.
Illumina Library Construction
Illumina cDNA libraries were constructed in a similar manner to Mandlik et al. [31]. Five micrograms of RNA was depleted of 16S and 23S rRNA species using the Gram Negative Ribo-Zero™ rRNA Removal Kit (Epicentre, Madison, WI). Removal of contaminating RNA species was assessed with the Agilent Bioanalyzer RNA 6000 nano chip (Agilent Technologies, Santa Clara, CA). Depleted mRNA was fragmented into 100-500 bp species with fragmentation buffer from the GeneChip® clean up module kit (Affymetrix, Santa Clara, CA). First-strand cDNA was synthesized with random hexamers, Actinomycin D and SuperScript III (Life Technologies, Grand Island, NY). Second strand cDNA was synthesized with dUTP replacing dTTP as described by Levin et al [89]. Double stranded cDNA ends were repaired and adenylated as described in the Illumina Truseq™ RNA sample preparation low throughput (LT) protocol (Illumina, San Diego, CA). Bar-coded Illumina adapters were ligated to the ends of the cDNA libraries and adapter-cDNA libraries were treated with Uracil-N-glycosylase (UNG) for 15 min at 37°C, followed by 95°C for 5 min. UNG-treaded cDNA was enriched by low-cycle PCR (8-cylces) with Illumina adapter specific primers. Final cDNA libraries were cleaned with two treatments of AMPureXP beads (Beckman Coulter, Brea, CA), and sequenced using 50 bp paired-end reads on an Illumina HiSeq2000 platform at the University of Michigan Sequencing Core.
Quantitative reverse transcriptase polymerase chain reaction
PCR primers used in this study are listed in Table S8 and were used to amplify nucleotide fragments of genes of interest between 100 and 150 bp. Isolation of RNA from in vitro and in vivo cultures (independent from RNAseq library preparations) was performed as mentioned previously. qRT-PCR was performed by using the Quantitect Sybr, Green RT-PCR kit (Qiagen, Valencia, CA) as by Taveirne et al. [17] and analyzed using the 2(-Delta Delta C(T)) method [90].
RNAseq analysis
Reads were aligned to the C. jejuni chromosome, pTet, and pVir sequence files (RefSeq NC_008787, NC_008770, and NC_008790) using BWA [91] version 5.9. Gene annotations were obtained from RefSeq and Rfam [75]. The overall fragment coverage of genomic regions corresponding to features such as ORFs and rRNAs was conducted as described [31]. Differential expression analysis was conducted using DESeq [45].
Chick ceca microbiota analysis
DNA extraction was done on untreated chick cecal content, in PBS, using Roche MagNA Pure Compact system according to the Roche MagNA Pure Nucleic Acid Isolation Kit I protocol instructions (Roche, Madison, WI). Amplification of the V3-V5 region of the 16S rRNA gene was accomplished using the Broad HMP protocol (HMP MDG Default Protocol v 4.2). Briefly, extracted DNA was used to construct DNA libraries targeting the V3-V5 hypervariable regions of the 16S rRNA gene using primers 357F (5’-CCTACGGGAGGCAGCAG-3’) with a B adaptor (5’-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-3’) and 926R (5’-CCGTCAATTCMTTTRAGT-3’) with a unique bar code to identify the target and an A adaptor (5’-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3’). A PCR reaction mixture of 1X AccuPrime PCR Buffer II (Invitrogen), 0.15 µl AccuPrime Taq DNA Polymerase High Fidelity, 0.2 µM Primer B, 0.2 µM uniquely bar-coded Primer A was used. DNA was amplified using the following conditions: 95°C for 2 min, 30 cycles of 95°C for 20 sec, 50°C for 30 sec and 72°C for 5 min. Amplicons were cleaned with the Agencourt AMPure XP and quantified using the Quant-it PicoGreen dsDNA kit (Invitrogen). Samples were pooled in equal proportion, cleaned again with AMPureXP, and sequenced on the Roche 454 GS Junior Titanium platform according to the manufacturer’s specifications. Analysis of 454 Pyrosequencing data was done using mothur (version 1.30.2) [43]. Mothur was used to group or assign bacterial 16S rRNA gene sequences into Operational Taxonomic Units (OTUs) using a 3% species level definition. Classifications were determined by comparing sequences to the Ribosomal Database Project [92]. Classified OTUs were used to determine the relative abundance of bacterial phyla and family in each sample. Principal coordinates analysis (PCoA) was used to assess community similarity among all samples and was calculated based on Yue and Clayton-based distance matrix [93]. An AMOVA (analysis of molecular variance) was performed to determine statistical differences between the community structures of each treatment group. Heatmaps of microbiome data (OTUs) were made using the heatmap command in R [94]. The heatmap command re-orders the data in the rows and the columns separately, so that similar data are grouped together by hierarchical clustering, as shown by dendrograms for the rows and the columns. The Euclidean distance was used in the clustering [95].
