

Abstract
Oxysterols bind the seven-spanner transmembrane protein Smoothened and potently activate vertebrate Hedgehog signaling, a pathway essential in embryonic development, adult stem cell maintenance and cancer. It is unknown, however, if oxysterols are important for normal vertebrate Hedgehog signaling, and whether antagonizing oxysterols can inhibit the Hedgehog pathway. We developed azasterols that block Hedgehog signaling by binding the oxysterol-binding site of Smoothened. We show that the binding site for oxysterols and azasterols maps to the extracellular, cysteine-rich domain of Smoothened, and is completely separable from the site bound by other small molecule modulators, located within the heptahelical bundle of Smoothened. Smoothened mutants in which oxysterol binding is abolished no longer respond to oxysterols, and cannot be maximally activated by the Hedgehog ligand. Our results show that oxysterol binding to vertebrate Smoothened is required for normal Hedgehog signaling, and that targeting the oxysterol binding site is an effective strategy to inhibit Smoothened.
Introduction
Cell-cell signaling via the Hedgehog (Hh) pathway is critical for numerous aspects of metazoan embryonic development and regeneration, while excessive Hh activity is involved in many cancers1,2. Among poorly understood aspects of Hh signal transduction is the question of how Hh signals are relayed across the plasma membrane, via the functional interaction between the multi-spanning membrane protein Patched (Ptch), which functions as the Hh receptor, and the seven-spanner Smoothened (Smo), a member of the Frizzled family of membrane proteins. In the absence of the Hh ligand, Ptch inhibits Smo through an unknown mechanism, ensuring that signals are not relayed to the cytoplasm. Hh signaling is initiated by binding of the Hh ligand to Ptch, leading to Smo activation and the consequent initiation of a specific transcriptional program driven by the Gli transcription factors.
A major unanswered question in Hh signaling is the mechanism of Smo regulation. Like other seven-spanners, Smo equilibrates between active and inactive conformations, and it is thought that this equilibrium is controlled by a ligand3, whose identity has remained elusive. Consistent with this hypothesis, vertebrate Smo harbors within its heptahelical bundle a binding site4 (hereby “Site A”) reminiscent of G protein-coupled receptors (GPCRs). Site A is targeted by numerous small molecules, including Smo inhibitors (such as the alkaloid cyclopamine4, SANT15, or the FDA-approved Smo inhibitor GDC04496) and activators (such as SAG5,7 and purmorphamine8); however, no endogenous small molecule that binds Site A has been identified so far.
The only natural molecules that activate Smo are oxysterols, oxidized cholesterol derivatives with potent effects on many cellular processes, including signaling and metabolism. Vertebrate Hh signaling is stimulated by oxysterols carrying hydroxyl groups on the isooctyl side chain of the molecule9,10, the most potent being 20(S)-hydroxycholesterol (20-OHC, Fig. 1a)11,12. Oxysterols activate Smo allosterically, by binding to a second site, distinct from Site A12 (hereby “Site B”). Several important questions about the participation of oxysterols in Hh signaling are open. First, it is unknown where Site B is located in Smo, and whether it is separable from Site A. Second, while Site A binds both Smo activators and inhibitors, we only know of oxysterol activators that bind Site B, raising the question of whether Site B can also be targeted by inhibitors. Finally, although oxysterols activate Smo, it is unknown if their binding to Smo is required for Smo activation during normal Hh signaling.
Figure 1.

22-azacholesterol inhibits vertebrate Hh signaling
(a) Structure of 22(S)-azacholesterol (22-NHC, 1) and 20(S)-hydroxycholesterol (20(S)-OHC).
(b) Shh Light II cells were treated with various concentrations of Shh, in the presence of increasing amounts of 22-NHC, and Hh pathway activation was measured by luciferase assay. Error bars represent standard deviation (n=4 independent experiments). 22-NHC inhibits Hh pathway activation by Shh but does not significantly change the EC50 of Shh.
(c) As in (b) but Hh signaling was activated by various concentrations of the 20-OHC analog, 20-OHC-Pent. 22-NHC inhibits Hh pathway activation by 20-OHC-Pent, without significantly changing the EC50.
(d) As in (b) but Hh signaling was activated by various concentrations of SAG. 22-NHC does not inhibit Hh pathway activation by SAG but decreases the EC50 for SAG.
(e) Smo−/− MEFs were rescued by stable expression of mSmo or the constitutively active mutant mSmoM2. Transcription of the Hh target gene, Gli1, was measured by Q-PCR in the absence or presence of 22-NHC (20 μM) or SANT1 (2 μM). Error bars indicate standard deviation (n=3 independent experiments). 22-NHC does not inhibit SmoM2.
(f) Shh Light II cells were stimulated with Shh, in the presence of increasing amounts of 22-NHC, with the addition of cylopamine or cyclopamine-KAAD. Hh pathway activity was assayed as in (b)
(g) As in (f) but with addition of SANT1, GDC0449 or itraconazole.
We have developed azasterols that block Hh signaling triggered by the Hh ligand and by 20-OHC. These compounds compete with 20-OHC for binding Smo, indicating that they bind Site B; in contrast, azasterols do not compete with small molecules that bind Site A. We used azasterol and oxysterol affinity probes to map Site B to the extracellular, cysteine-rich domain (CRD) of vertebrate Smo; furthermore, we show that Site A and B are completely separable. When Site B is mutated, 20-OHC binding to Smo is abolished and 20-OHC can no longer activate Smo, thus validating functionally the surprising identification of Site B within SmoCRD. These oxysterol-insensitive mutants have greatly decreased responsiveness to Hh compared to wild-type Smo, indicating that binding of endogenous oxysterols to Smo is necessary for high level vertebrate Hh signaling.
Results
A Hedgehog inhibitor that mimics sterol depletion
20-OHC strongly activates vertebrate Hh signaling11,12; however, the consequences for Hh signaling of blocking 20-OHC are not known. To obtain a potential 20-OHC inhibitor, we synthesized 22-azacholesterol (22-NHC, Fig. 1a). When tested in Hh-responsive NIH-3T3 cells, 22-NHC inhibited signaling by Sonic Hedgehog (Shh) in a dose-dependent manner, with an IC50 of about 3 μM (Fig. 1b); importantly, cells exposed to up to 20 μM 22-NHC for 36 hours did not show signs of toxicity. Synthesis of 22-NHC generates a C-20 stereocenter, resulting in 2 possible diastereomers, 22(S)-NHC and 22(R)-NHC. Our synthesis generated predominantly 22(S)-NHC, and pure 22(S)-NHC recapitulated the inhibitory activity of the mix. 22-NHC did not affect the EC50 of Shh but it decreased maximum stimulation (Fig. 1b), indicative of non-competitive inhibition.