Supporting Information
Structural changes to the chicken cecal microbiota. A heatmap of the top 25 (>1%) operational taxonomic units (OTUs) found in the chick cecal microbiome shows differences between the four-treatment groups (PBS control n=5; brooder 1 n=5; brooder 2 n=5; brooder 3 n=5). The distance between treatment groups was measured by a dendrogram in R statistical program. The PBS controls (blue bars) group very distinctly from chicks colonized with C. jejuni in brooder 1 (black bars) and brooders 2 and 3 (green and red bars). Bacterial phylum, family and OTU number are listed on the heatmap. The heatmap scale ranges from 0 to 100% relative OTU abundance.
(TIF)
Comparison of gene expression between biological replicates used in DESeq analysis. Scatter plots of the log2 (FPKMO) abundance of open reading frames obtained from RNAseq libraries created from in vitro mid-log samples (A–C), in vitro stationary phase samples (D–F) and in vivo chick samples (G–I). R2 (coefficient of determination) was determined by linear regression, represented by the sold line.
(TIF)
Genes increased in abundance in vivo compared to in vitro mid-log phase cultures. Listed are genes with increased abundance during in vivo colonization compared to in vitro mid-exponential phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vivo compared to in vitro mid-log phase cultures. Listed are genes with decreased abundance during in vivo colonization compared to in vitro mid-exponential phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes increased in abundance in vivo compared to in vitro stationary phase cultures. Listed are genes with increased abundance during in vivo colonization compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vivo compared to in vitro stationary phase cultures. Listed are genes with decreased abundance during in vivo colonization compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes increased in abundance in vitro mid-log compared to in vitro stationary phase cultures. Listed are genes with increased abundance during in vitro mid-exponential phase broth grown cultures compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vitro mid-log compared to in vitro stationary phase cultures. Listed are genes with decreased abundance during in vitro mid-exponential phase broth grown cultures compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Non-coding RNAs that align to pTet. Location of non-coding RNA species identified by RNAseq that map to the pTET plasmid.
(DOCX)
Primers used in qRT-PCR experiments. Listed are the primer sets, along with their sequence, used in qRT-PCR experiments. Each primer set is annotated by which gene it will amplify.
(DOCX)
Results of DEseq analysis of RNAseq data for all annotated C. jejuni ORFs. Each annotated gene in the C. jejuni genome, and two plasmids, is listed in locus number order. DESeq analysis for each gene under each growth condition comparison is given, including the base mean reads for each growth condition and the fold change (and log2 fold change) between each condition. padj is the adjusted P values for each gene and resVarA/B are the variance values within each set of biological replicates.
(XLS)
Acknowledgments
The authors would like to thank Bob Lyons and his staff at the University of Michigan DNA Sequencing Core for their help and guidance with the sequencing of RNAseq libraries. We would also like to thank Susan Foltin at the University of Michigan for her help and guidance with the 454-pysrocequencing. Finally we would like to thank Thomas Abeel from the Broad Institute of MIT and Harvard and the VIB Department of Plant Systems Biology at Ghent University for his help in creating the C. jejuni RNAseq transcriptome browser.