We first sought to determine the step in Hh signal transduction inhibited by 22-NHC. Binding of Shh to Patched1 (Ptch1) causes its removal from cilia13, which triggers Smoothened (Smo) activation. 22-NHC had no effect on the disappearance of Ptch1 from cilia in response to Shh (Supplementary Results, Supplementary Fig. 1a), suggesting that 22-NHC inhibits Hh signaling downstream of Ptch1.
We next asked if 22-NHC inhibits two Smo activators, oxysterols and SAG. As oxysterol we used 20-OHC-Pent (see below), a 20-OHC analog that we synthesized, which is slightly more potent than 20-OHC; similar results were obtained with 20-OHC. 22-NHC inhibited 20-OHC-Pent in a dose-dependent manner and non-competitively (Fig. 1c): 22-NHC did not significantly change the EC50 but decreased the maximal stimulation level. In contrast, 22-NHC did not inhibit SAG (Fig. 1d); in fact, 22-NHC caused an increase in SAG responsiveness (note the decreased EC50 in the presence of 22-NHC). While we do not understand the basis for this, one possibility is that it is caused by 22-NHC promoting Smo translocation to cilia (see below), which might sensitize cells to SAG. Additionally, 22-NHC did not inhibit mSmoM2 (Fig. 1e), an oncogenic mouse Smo (mSmo) mutant locked in active conformation14, or constitutive Hh signaling in SuFu−/− MEFs15 (Supplementary Fig. 1b), in which the pathway is activated downstream of Smo. Together, these results indicate that 22-NHC targets either Smo or an unknown component between Ptch1 and Smo. The inhibition profile of 22-NHC mirrors the effect of sterol depletion, which blocks activation of the vertebrate Hh pathway by Shh but not by SAG, mSmoM2 or by loss of SuFu16.
Finally, we asked if 22-NHC synergizes with inhibitors of Smo. 22-NHC did not synergize with cyclopamine (Cyc), cyclopamine-KAAD, SANT1, GDC0449 or itraconazole to inhibit Shh (Fig. 1f and 1g); conversely, these inhibitors did not significantly affect the IC50 of 22-NHC. Thus 22-NHC does not interact with other Smo inhibitors, suggesting a different mechanism for 22-NHC.
22-NHC binds Smoothened at site distinct from cyclopamine
Shh stimulation causes rapid accumulation of Smo in primary cilia17, a process blocked by some Site A Smo inhibitors, such as SANT1. We tested the possibility that 22-NHC inhibit this early event in Hh signaling. 22-NHC had no effect on Smo accumulation at cilia in response to Shh (Supplementary Fig. 2a), suggesting that 22-NHC can inhibit cilia-localized Smo. Interestingly, 22-NHC by itself caused ciliary accumulation of Smo (Supplementary Fig. 2b), although to lower levels than Shh (Supplementary Fig. 2a); this effect occurred rapidly, with ciliary Smo reaching maximum after 3-hour treatment with 22-NHC (Supplementary Fig. 2c). This behavior of 22-NHC is reminiscent of Cyc18,19, a Hh inhibitor that binds Smo at Site A and causes Smo accumulation in cilia. To test if 22-NHC also binds Site A, we performed binding assays in cells with the fluorescent derivative, BODIPY-cyclopamine4 (BODIPY-Cyc). 22-NHC did not compete binding of BODIPY-Cyc to mSmo, similar to 20-OHC (Supplementary Fig. 3a); as expected, SANT1 abolished BODIPY-Cyc binding. We also synthesized a fluorescent derivative of SANT1, BODIPY-SANT1 (Supplementary Fig. 3b), which retains SANT1 activity (Supplementary Fig. 3c). 22-NHC did not compete binding of BODIPY-SANT1 to mSmo, while SANT1 did (Supplementary Fig. 3a). Together, these results indicate that 22-NHC does not bind Site A of mSmo, in contrast to Cyc.
To test if 22-NHC binds mSmo at a different site, we developed a ligand affinity assay. We focused on the alkyl side chain of 22-NHC as potential site for covalent attachment to beads, and assayed analogs with modified side chains (compounds 2–5, Supplementary Fig. 4a). Analogs bearing N-propyl or N-ethyl groups retain the inhibitory activity of 22-NHC, albeit they are less potent (Supplementary Fig. 4b). Unexpectedly, C-20 stereochemistry does not matter in these analogs: both S and R diastereomers inhibit Hh signaling, in contrast to the strict C-20 stereochemistry required for Hh activation by oxysterols12 (see also below). For analogs bearing N-hydroxypropyl or N-hydroxyethyl groups, the S diastereomers are modestly active, while the R diastereomers are inactive as Hh inhibitors (Supplementary Fig. 4b).
Based on the structure-function analysis above, we synthesized 22-NHC-PEG-NH2 (compound 6, Supplementary note), a derivative with a polyethylene glycol (PEG) linker, which was covalently attached to amine-reactive beads (Fig. 2a). As source of mSmo protein, we used detergent extracts of 293T cells stably expressing mSmo tagged with mCherry; this fusion protein is active, rescuing Hh signaling in Smo−/− mouse embryonic fibroblasts (MEFs) (see below). 22-NHC beads efficiently captured mSmo (Fig. 2b), preferentially the glycosylated species with low electrophoretic mobility, representing post-Golgi mSmo; this suggests that it is mainly mature mSmo that binds 22-NHC. Binding of mSmo to 22-NHC beads was competed in a dose-dependent manner by free 22-NHC added to the binding reaction (Fig. 2b), suggesting that it was specific. SAG, SANT1 and GDC0449 had no effect on mSmo binding to 22-NHC beads (Fig. 2b), consistent with 22-NHC not binding Site A. Itraconazole, a Smo inhibitor that does not bind Site A20 also had no effect (Fig. 2b). As negative control, we used beads derivatized with the PEG linker incorporated into 22-NHC-PEG-NH2 (Fig. 2a); these beads captured negligible amounts of mSmo (Fig. 2b). As further proof of specificity, 22-NHC beads did not bind a seven-spanner related to Smo, mouse Frizzled-7 (mFz7) (Supplementary Fig. 5a). Finally, binding was specific for vertebrate Smo: 22-NHC beads did not bind Drosophila Smo (DrSmo, Supplementary Fig. 5a) but bound Xenopus laevis Smo (xSmo, Supplementary Fig. 5b). Together, these results demonstrate that 22-NHC binds vertebrate Smo at a different site from Site A and from the hypothetical itraconazole site.
Figure 2.
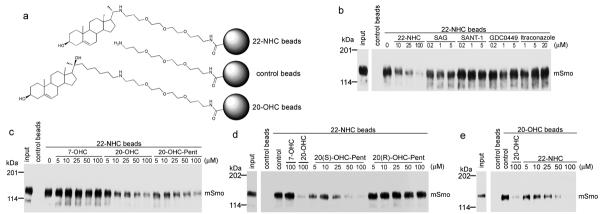
22-NHC binds Smo at the oxysterol-binding site
(a) Schematic of 22-NHC and 20-OHC affinity matrices. 22-NHC and 20-OHC are covalently attached to agarose beads via a polyethylene glycol (PEG) linker. Control beads carry only the PEG linker.