Funding Statement
This project was supported in part with federal funds from the USDA to MET [Grant Number: 2012- 67012-19704], the NIAID to VJD [Grant Number: AI-076608] and from the NHLBI to CMT [Grant Number: T32-HL007749]. Additionally, this project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No.: HHSN272200900018C to JL. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the USDA, the NIAID, the DHHS or the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Altekruse SF, Stern NJ, Fields PI, Swerdlow DL (1999) Campylobacter jejuni--an emerging foodborne pathogen. Emerg Infect Dis 5: 28-35. doi:10.3201/eid0501.990104. PubMed: 10081669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park SF (2002) The physiology of Campylobacter species and its relevance to their role as foodborne pathogens. Int J Food Microbiol 74: 177-188. doi:10.1016/S0168-1605(01)00678-X. PubMed: 11981968. [DOI] [PubMed] [Google Scholar]
- 3. Jacobs-Reitsma WL, [!(surname)!], Wagenaar J (2000) Campylobacter in the Food Supply; Nachamkin I, Szymanski CM, Blaser MJ. Washington, DC: ASM Press. [Google Scholar]
- 4. Davis LM, Kakuda T, DiRita VJ (2009) A Campylobacter jejuni znuA orthologue is essential for growth in low-zinc environments and chick colonization. J Bacteriol 191: 1631-1640. doi:10.1128/JB.01394-08. PubMed: 19103921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hendrixson DR, DiRita VJ (2004) Identification of Campylobacter jejuni genes involved in commensal colonization of the chick gastrointestinal tract. Mol Microbiol 52: 471-484. doi:10.1111/j.1365-2958.2004.03988.x. PubMed: 15066034. [DOI] [PubMed] [Google Scholar]
- 6. Wassenaar TM, van der Zeijst BA, Ayling R, Newell DG (1993) Colonization of chicks by motility mutants of Campylobacter jejuni demonstrates the importance of flagellin A expression. J Gen Microbiol 139 6: 1171-1175. doi:10.1099/00221287-139-6-1171. PubMed: 8360610. [DOI] [PubMed] [Google Scholar]
- 7. Weerakoon DR, Borden NJ, Goodson CM, Grimes J, Olson JW (2009) The role of respiratory donor enzymes in Campylobacter jejuni host colonization and physiology. Microb Pathog 47: 8-15. doi:10.1016/j.micpath.2009.04.009. PubMed: 19397993. [DOI] [PubMed] [Google Scholar]
- 8. Weingarten RA, Grimes JL, Olson JW (2008) Role of Campylobacter jejuni respiratory oxidases and reductases in host colonization. Appl Environ Microbiol 74: 1367-1375. doi:10.1128/AEM.02261-07. PubMed: 18192421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C et al. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665-668. doi:10.1038/35001088. PubMed: 10688204. [DOI] [PubMed] [Google Scholar]
- 10. Pearson BM, Gaskin DJ, Segers RP, Wells JM, Nuijten PJ et al. (2007) The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828). J Bacteriol 189: 8402-8403. doi:10.1128/JB.01404-07. PubMed: 17873037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fouts DE, Mongodin EF, Mandrell RE, Miller WG, Rasko DA et al. (2005) Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. PLOS Biol 3: e15. doi:10.1371/journal.pbio.0030015. PubMed: 15660156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brás AM, Chatterjee S, Wren BW, Newell DG, Ketley JM (1999) A novel Campylobacter jejuni two-component regulatory system important for temperature-dependent growth and colonization. J Bacteriol 181: 3298-3302. PubMed: 10322038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caldwell MB, Guerry P, Lee EC, Burans JP, Walker RI (1985) Reversible expression of flagella in Campylobacter jejuni . Infect Immun 50: 941-943. PubMed: 4066041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palyada K, Threadgill D, Stintzi A (2004) Iron acquisition and regulation in Campylobacter jejuni . J Bacteriol 186: 4714-4729. doi:10.1128/JB.186.14.4714-4729.2004. PubMed: 15231804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sampathkumar B, Napper S, Carrillo CD, Willson P, Taboada E et al. (2006) Transcriptional and translational expression patterns associated with immobilized growth of Campylobacter jejuni . Microbiology 152: 567-577. doi:10.1099/mic.0.28405-0. PubMed: 16436444. [DOI] [PubMed] [Google Scholar]
- 16. Stintzi A (2003) Gene expression profile of Campylobacter jejuni in response to growth temperature variation. J Bacteriol 185: 2009-2016. doi:10.1128/JB.185.6.2009-2016.2003. PubMed: 12618466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taveirne ME, Sikes ML, Olson JW (2009) Molybdenum and tungsten in Campylobacter jejuni: their physiological role and identification of separate transporters regulated by a single ModE-like protein. Mol Microbiol 74: 758-771. doi:10.1111/j.1365-2958.2009.06901.x. PubMed: 19919002. [DOI] [PubMed] [Google Scholar]