(b) 22-NHC beads were incubated with detergent extracts of 293T cells expressing mSmo-Cherry, in the presence of the indicated concentrations of competitor compounds. The beads were washed and bound protein was eluted, separated by SDS-PAGE and immunoblotted with anti-Cherry antibodies. A portion of the extract was analyzed in parallel, to show input. MSmo binds specifically to 22-NHC beads, and binding is not competed by other Smo inhibitors.
(c) As in (b) but with the addition of the inactive oxysterol 7-OHC, or the active oxysterols, 20-OHC and 20-OHC-Pent. Binding of mSmo to 22-NHC beads is competed in a dose-dependent manner by 20-OHC and 20-OHC-Pent, but not by 7-OHC.
(d) As in (b) but with the addition of 20(S)-OHC-Pent and 20(R)-OHC-Pent. Binding of mSmo to 22-NHC beads is competed by the active S diastereomer but not by the inactive R diastereomer.
(e) As in (b) but using 20-OHC beads, in the presence of free 22-NHC or 20-OHC. Binding of mSmo to 20-OHC beads is competed in a dose-dependent manner by free 22-NHC and 20-OHC.
The full immunoblots for this figure are shown in Supplementary figure 11.
22-NHC binds the oxysterol-binding site of Smoothened
Oxysterols are allosteric activators of Smo that bind to Site B, which is distinct from Site A and the itraconazole site12. We asked if 22-NHC binds Smo at Site B by oxysterol competition experiments. Binding of mSmo to 22-NHC beads was competed in a dose-dependent manner by two active oxysterols, 20-OHC and 20-OHC-Pent, while 7-hydroxycholesterol (7-OHC), an inactive oxysterol9, had no effect (Fig. 2c); similar results were obtained for xSmo (Supplementary Fig. 5b). Thus 22-NHC and oxysterols that activate the Hh pathway compete for binding to vertebrate Smo.
Binding of 20-OHC is strictly stereospecific: 20(S)-OHC binds Smo while 20(R)-OHC does not12. If 22-NHC binds Site B it would be expected that oxysterol competition be also stereospecific. To test this prediction, we prepared pure diastereomers of 20-OHC-Pent (Supplementary Fig. 5c). Only the active diastereomer 20(S)-OHC-Pent (see below) competed mSmo binding to 22-NHC beads, while the inactive diastereomer 20(R)-OHC-Pent had no effect (Fig. 2d). Thus oxysterol C-20 stereochemistry is critical for competing binding of 22-NHC to Smo.
To perform reciprocal binding experiments, we generated 20-OHC beads (Fig. 2a) using an amine derivative of 20-OHC that incorporates the same PEG linker used for 22-NHC beads. These 20-OHC beads bound mSmo (Fig. 2e), as described for a similar 20-OHC affinity matrix12. Importantly, 22-NHC inhibited mSmo binding to 20-OHC beads in a dose-dependent manner (Fig. 2e). Like 22-NHC beads, 20-OHC beads did not bind DrSmo (Supplementary Fig. 5d), but bound xSmo (Supplementary Fig. 5e), consistent with oxysterols activating the vertebrate, but not the Drosophila Hh pathway. Furthermore, in a variety of Smo binding experiments (see below) 22-NHC and 20-OHC beads behaved virtually identically. Together, these results indicate that 22-NHC and 20-OHC bind to the same site (Site B), and suggest that 22-NHC inhibits Hh signaling by competing with binding of 20-OHC to mSmo.
20-OHC and 22-NHC bind to Smoothened extracellular domain
The location of Site B has not been determined, and is unknown if Site B and Site A are separable, particularly in view of their allosteric interaction. We used mSmo deletion analysis (Supplementary Fig. 6a) and 22-NHC and 20-OHC ligand affinity to map location of Site B. MSmo lacking the extracellular, cysteine-rich domain (mSmoΔCRD) did not bind to 22-NHC beads (Supplementary Fig. 6b) or to 20-OHC beads (Supplementary Fig. 6c), in contrast to full-length mSmo and to mSmo lacking the intracellular C-terminal domain (mSmoΔICD) (Supplementary Fig. 6d). Importantly, mSmoΔCRD bound BODIPY-Cyc (Supplementary Fig. 7a) and was functional in Smo−/− MEFs (see below), indicating it was properly folded. These results show that the CRD is required for mSmo binding to 20-OHC and 22-NHC.
We next asked if mSmoCRD, produced in cultured cells as a soluble, secreted protein, is sufficient to bind 22-NHC and 20-OHC. MSmoCRD bound to 22-NHC beads and was competed by free 22-NHC, similar to full-length mSmo (Fig. 3a). Importantly, secreted DrSmoCRD did not bind 22-NHC beads (Supplementary Fig. 7b), as expected from the lack of binding of DrSmo. Binding of mSmoCRD to 22-NHC beads was competed by 20-OHC and 20-OHC-Pent, but not by 7-OHC (Fig. 3b), suggesting that, like full-length mSmo, mSmoCRD binds oxysterols that activate Hh signaling. Furthermore, the stereochemistry of oxysterol competition was identical as for mSmo: 20(S)-OHC-Pent but not 20(R)-OHC-Pent competed mSmoCRD binding to 22-NHC beads (Fig. 3c). We obtained identical results using 20-OHC beads: mSmoCRD bound 20-OHC beads with an affinity similar to that of mSmo, and was competed by 20(S)-OHC-Pent but not by 20(R)-OHC-Pent (Fig. 3d). Furthermore, binding of mSmo and mSmoCRD to 20-OHC beads was competed by the active oxysterol 25-OHC, but not by the inactive 7-ketocholesterol (7-KC)9 (Supplementary Fig. 7c). Together, these results demonstrate that Site B resides in SmoCRD and is completely separable from Site A. Interestingly, it was proposed that SmoCRD might bind sterols, based on structural similarity between the sterol-binding protein NPC2 and the CRD of Frizzled proteins21; our findings confirm this prediction.
Figure 3.
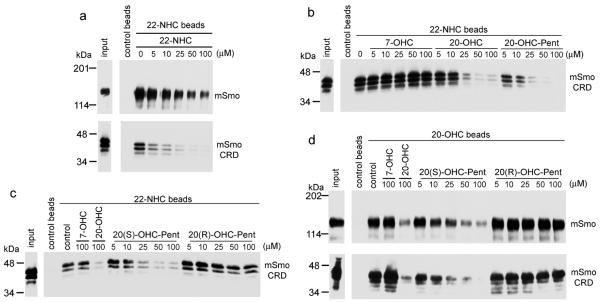
Oxysterols and 22-NHC bind the extracellular cysteine-rich domain (CRD) of vertebrate Smo
(a) Secreted HA-tagged mSmoCRD and detergent extracts of 293T cells expressing mSmo-Cherry were tested for binding to 22-NHC beads, in the presence of free 22-NHC. MSmoCRD binds 22-NHC beads, similar to mSmo.
(b) MSmoCRD binding to 22-NHC beads was assayed as in (a), in the presence of 7-OHC, 20-OHC or 20-OHC-Pent. Only the active sterols 20-OHC and 20-OHC-Pent compete mSmoCRD binding to 22-NHC beads, while the inactive 7-OHC does not.
(c) As in (b) but in the presence of the diastereomers 20(S)-OHC-Pent and 20(R)-OHC-Pent. Only the active S diastereomer competes mSmoCRD binding to 22-NHC beads.
(d) MSmoCRD and mSmo binding to 20-OHC beads was assayed as in (c). Both mSmoCRD and mSmo bind 20-OHC beads and are competed by 20(S)-OHC-Pent, but not 20(R)-OHC-Pent. The binding affinities of mSmoCRD and mSmo to 20-OHC beads are similar.
The full immunoblots for this figure are shown in Supplementary figure 11.
Oxysterol structural requirements in Hedgehog signaling
To better understand the effect of oxysterols on Hh signaling, we asked what structural aspects of 20-OHC are important for mSmo activation and ciliary recruitment. We first focused on the isooctyl tail, and synthesized 20-OHC analogs with progressively shorter tails (Fig. 4a). Shortening the tail by 1 or 2 carbons (20-OHC-Pent and 20-OHC-Bu) preserved activity (Fig. 4b). As for 20-OHC12, the diastereomers 20(S)-OHC-Pent and 20(S)-OHC-Bu were active (Fig. 4b) while 20(R)-OHC-Pent and 20(R)-OHC-Bu were inactive (Supplementary Fig. 8a); this correlated with their ability to recruit mSmo to cilia (Fig. 4c and 4d). Analogs of 20-OHC with shorter tails (20-OHC-Pr, 20-OHC-Et and 20-OHC-Me) did not stimulate Hh signaling (Supplementary Fig. 8a), indicating that a tail of 6 carbon atoms is the minimum for mSmo activation. These shorter tail analogs did not inhibit pathway activation by Shh (Supplementary Fig. 8b). Interestingly, 20(S)-OHC-Pr caused accumulation of mSmo at cilia, while 20-OHC-Me, 20-OHC-Et (R and S) and 20(R)-OHC-Pr had no effect (Fig. 4c and 4d). Furthermore, 20(S)-OHC-Pr potentiated Shh activity (Supplementary Fig. 8b), an effect similar to the Shh sensitization seen with glucocorticoids which cause mSmo accumulation in cilia without activating transcription22. Thus 20(S)-OHC-Pr is sufficient to localize mSmo to cilia but cannot activate it, suggesting that it induces a mSmo conformation distinct from the active one induced by analogs with longer tails.
Figure 4.

Structural requirements for oxysterol activation of Smo
(a) Structures of 20-OHC analogs used in this study (compounds 7–13). All analogs except 20-OHC-Me (7) have a C-20 stereocenter, and pure S and R diastereomers were isolated and assayed separately.
(b) Shh Light II cells were treated with varying concentrations of the oxysterols 20(S)-OHC, 20(S)-OHC-Pent, 20(S)-OHC-PentSat and 20(S)-OHC-Bu, followed by measuring Hh pathway activity by luciferase assay. The inactive oxysterol, 7-OHC, was used as negative control. Error bars represent standard deviation (n=4 independent experiments).
(c) NIH-3T3 cells were incubated overnight with the indicated oxysterols (10 μM). Cells were then fixed and processed for immunofluorescence with rabbit anti-Smo antibodies (to detect endogenous Smo) and mouse anti-acetylated tubulin antibodies (to visualize primary cilia). SAG (1 μM) and 7-OHC (10 μM) were used as positive and negative control, respectively. The graph shows the percentage of Smo-positive cilia. At least 150 cilia were analyzed per condition. Error bars represent the sub-sampling standard deviation of the fraction of positive cilia (see Online Methods).
(d) As in (c), but with box plots showing the fluorescence intensity of endogenous Smo at cilia. For each condition, the Smo signal was normalized to the intensity of the 20(S)-OHC treatment. The lower and upper bounds of each box represent the 25th and 75th percentile of the distribution of Smo intensity at cilia, while the horizontal line represents the median intensity across the entire population of cilia.
Finally, we asked if the Δ5 double bond and the 3Δ-OH group are important. The saturated analog, 20-OHC-PentSat (Fig. 4a), activated Hh signaling as the S diastereomer (Fig. 4b), while the R diastereomer was inactive (Supplementary Fig. 8a); thus the Δ5 double bond is not required for activity. A 3Δ-methyl ether analog of 20-OHC-Pent (20-OHC-Pent-3Δ–OMe, Fig. 4a) did not activate or inhibit Hh signaling (Supplementary Fig. 8c), and had no effect on mSmo binding to 22-NHC beads (Supplementary Fig. 8d). These results indicate that a free 3Δ-OH group is absolutely required for 20-OHC activity.
Role of oxysterols in vertebrate Hedgehog signaling
Although oxysterols activate Smo, it is not known what role their interaction with Smo plays in Hh signaling. To generate mSmo mutants that do not bind oxysterols, we relied on the homology between the CRDs of Smo and Frizzled (Fz) proteins. It was proposed that SmoCRD binds sterols similar to how FzCRD binds the palmityl residue of Wnt proteins21. Based on the crystal structure of mFz8CRD bound to Xwnt823, we focused on a stretch of 8 amino acids in mSmo, whereby the corresponding sequence in mFz8 includes 5 amino acids that form contacts with the palmityl residue23 (Fig. 5a). This stretch is conserved among vertebrate Smo orthologs but not in DrSmo, which does not bind oxysterols. To test if this stretch is important for oxysterol binding, we swapped amino acids 112–119 of mSmo for the corresponding amino acids of DrSmo. The resulting mSmo mutant (mSmoDrSmoCRDmut) did not bind 22-NHC beads, (Supplementary Fig. 9a), while the secreted mSmoCRDDrSmoCRDmut did not bind 20-OHC beads (Supplementary Fig. 9b). Importantly, mSmoDrSmoCRDmut bound BODIPY-Cyc (Supplementary Fig. 9c), indicating proper folding and an intact Site A.
Figure 5.

Oxysterol binding to Smo is required for high level Hh signaling
(a) Alignment of a portion of CRDs of mouse Smo (mSmo), chicken Smo (gSmo), Xenopus Smo (xSmo), zebrafish Smo (zfSmo), Drosophila Smo (DrSmo) and mouse Fz8 (mFz8). The stretch indicated with red lines contains 5 residues that, in mFz8, contact the Xwnt8 palmityl moiety (Q71, F72, P74, L75 and I78).