- 18. Woodall CA, Jones MA, Barrow PA, Hinds J, Marsden GL et al. (2005) Campylobacter jejuni gene expression in the chick cecum: evidence for adaptation to a low-oxygen environment. Infect Immun 73: 5278-5285. doi:10.1128/IAI.73.8.5278-5285.2005. PubMed: 16041056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hendrixson DR (2006) A phase-variable mechanism controlling the Campylobacter jejuni FlgR response regulator influences commensalism. Mol Microbiol 61: 1646-1659. doi:10.1111/j.1365-2958.2006.05336.x. PubMed: 16899076. [DOI] [PubMed] [Google Scholar]
- 20. Karlyshev AV, Linton D, Gregson NA, Wren BW (2002) A novel paralogous gene family involved in phase-variable flagella-mediated motility in Campylobacter jejuni . Microbiology 148: 473-480. PubMed: 11832511. [DOI] [PubMed] [Google Scholar]
- 21. Guerry P, Szymanski CM, Prendergast MM, Hickey TE, Ewing CP et al. (2002) Phase variation of Campylobacter jejuni 81-176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro . Infect Immun 70: 787-793. doi:10.1128/IAI.70.2.787-793.2002. PubMed: 11796612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bacon DJ, Szymanski CM, Burr DH, Silver RP, Alm RA et al. (2001) A phase-variable capsule is involved in virulence of Campylobacter jejuni 81-176. Mol Microbiol 40: 769-777. doi:10.1046/j.1365-2958.2001.02431.x. PubMed: 11359581. [DOI] [PubMed] [Google Scholar]
- 23. Hänninen ML, Hakkinen M, Rautelin H (1999) Stability of related human and chicken Campylobacter jejuni genotypes after passage through chick intestine studied by pulsed-field gel electrophoresis. Appl Environ Microbiol 65: 2272-2275. PubMed: 10224037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Boer P, Wagenaar JA, Achterberg RP, van Putten JP, Schouls LM et al. (2002) Generation of Campylobacter jejuni genetic diversity in vivo. Mol Microbiol 44: 351-359. doi:10.1046/j.1365-2958.2002.02930.x. PubMed: 11972775. [DOI] [PubMed] [Google Scholar]
- 25. Gaynor EC, Wells DH, MacKichan JK, Falkow S (2005) The Campylobacter jejuni stringent response controls specific stress survival and virulence-associated phenotypes. Mol Microbiol 56: 8-27. doi:10.1111/j.1365-2958.2005.04525.x. PubMed: 15773975. [DOI] [PubMed] [Google Scholar]
- 26. Yun J, Jeon B, Barton YW, Plummer P, Zhang Q et al. (2008) Role of the DksA-like protein in the pathogenesis and diverse metabolic activity of Campylobacter jejuni . J Bacteriol 190: 4512-4520. doi:10.1128/JB.00105-08. PubMed: 18456813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stintzi A, Marlow D, Palyada K, Naikare H, Panciera R et al. (2005) Use of genome-wide expression profiling and mutagenesis to study the intestinal lifestyle of Campylobacter jejuni . Infect Immun 73: 1797-1810. doi:10.1128/IAI.73.3.1797-1810.2005. PubMed: 15731081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cattoir V, Narasimhan G, Skurnik D, Aschard H, Roux D et al. (2013) Transcriptional response of mucoid Pseudomonas aeruginosa to human respiratory mucus. mBio 3: e00410-00412 PubMed: 23143799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chaudhuri RR, Yu L, Kanji A, Perkins TT, Gardner PP et al. (2011) Quantitative RNA-seq analysis of the Campylobacter jejuni transcriptome. Microbiology 157: 2922-2932. doi:10.1099/mic.0.050278-0. PubMed: 21816880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dugar G, Herbig A, Förstner KU, Heidrich N, Reinhardt R et al. (2013) High-Resolution Transcriptome Maps Reveal Strain- Specific Regulatory Features of Multiple Campylobacter jejuni Isolates. PLOS Genet 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mandlik A, Livny J, Robins WP, Ritchie JM, Mekalanos JJ et al. (2011) RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10: 165-174. doi:10.1016/j.chom.2011.07.007. PubMed: 21843873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S et al. (2010) The primary transcriptome of the major human pathogen Helicobacter pylori . Nature 464: 250-255. doi:10.1038/nature08756. PubMed: 20164839. [DOI] [PubMed] [Google Scholar]