(b) Cherry-tagged mSmo, mSmoL112D, mSmoW113Y or mSmoS114Y were tested for binding to 22-NHC and 20-OHC beads, in the presence or absence of 20-OHC (100 μM). MSmoL112D and mSmoW113Y do not bind 22-NHC and 20-OHC beads. The full immunoblot is shown in Supplementary figure 11.
(c) Smo−/− MEFs rescued with mSmo, mSmoL112D, mSmoW113Y or mSmoS114Y were incubated overnight with DMSO control, SAG (1 μM), 20-OHC (10 μM) or Shh. The graph shows the percentage of Smo-positive cilia. Error bars represent the sub-sampling standard deviation of the fraction of positive cilia (see Online Methods). Between 131–207 cilia were analyzed per condition. MSmoL112D and mSmoW113Y have a defective response to 20-OHC and Shh.
(d) As in (c), but with box plots showing Smo fluorescence intensity at cilia. For each condition, the Smo signal was normalized to the intensity of the SAG treatment for the respective cell line.
(e) As in (c), but cells were processed for Q-PCR, to measure Gli1 transcription. For each treatment, Gli1 levels were normalized to the level induced by SAG in the respective cell line. Error bars show standard deviation (n=3 independent experiments). MSmoL112D and mSmoW113Y do not respond to 20-OHC and have a reduced responsiveness to Shh.
To determine if mSmoDrSmoCRDmut and mSmoΔCRD respond to oxysterols, we generated Smo−/− MEFs that stably express the proteins at low levels, and assayed their activation by SAG and 20-OHC, by immunofluorescence (to measure ciliary recruitment of mSmo, Supplementary Fig. 9d and 9e) and by Q-PCR (to measure the transcriptional output of the Hh pathway, Supplementary Fig. 9f). Both mSmoDrSmoCRDmut and mSmoΔCRD rescued the response of Smo−/− MEFs to SAG, indicating that they are fully functional in activating the downstream steps of Hh signaling; however, mSmoDrSmoCRDmut and mSmoΔCRD did not respond to 20-OHC, in contrast to mSmo. These results validate our mapping of the oxysterol-binding site within mSmoCRD.
We identified two mSmo point mutants defective in oxysterol binding by individually mutating residues in the LWS sequence (amino acids 112–114) to the corresponding DYY sequence in DrSmo. As shown in figure 5b, mSmoL112D and mSmoW113Y no longer bound 22-NHC and 20-OHC beads, in contrast to mSmoS114Y and mSmo. MSmoL112D and mSmoW113Y showed a prominent post-Golgi band on SDS-PAGE (Fig. 5b) and bound BODIPY-Cyc (Supplementary Fig. 9g), indicating proper folding.
To test the function of oxysterol binding to mSmo, we compared Hh signaling in Smo−/− MEFs stably expressing mSmo, mSmoL112D, mSmoW113Y or mSmoS114Y (Fig. 5c–e). As expected, all 4 proteins rescued the response to SAG. Consistent with loss of oxysterol binding, mSmoL112D and mSmoW113Y did not respond to 20-OHC, in contrast to mSmo and mSmoS114Y. Interestingly, mSmoL112D and mSmoW113Y had a greatly reduced response to Shh compared to mSmo and mSmoS114Y. Similarly, a significantly lower response to Shh was observed for mSmoΔCRD (Supplementary Fig. 9h and 9i) and the double mutant mSmoL112D/W113Y (Supplementary Fig. 9j). In conclusion, binding of oxysterols to the CRD is required for high-level mSmo activation by Shh; in contrast, low-level mSmo activation by Shh still occurs in absence of oxysterol binding. This suggests that Hh signaling controls mSmo via Site A, or possibly via both Site A and Site B (see Discussion).
Conservation and divergence in Smoothened regulation
In spite of Smo conservation, only vertebrate Smo binds oxysterols, raising the question of whether Smo regulation is conserved. To begin addressing this issue, we asked if DrSmo retains any signaling activity in vertebrate cells. When stably expressed in Smo−/− MEFs, DrSmo did not localize to cilia (Supplementary Fig. 10a) and was thus inactive (Supplementary Fig. 10b); this result was not surprising, as cilia are not involved in Hh signaling in Drosophila.
To direct DrSmo to cilia, we examined the ciliary localization determinants of mSmo. Since mSmoΔICD does not localize to cilia24, we tested if mSmoICD is sufficient for ciliary localization. We generated chimeras in which mSmoICD replaced the cytoplasmic tail of two seven-spanners that do not traffic to cilia, mouse Frizzled7 (mFz7) and the rat muscarinic acetylcholine receptor M2 (rMAChR). Stably expressed mFz7mSmoICD localized to cilia in Smo−/−MEFs (Supplementary Fig. 10c), while rMAChRmSmoICD did not but was strongly recruited to cilia by treatment with either agonist (acetylcholine) or antagonist (scopolamine) (Supplementary Fig. 10c). We interpret this behavior of rMAChRmSmoICD as result of improved folding due to agonist or antagonist binding. Both mFz7mSmoICD and rMAChRmSmoICD were inactive in Hh signaling, even in the presence of agonists Wnt3a (Fig. 6a) and acetylcholine, respectively (Fig. 6b). These results show that mSmoICD is sufficient for ciliary localization but not for activating Hh signaling.
Figure 6.

Conserved and divergent aspects of Smo signaling
(a) Smo−/− MEFs, stably expressing mSmo or the cilia-localized chimera mFz7mSmoICD were incubated with control media, 20-OHC (10 μM), Shh, or wnt3a. The cells were processed for Q-PCR, to measure Gli1 transcription. Error bars show standard deviation (n=3 independent experiments). MFz7mSmoICD did not rescue Hh signaling in Smo−/− cells, irrespective of the presence of Wnt3a.
(b) As in (a), but with stable expression of the chimera rMAcChRmSmoICD, which is recruited to cilia by treatment with acetylcholine (AcCh, 100 μM). RMAcChRmSmoICDdoes not rescue Hh signaling, in the presence or absence of AcCh.
(c) As in (a) but with stable expression of low levels of DrSmomSmoICD. DrSmomSmoICD is constitutively active, and is not further activated by 20-OHC (10 μM), Shh, or SAG (400 nM). In contrast, DrSmo is inactive in Smo−/− MEFs.
(d) As in (c), but with addition of 20 μM forskolin (FSK), to block Hh signaling downstream of Smo. Signaling by both mSmo and DrSmomSmoICD is blocked by FSK.
(e) Schematic of the mSmo protein and of the location of Sites A and B. For each site, activators are in blue, while inhibitors are in red.
(f) Regulation of vertebrate Smo. Inhibition of Ptch by Shh results in Smo Site A activation; it is unclear if Site B is also activated by Shh. The oxysterol 20-OHC, which binds to Site B in the extracellular domain of Smo, potentiates Site A activation. Active Smo then signals to the cytoplasm.
To determine if DrSmo can signal at cilia, we generated a chimera (DrSmomSmoICD) that consists of DrSmo CRD and heptahelical bundle, followed by mSmoICD; as expected, DrSmomSmoICD localized to cilia in Smo−/−MEFs (Supplementary Fig. 10d). Interestingly, DrSmomSmoICD strongly activated Hh signaling (Fig. 6c), indicating that the DrSmo portion of the chimera is active in vertebrate cells. DrSmomSmoICD was constitutively active, and could not be further stimulated by 20-OHC, SAG or Shh (Fig. 6c); it was, however, inhibited by forskolin, which blocks Hh signaling downstream of Smo (Fig. 6d). The lack of response to 20-OHC and SAG is consistent with DrSmomSmoICD not binding these two molecules. The inability of Shh to stimulate DrSmomSmoICD indicates that mPtch cannot repress DrSmomSmoICD at cilia, thus suggesting DrSmo is regulated differently from mSmo. Finally, in contrast to mSmo, DrSmomSmoICD was not inhibited by sterol depletion (Supplementary Fig. 10e and 10f), indicating that oxysterols are not required for DrSmomSmoICD activation.
Discussion
Our findings suggest a mechanism for how the vertebrate Hh pathway is modulated by oxysterols. During Hh signaling, Shh relieves the inhibition exerted by Ptch on Smo, and an open question is how Smo is regulated. Endogenous small molecules are hypothesized to control the equilibrium between the active and the inactive conformation of Smo3, in turn determining the output of Hh signaling at the membrane. Oxysterols9,10, particularly 20-OHC11,12, are so far the only metabolites that activate Hh signaling, acting as Smo allosteric activators12. We describe 22-NHC, a Hh inhibitor that acts by a novel mechanism, competing 20-OHC binding to Smo. 22-NHC inhibits Shh non-competitively, consistent with 22-NHC binding the allosteric Smo Site B. Surprisingly, Site B maps to the extracellular CRD of Smo, and we show is completely separable from Site A (Fig. 6e). We demonstrate that 20-OHC binding to SmoCRD is required for high-level Smo activation. Interestingly, Shh can still activate Smo mutants lacking a functional Site B, although at greatly reduced levels; thus Site A is sufficient for Smo to respond to Shh. These results suggest that, during Hh signaling, Smo is activated by two synergistic inputs: Shh/Ptch-dependent activation of Site A and allosteric activation by oxysterols binding Site B (Fig. 6f). Many aspects of this mechanism remain to be elucidated, including measuring endogenous 20-OHC levels, elucidating its biosynthesis, and determining if 20-OHC binding to Site B is controlled by Shh/Ptch, or it represents an independent input.
Mapping the 20-OHC binding site to SmoCRD confirms the prediction that Frizzled-type CRDs are related to sterol binding proteins such as the lysosomal cholesterol carrier, NPC221. We used the crystal structure of the mFz8-Xwnt8 complex23 to mutate mSmoCRD residues that align with mFz8CRD residues that bind the palmitate on Xwnt8. These Smo mutants were defective in 20-OHC binding, suggesting that oxysterol binding likely resembles mFz8CRD binding to palmitate; structural studies will be needed to determine exactly how SmoCRD binds 20-OHC. Interestingly, for two other sterol-binding membrane proteins, the sterol binding sites also map to soluble portions: the Niemann-Pick protein 1 (NPC1) binds oxysterols via its N-terminal domain projecting in the lysosomal lumen25, and the SREBP cleavage-activating protein (SCAP) binds cholesterol via a large ER luminal loop26. It will be important to determine if this is a general feature among sterol-binding membrane proteins.
How are Sites A and B of Smo regulated during Hh signaling? No endogenous small molecule that binds Site A is known, and it is unclear if such a molecule would be an agonist (which Ptch would prevent from reaching Smo) or antagonist (which Ptch would deliver to Smo). Our results indicate that Ptch controls Smo at least in part through Site A, but they cannot distinguish between these two alternatives. The situation is clearer for Site B, which, during Hh signaling, needs be occupied by an endogenous activator, perhaps 20-OHC – this conclusion is based on the inhibitory effect of sterol depletion and of blocking 20-OHC binding to Smo. The endogenous concentration of oxysterols like 20-OHC is unknown, but is likely much lower than the micromolar EC50 for Hh activation. Although higher local concentrations might exist in cells, endogenous oxysterol levels are perhaps too low to activate Smo by themselves, but high enough to synergize with Site A activation by an endogenous agonist. One advantage of such a mechanism is that it allows a Shh-independent way to modulate signaling, while ensuring that activation remains dependent on Shh/Ptch. Different tissues might have different oxysterol levels and profiles, thus allowing for varying levels of Hh signaling. Alternatively, in some cells, oxysterols might reach levels at which they significantly activate Hh signaling in a ligand/Ptch-independent manner, a possibility with important implications in cancer. Whole animal studies will be important in determining if the Hh pathway is differentially modulated by oxysterols in various tissues.
How does the allosteric interaction between Sites B and A result in Smo activation? It seems likely that SmoCRD binds 20-OHC and the resulting complex interacts with the heptahelical bundle of Smo, contributing to stabilizing the active conformation of Site A. An active Site A is required for oxysterols to stimulate Hh signaling, as Site A inhibitors such as cyclopamine and SANT1 inhibit oxysterols9,10,12. SmoCRD, however, is not required for Smo activation by synthetic agonists that bind Site A like SAG, suggesting that Site B plays only a modulating role during Shh stimulation. Perhaps endogenous activation of Site A is weaker than that elicited by SAG, and thus oxysterols are required for high-level Smo activation. Detailed biochemical and structural studies will be needed to determine how the interaction between Sites A and B controls Smo activity.
The azasterol 22-NHC represents the first Site B inhibitor of Smo. 22-NHC inhibits Smo activated by Shh and 20-OHC, but not by SAG, and cannot inhibit the constitutively active SmoM2 mutant. Thus 22-NHC recapitulates the inhibitory effect of sterol depletion and of cholesterol biosynthesis defects on vertebrate Hh signaling16. The simplest interpretation is that sterol depletion removes the endogenous activator of Site B, perhaps 20-OHC. It should be pointed out that blocking HMG-CoA reductase with statins without also depleting sterols does not block Hh signaling16, thus the effect of 22-NHC is not explained by general inhibition of cholesterol biosynthesis. We cannot exclude the possibility that, in addition to binding Smo, 22NHC might also block conversion of cholesterol into an unknown metabolite, such as an oxysterol.