- 33. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57-63. doi:10.1038/nrg2484. PubMed: 19015660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown L, Elliott T (1996) Efficient translation of the RpoS sigma factor in Salmonella typhimurium requires host factor I, an RNA-binding protein encoded by the hfq gene. J Bacteriol 178: 3763-3770. PubMed: 8682778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajnsdorf E, Régnier P (2000) Host factor Hfq of Escherichia coli stimulates elongation of poly(A) tails by poly(A) polymerase I. Proc Natl Acad Sci U S A 97: 1501-1505. doi:10.1073/pnas.040549897. PubMed: 10677490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muffler A, Fischer D, Hengge-Aronis R (1996) The RNA-binding protein HF-I, known as a host factor for phage Qbeta RNA replication, is essential for rpoS translation in Escherichia coli . Genes Dev 10: 1143-1151. doi:10.1101/gad.10.9.1143. PubMed: 8654929. [DOI] [PubMed] [Google Scholar]
- 37. Muffler A, Traulsen DD, Fischer D, Lange R, Hengge-Aronis R (1997) The RNA-binding protein HF-I plays a global regulatory role which is largely, but not exclusively, due to its role in expression of the sigmaS subunit of RNA polymerase in Escherichia coli . J Bacteriol 179: 297-300. PubMed: 8982015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei BL, Brun-Zinkernagel AM, Simecka JW, Prüss BM, Babitzke P et al. (2001) Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli . Mol Microbiol 40: 245-256. doi:10.1046/j.1365-2958.2001.02380.x. PubMed: 11298291. [DOI] [PubMed] [Google Scholar]
- 39. Barnard FM, Loughlin MF, Fainberg HP, Messenger MP, Ussery DW et al. (2004) Global regulation of virulence and the stress response by CsrA in the highly adapted human gastric pathogen Helicobacter pylori . Mol Microbiol 51: 15-32. PubMed: 14651608. [DOI] [PubMed] [Google Scholar]
- 40. Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM (1993) Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175: 4744-4755. PubMed: 8393005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fields JA, Thompson SA (2008) Campylobacter jejuni CsrA mediates oxidative stress responses, biofilm formation, and host cell invasion. J Bacteriol 190: 3411-3416. doi:10.1128/JB.01928-07. PubMed: 18310331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G (2011) A cis-encoded antisense small RNA regulated by the HP0165-HP0166 two-component system controls expression of ureB in Helicobacter pylori . J Bacteriol 193: 40-51. doi:10.1128/JB.00800-10. PubMed: 20971914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537-7541. doi:10.1128/AEM.01541-09. PubMed: 19801464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, et al (2008) Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLOS ONE 3: e2945. doi:10.1371/journal.pone.0002945. PubMed: 18698407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. doi:10.1186/gb-2010-11-10-r106. PubMed: 20979621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Field LH, Headley VL, Payne SM, Berry LJ (1986) Influence of iron on growth, morphology, outer membrane protein composition, and synthesis of siderophores in Campylobacter jejuni . Infect Immun 54: 126-132. PubMed: 2944843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holmes K, Mulholland F, Pearson BM, Pin C, McNicholl-Kennedy J et al. (2005) Campylobacter jejuni gene expression in response to iron limitation and the role of Fur. Microbiology 151: 243-257. doi:10.1099/mic.0.27412-0. PubMed: 15632442. [DOI] [PubMed] [Google Scholar]
- 48. Palyada K, Threadgill D, Stintzi A (2004) Iron acquisition and regulation in Campylobacter jejuni . J Bacteriol 186: 4714-4729. doi:10.1128/JB.186.14.4714-4729.2004. PubMed: 15231804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu F, Zeng X, Haigh RD, Ketley JM, Lin J (2010) Identification and characterization of a new ferric enterobactin receptor, CfrB, in Campylobacter . J Bacteriol 192: 4425-4435. doi:10.1128/JB.00478-10. PubMed: 20585060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Vliet AH, Wooldridge KG, Ketley JM (1998) Iron-responsive gene regulation in a Campylobacter jejuni fur mutant. J Bacteriol 180: 5291-5298. PubMed: 9765558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Veen HW (1997) Phosphate transport in prokaryotes: molecules, mediators and mechanisms. Antonie Van Leeuwenhoek 72: 299-315. doi:10.1023/A:1000530927928. PubMed: 9442271. [DOI] [PubMed] [Google Scholar]
- 52. Wösten MM, Parker CT, van Mourik A, Guilhabert MR, van Dijk L et al. (2006) The Campylobacter jejuni PhosS/PhosR operon represents a non-classical phosphate-sensitive two-component system. Mol Microbiol 62: 278-291. doi:10.1111/j.1365-2958.2006.05372.x. PubMed: 16956379. [DOI] [PubMed] [Google Scholar]