Unlike vertebrate Smo, no small molecules are known that bind DrSmo. Furthermore, we find DrSmo does not bind oxysterols and is not inhibited by sterol depletion. How is then DrSmo activated, and is this mechanism conserved? We assayed the activity of a portion of DrSmo consisting of CRD and heptahelical bundle in mammalian cells, after targeting it to cilia by fusion with the mSmoICD. This DrSmo construct is active and refractory to inhibition by Ptch, suggesting that DrSmo might be regulated differently from vertebrate Smo, in spite of Ptch conservation. Interestingly, DrSmoΔCRD is completely inactive in Drosophila27, in contrast to vertebrate SmoΔCRD3,28. Thus DrSmoCRD does not bind sterols but is absolutely required for function, perhaps by playing a critical role in stabilizing the active conformation of DrSmo. Understanding the mechanistic basis for the seeming evolutionary divergence in Smo regulation is an important future goal.
Online methods
Cell culture and reagents
The following compounds were obtained from commercial sources: cyclopamine from LC Laboratories (>99%), BODIPY-cyclopamine from Toronto Research Chemicals; SAG from Axxora (≥98%); forskolin from Sigma (≥98%); itraconazole from Sigma (≥98%); SANT1 from Calbiochem (≥95%); GDC0449 from LC Laboratories (>99%); 20-hydroxycholesterol, 7-hydroxycholesterol, 25-hydroxycholesterol and 7-ketocholesterol from Steraloids (≥98%).
Chemical synthesis
A complete description of the synthesis and characterization of sterol derivatives and BODIPY-SANT1 is provided in the Supplementary Note.
Antibodies
Polyclonal antibodies against Cherry were generated in rabbits (Cocalico Biologicals) and were affinity purified against recombinant Cherry immobilized on Affigel-10 beads (BioRad). Rabbit anti-Smo polyclonal antibodies were described before29. The monoclonal anti-acetylated tubulin antibody was obtained from Sigma.
DNA constructs
Expression constructs were assembled by PCR in the mammalian expression vector pCS2+, from which they were subcloned into a vector for lentiviral production30. Constructs encoding membrane proteins were tagged with Cherry at their C-terminus. These constructs were: full-length mouse Smo (mSmo), full-length Drosophila Smo (DrSmo), full-length mouse Frizzled7 (mFz7), mSmoΔICD (aminoacids 1–554 of mSmo), mSmoM2 (constitutively active point mutant W539L), mSmoΔCRD (amino acids 183–793 of mSmo, preceded by the signal sequence of human calreticulin), DrSmomSmoICD (amino acids 1–556 of DrSmo fused to amino acids 543–793 of mSmo), mSmoDrSmoCRDmut (amino acids 112–119 of mSmo, LWSGLRNA, replaced by amino acids 129–136 of DrSmo, DYYALKHV), mSmoL112D, mSmoW113Y, mSmoS114Y, mSmoL112D/W113Y, mFz7mSmoICD (amino acids 1–548 of mouse Frizzled7 fused to amino acids 543–793 of mSmo), rMAChRmSmoICD (amino acids 1–442 of the rat muscarinic acetylcholine receptor M2 fused to amino acids 543–793 of mSmo). Full-length Xenopus Smo (xSmo) was tagged with eGFP at the C-terminus and was expressed in Sf9 cells by baculoviral infection. The baculovirus was generated using the Bac-to-bac system (Life Sciences), according to the manufacturer's instructions. Constructs for expressing secreted extracellular cysteine-rich domains (CRDs) of Smo proteins contained the signal sequence of human calreticulin, followed by the CRD sequence lacking the Smo signal sequence, followed by a hemagglutinin (HA) tag and 8 histidine residues. The CRD sequences were: mSmoCRD (amino acids 32–236 of mSmo), DrSmoCRD (amino acids 32–257 of DrSmoSmo) and mSmoCRDDrSmoCRDmut (amino acids 32–236 of mSmoDrSmoCRDmut).
Cell culture and generation of stable cell lines
NIH-3T3 and Shh Light II cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% bovine calf serum, penicillin and streptomycin. Mouse embryonic fibroblasts (MEFs) and 293T cells were grown in DMEM with 10% fetal bovine serum, penicillin and streptomycin. Stable cell lines were generated by lentiviral transduction, followed by selection with blasticidin, as described30. Smo−/− MEFs expressing low amounts of various Cherry-tagged Smo proteins were isolated by fluorescence-activated cell sorting. Expression of the tagged construct was confirmed by immunofluorescence and by Q-PCR assays of Hh pathway activity.
Hh ligand production
Hh ligand was produced by transiently transfecting 293T cells with an expression plasmid encoding amino acids 1–198 of human Shh. Shh was collected for 48 hours into starvation medium (DMEM supplemented with penicillin and streptomycin). For maximal stimulation of the Hh pathway, Shh-conditioned medium was used diluted 1:3 −11:4 into fresh starvation medium.
Reporter assays
Hh activity assays were performed in Shh Light II cells14 (obtained from ATCC), a line of NIH-3T3 cells expressing firefly luciferase from a Gli-responsive promoter and Renilla luciferase from a constitutive promoter14. Confluent Shh Light II cultures were starved overnight in DMEM. The medium was then replaced with DMEM supplemented with the appropriate Hh pathway agonist, antagonist, and/or test compound. All small molecules were added to cellular media from concentrated stocks in DMSO, except 20-OHC-Pent-3β-MeO, which was added as a soluble complex with methyl-β-cyclodextrin (MCD), prepared as described31. After 30 hours, Renilla and firefly luciferase levels were measured using the Dual-Glo kit (Promega). Hh pathway activity was expressed as the ratio of firefly to Renilla luciferase, normalized to 100% for maximally stimulated cells (cells treated with 1μM SAG, 10 μM oxysterol, or 1:3 Shh ligand, depending on the experiment), or normalized to 1 for unstimulated cells. For all reporter assays, each data point represents the mean from quadruplicate wells of Shh Light II cells, and error bars represent the standard deviation. All experiments in the paper were performed at least twice. Dose-response curves were plotted using Prism (GraphPad), by non-linear regression to a four-parameter curve.
Real-time PCR assays of the Hh pathway
Confluent NIH-3T3 cells or MEFs were starved overnight in starvation medium, after which they were incubated for 24 hours in starvation medium supplemented with the desired compounds. Total RNA was isolated from cells with RNA-Bee (TelTest), treated with RNase-free DNase (Promega), and purified using the GenCatch total RNA Extraction System (Epoch Biolabs). Reverse transcription was performed using random hexamers and Transcriptor reverse transcriptase (Roche). Transcription of the Hh target gene Gli1 was measured by real-time PCR using FastStart SYBR Green Master reagent (Roche) on a Rotor-Gene 6000 (Corbett Robotics). Relative gene expression was calculated using a Two Standard Curve method in which the gene-of-interest was normalized to the Ribosomal Protein L27 gene. The sequences for gene-specific primers are: L27: 5'-GTCGAGATGGGCAAGTTCAT-3' and 5'-GCTTGGCGATCTTCTTCTTG-3', Gli1: 5'-GGCCAATCACAAGTCAAGGT-3' and 5'-TTCAGGAGGAGGGTACAACG -3'. For all real-time PCR experiments, each data point represents the mean from triplicate wells of cultured cells, and error bars represent the standard deviation.