- 53. Palyada K, Sun YQ, Flint A, Butcher J, Naikare H et al. (2009) Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni . BMC Genomics 10: 481. doi:10.1186/1471-2164-10-481. PubMed: 19835633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Day WA Jr., Sajecki JL, Pitts TM, Joens LA (2000) Role of catalase in Campylobacter jejuni intracellular survival. Infect Immun 68: 6337-6345. doi:10.1128/IAI.68.11.6337-6345.2000. PubMed: 11035743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grant KA, Park SF (1995) Molecular characterization of katA from Campylobacter jejuni and generation of a catalase-deficient mutant of Campylobacter coli by interspecific allelic exchange. Microbiology 141(6): 1369-1376. doi:10.1099/13500872-141-6-1369. [DOI] [PubMed] [Google Scholar]
- 56. Elvers KT, Wu G, Gilberthorpe NJ, Poole RK, Park SF (2004) Role of an inducible single-domain hemoglobin in mediating resistance to nitric oxide and nitrosative stress in Campylobacter jejuni and Campylobacter coli . J Bacteriol 186: 5332-5341. doi:10.1128/JB.186.16.5332-5341.2004. PubMed: 15292134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith CK, Abuoun M, Cawthraw SA, Humphrey TJ, Rothwell L et al. (2008) Campylobacter colonization of the chicken induces a proinflammatory response in mucosal tissues. FEMS Immunol Med Microbiol 54: 114-121. doi:10.1111/j.1574-695X.2008.00458.x. PubMed: 18647351. [DOI] [PubMed] [Google Scholar]
- 58. Smith CK, Kaiser P, Rothwell L, Humphrey T, Barrow PA et al. (2005) Campylobacter jejuni-induced cytokine responses in avian cells. Infect Immun 73: 2094-2100. doi:10.1128/IAI.73.4.2094-2100.2005. PubMed: 15784550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van Deun K, Pasmans F, Ducatelle R, Flahou B, Vissenberg K et al. (2008) Colonization strategy of Campylobacter jejuni results in persistent infection of the chicken gut. Vet Microbiol 130: 285-297. doi:10.1016/j.vetmic.2007.11.027. PubMed: 18187272. [DOI] [PubMed] [Google Scholar]
- 60. Connell S, Meade KG, Allan B, Lloyd AT, Kenny E et al. (2012) Avian resistance to Campylobacter jejuni colonization is associated with an intestinal immunogene expression signature identified by mRNA sequencing. PLOS ONE 7: e40409. doi:10.1371/journal.pone.0040409. PubMed: 22870198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Corcionivoschi N, Alvarez LA, Sharp TH, Strengert M, Alemka A et al. (2012) Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 12: 47-59. doi:10.1016/j.chom.2012.05.018. PubMed: 22817987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Zoete MR, Keestra AM, Roszczenko P, van Putten JP (2010) Activation of human and chicken toll-like receptors by Campylobacter spp. Infect Immun 78: 1229-1238. doi:10.1128/IAI.00897-09. PubMed: 20038539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Batchelor RA, Pearson BM, Friis LM, Guerry P, Wells JM (2004) Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150: 3507-3517. doi:10.1099/mic.0.27112-0. PubMed: 15470128. [DOI] [PubMed] [Google Scholar]
- 64. Guerry P, Pope PM, Burr DH, Leifer J, Joseph SW et al. (1994) Development and characterization of recA mutants of Campylobacter jejuni for inclusion in attenuated vaccines. Infect Immun 62: 426-432. PubMed: 8300203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bacon DJ, Alm RA, Burr DH, Hu L, Kopecko DJ et al. (2000) Involvement of a plasmid in virulence of Campylobacter jejuni 81-176. Infect Immun 68: 4384-4390. doi:10.1128/IAI.68.8.4384-4390.2000. PubMed: 10899834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fani R, Brilli M, Fondi M, Lió P (2007) The role of gene fusions in the evolution of metabolic pathways: the histidine biosynthesis case. BMC Evol Biol 7 Suppl 2: S4. doi:10.1186/1471-2148-7-S1-S4. PubMed: 17767732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McNally DJ, Hui JP, Aubry AJ, Mui KK, Guerry P et al. (2006) Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81-176 using a focused metabolomics approach. J Biol Chem 281: 18489-18498. doi:10.1074/jbc.M603777200. PubMed: 16684771. [DOI] [PubMed] [Google Scholar]
- 68. Pei ZH, Ellison3rd, Blaser MJ (1991) Identification, purification, and characterization of major antigenic proteins of Campylobacter jejuni. The Journal of biological chemistry 266: 16363-16369
- 69. Linton D, Allan E, Karlyshev AV, Cronshaw AD, Wren BW (2002) Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni . Mol Microbiol 43: 497-508. doi:10.1046/j.1365-2958.2002.02762.x. PubMed: 11985725. [DOI] [PubMed] [Google Scholar]
- 70. Sellars MJ, Hall SJ, Kelly DJ (2002) Growth of Campylobacter jejuni supported by respiration of fumarate, nitrate, nitrite, trimethylamine-N-oxide, or dimethyl sulfoxide requires oxygen. J Bacteriol 184: 4187-4196. doi:10.1128/JB.184.15.4187-4196.2002. PubMed: 12107136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Weerakoon DR, Olson JW (2008) The Campylobacter jejuni NADH:ubiquinone oxidoreductase (complex I) utilizes flavodoxin rather than NADH. J Bacteriol 190: 915-925. doi:10.1128/JB.01647-07. PubMed: 18065531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jackson RJ, Elvers KT, Lee LJ, Gidley MD, Wainwright LM et al. (2007) Oxygen reactivity of both respiratory oxidases in Campylobacter jejuni: the cydAB genes encode a cyanide-resistant, low-affinity oxidase that is not of the cytochrome bd type. J Bacteriol 189: 1604-1615. doi:10.1128/JB.00897-06. PubMed: 17172349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Myers JD, Kelly DJ (2005) A sulphite respiration system in the chemoheterotrophic human pathogen Campylobacter jejuni . Microbiology 151: 233-242. doi:10.1099/mic.0.27573-0. PubMed: 15632441. [DOI] [PubMed] [Google Scholar]
- 74. Stintzi A (2003) Gene expression profile of Campylobacter jejuni in response to growth temperature variation. J Bacteriol 185: 2009-2016. doi:10.1128/JB.185.6.2009-2016.2003. PubMed: 12618466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL et al. (2009) Rfam: updates to the RNA families database. Nucleic Acids Res 37: D136-D140. doi:10.1093/nar/gkp725. PubMed: 18953034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gundogdu O, Bentley SD, Holden MT, Parkhill J, Dorrell N et al. (2007) Re-annotation and re-analysis of the Campylobacter jejuni NCTC11168 genome sequence. BMC Genomics 8: 162. doi:10.1186/1471-2164-8-162. PubMed: 17565669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wurtzel O, Sesto N, Mellin JR, Karunker I, Edelheit S et al. (2012) Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol 8: 583 PubMed: 22617957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tjaden B (2008) TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 36: W109-W113. doi:10.1093/nar/gkn264. PubMed: 18477632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bouvier M, Sharma CM, Mika F, Nierhaus KH, Vogel J (2008) Small RNA binding to 5' mRNA coding region inhibits translational initiation. Mol Cell 32: 827-837. doi:10.1016/j.molcel.2008.10.027. PubMed: 19111662. [DOI] [PubMed] [Google Scholar]
- 80. Massé E, Escorcia FE, Gottesman S (2003) Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli . Genes Dev 17: 2374-2383. doi:10.1101/gad.1127103. PubMed: 12975324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Morita T, Maki K, Aiba H (2005) RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev 19: 2176-2186. doi:10.1101/gad.1330405. PubMed: 16166379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Prévost K, Salvail H, Desnoyers G, Jacques JF, Phaneuf E et al. (2007) The small RNA RyhB activates the translation of shiA mRNA encoding a permease of shikimate, a compound involved in siderophore synthesis. Mol Microbiol 64: 1260-1273. doi:10.1111/j.1365-2958.2007.05733.x. PubMed: 17542919. [DOI] [PubMed] [Google Scholar]
- 83. Urban JH, Vogel J (2008) Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLOS Biol 6: e64. doi:10.1371/journal.pbio.0060064. PubMed: 18351803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Waters LS, Storz G (2009) Regulatory RNAs in bacteria. Cell 136: 615-628. doi:10.1016/j.cell.2009.01.043. PubMed: 19239884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Livny J, Teonadi H, Livny M, Waldor MK (2008) High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLOS ONE 3: e3197. doi:10.1371/journal.pone.0003197. PubMed: 18787707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y (2012) GenomeView: a next-generation genome browser. Nucleic Acids Res 40: e12. doi:10.1093/nar/gkr995. PubMed: 22102585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hendrixson DR, Akerley BJ, DiRita VJ (2001) Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol 40: 214-224. doi:10.1046/j.1365-2958.2001.02376.x. PubMed: 11298288. [DOI] [PubMed] [Google Scholar]
- 88. Jerome JP, Bell JA, Plovanich-Jones AE, Barrick JE, Brown CT et al. (2011) Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLOS ONE 6: e16399. doi:10.1371/journal.pone.0016399. PubMed: 21283682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA et al. (2010) Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7: 709-715. doi:10.1038/nmeth.1491. PubMed: 20711195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. doi:10.1006/meth.2001.1262. PubMed: 11846609. [DOI] [PubMed] [Google Scholar]
- 91. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754-1760. doi:10.1093/bioinformatics/btp324. PubMed: 19451168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cole JR, Wang Q, Cardenas E, Fish J, Chai B et al. (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141-D145. doi:10.1093/nar/gkp353. PubMed: 19004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yue JC, Clayton MK (2005) A similarity measure based on species proportions. Commun Stat Theor Methods 34: 2123-2131. doi:10.1080/STA-200066418. [Google Scholar]
- 94. R Development Core Team (2010) R: A language and environment for statisitcal programing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 95. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95: 14863-14868. doi:10.1073/pnas.95.25.14863. PubMed: 9843981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structural changes to the chicken cecal microbiota. A heatmap of the top 25 (>1%) operational taxonomic units (OTUs) found in the chick cecal microbiome shows differences between the four-treatment groups (PBS control n=5; brooder 1 n=5; brooder 2 n=5; brooder 3 n=5). The distance between treatment groups was measured by a dendrogram in R statistical program. The PBS controls (blue bars) group very distinctly from chicks colonized with C. jejuni in brooder 1 (black bars) and brooders 2 and 3 (green and red bars). Bacterial phylum, family and OTU number are listed on the heatmap. The heatmap scale ranges from 0 to 100% relative OTU abundance.
(TIF)
Comparison of gene expression between biological replicates used in DESeq analysis. Scatter plots of the log2 (FPKMO) abundance of open reading frames obtained from RNAseq libraries created from in vitro mid-log samples (A–C), in vitro stationary phase samples (D–F) and in vivo chick samples (G–I). R2 (coefficient of determination) was determined by linear regression, represented by the sold line.
(TIF)
Genes increased in abundance in vivo compared to in vitro mid-log phase cultures. Listed are genes with increased abundance during in vivo colonization compared to in vitro mid-exponential phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vivo compared to in vitro mid-log phase cultures. Listed are genes with decreased abundance during in vivo colonization compared to in vitro mid-exponential phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes increased in abundance in vivo compared to in vitro stationary phase cultures. Listed are genes with increased abundance during in vivo colonization compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vivo compared to in vitro stationary phase cultures. Listed are genes with decreased abundance during in vivo colonization compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes increased in abundance in vitro mid-log compared to in vitro stationary phase cultures. Listed are genes with increased abundance during in vitro mid-exponential phase broth grown cultures compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Genes decreased in abundance in vitro mid-log compared to in vitro stationary phase cultures. Listed are genes with decreased abundance during in vitro mid-exponential phase broth grown cultures compared to in vitro stationary phase broth grown cultures, as determined by DESeq analysis (materials and methods). Only genes significantly differentially regulated (>4-fold difference in abundance, padj<0.05) are listed. padj<0.05, is a corrected p-value analogous to a false detection rate of < 5%. Genes are grouped by functional classification and by their C. jejuni 81-176 locus numbers and gene name or function.
(DOCX)
Non-coding RNAs that align to pTet. Location of non-coding RNA species identified by RNAseq that map to the pTET plasmid.
(DOCX)
Primers used in qRT-PCR experiments. Listed are the primer sets, along with their sequence, used in qRT-PCR experiments. Each primer set is annotated by which gene it will amplify.
(DOCX)
Results of DEseq analysis of RNAseq data for all annotated C. jejuni ORFs. Each annotated gene in the C. jejuni genome, and two plasmids, is listed in locus number order. DESeq analysis for each gene under each growth condition comparison is given, including the base mean reads for each growth condition and the fold change (and log2 fold change) between each condition. padj is the adjusted P values for each gene and resVarA/B are the variance values within each set of biological replicates.
(XLS)