Immunofluorescence
Cells were grown on glass coverslips and were fixed in PBS with 4% formaldehyde, followed by permeabilization with TBST. Non-specific binding sites were blocked by incubation in TBST with 50 mg/mL bovine serum albumin (TBST-BSA). Primary and secondary antibodies were used diluted in TBST-BSA. The primary antibodies were: rabbit polyclonal against Cherry (final concentration 2 μg/mL), rabbit polyclonal against mSmo29 (final concentration 2 μg/mL), mouse anti-acetylated tubulin monoclonal antibody (Sigma, final dilution dilution of 1:5,000). Alexa-594- and Alexa-488-conjugated secondary antibodies (Life Sciences) were used at a final concentration of 1 μg/mL. The coverslips were mounted on glass slides in mounting media (0.5% p-phenylenediamine, 20 mM Tris pH 8.8, 90% glycerol). The cells were imaged by epifluorescence on a Nikon TE2000E microscope equipped with an OrcaER camera (Hammamatsu) and 40× PlanApo 0.95NA or 100× PlanApo 1.4NA oil objective (Nikon). Images were acquired using the Metamorph software (Applied Precision). Ciliary localization of Smo was measured using custom image analysis software implemented in MATLAB. Briefly, the software first identifies cilia by local adaptive thresholding of acetylated tubulin images. The segmented images are cleaned by automatic removal of objects whose size and shape fall outside the normal range for a typical cilium. Next, the pixel intensity of the protein of interest (Smo) in each cilium is corrected by subtracting the local background, defined as the median intensity of the pixels surrounding the cilium. Ciliary Smo is then quantified as the total corrected intensity in each cilium, normalized to the area of the cilium. To count Smo-positive cilia, fluorescence in the Smo channel is first calculated for the cilia in the negative control sample (untreated cells in case of scoring endogenous Smo, or Smo−/−cells in case of scoring Cherry-tagged fusions proteins stably expressed in Smo−/− MEFs). This data is used to calculate a threshold value that is above the fluorescence intensity for >95% of cilia in the negative control sample; note that this method overestimates the number of Smo-positive cilia in the negative control, by allowing a false positive rate of up to 5%. Using the calculated threshold, cilia are then scored in all remaining samples, and the fraction of Smo-positive cilia is graphed. The error bars represent the sub-sampling standard deviation of the fraction of positive cilia; this is calculated by dividing the cilia population for each experimental sample into five random and non-overlapping sub-samples, then calculating the fraction of positive cilia in each sub-sample, and finally calculating the standard deviation of the fraction of positive cilia across the sub-samples. Smo intensity at cilia is also graphed using box plots; for each condition, the lower and upper bounds of the box represent the 25th and 75th percentile of the Smo intensity distribution, while the horizontal line represents the median intensity across the entire cilia population. For the experiments presented in this paper, between 130–600 cilia were analyzed per condition. For some experiments, ciliary localization of Smo was measured manually, by scoring the presence or absence of Smo in 150 cilia per condition.
Sterol depletion
Sterol depletion was performed on starved, confluent cultures of stable lines derived from Smo−/− MEFs. The cultures were incubated for 30 minutes with 1% methyl-β-cyclodextrin (MCD) in DMEM (to remove sterols), after which all subsequent incubations were in DMEM with 20 μM pravastatin (to block new sterol synthesis), with or without the indicated additives. For rescue experiments, cholesterol was added back by incubating the cells for 1 hour with soluble cholesterol-MCD complexes (100 μM in DMEM supplemented with 40 μM pravastatin). After overnight incubation with the desired compounds, the cells were processed for immunofluorescence or for Q-PCR, as described above.
BODIPY-cyclopamine and BODIPY-SANT1 binding assays
Various Smo proteins, tagged at their C-termini with Cherry, were expressed in 293T cells either stably or by transient transfection. The cells were incubated for 1 hour in DMEM with 10 nM BODIPY-cyclopamine or 10 nM BODIPY-SANT1, in the presence or absence of the indicated concentration of competitor drug. The cells were washed with DMEM, fixed in PBS with 4% formaldehyde for 20 minutes, followed by 5 washes with TBST (10 mM Tris pH 7.5, 150 mM NaCl, 0.2% Triton X-100). The cells were then imaged by epifluorescence microscopy, capturing for each field of view an image of the Smo-Cherry fusion and one of the BODIPY compound.
Preparation of ligand affinity matrices
Free amine derivatives of 22-NHC or 20-OHC were dissolved in dry isopropanol (20 mM final concentration), and were added to amine-reactive Affigel-10 beads (BioRad). After addition of dry triethylamine (100 mM final), the beads were incubated at room temperature overnight, with end-over-end rotation. Unreacted sites on the beads were consumed by incubation with ethanolamine (1M final in isopropanol), after which the beads were washed extensively with isopropanol. The beads were then washed extensively with water, followed by 3 washes with the wash buffer used in ligand affinity experiments (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% dodecyl-β-maltoside). Control beads were generated in parallel, by reacting Affigel-10 beads with the ethylene glycol diamine linker (4,7,10-trioxa-1,13-tridecanediamine, 200 mM final) used in the synthesis of 22-NHC or 20-OHC amine derivatives.
Ligand affinity assays
Recombinant protein for ligand affinity assays was produced by stable or transient expression in 293T cells, except for xSmo-eGFP, which was produced in Sf9 cells by baculovirus infection. Cell expressing various transmembrane Smo constructs, C-terminally tagged with Cherry or eGFP, were harvested and lysed on ice for 30 minutes in lysis buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% dodecyl-β-maltoside), supplemented with protease inhibitors (leupeptin, pepstatin and chymostatin at 10 μg/mL final concentration). The detergent extract was clarified by centrifugation at 20,000g, for 30 min at 4C. The supernatant was first incubated with the desired competitor compound or DMSO control for 5 minutes on ice. All compounds were added to binding reactions from DMSO stock solutions. After this incubation, 22-NHC beads, 20-OHC beads or control beads were added, followed by end-over-end rotation for 1 hour at 4C. The beads were washed three times with wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% dodecyl-β-maltoside), after which bound proteins were eluted in SDS-PAGE sample buffer with DTT (50 mM final) at 37C. The proteins were separated by SDS-PAGE, followed by immunoblotting with anti-Cherry or anti-GFP antibodies. A portion of the clarified detergent extract was used as input.
Cells expressing HA-tagged secreted SmoCRD contructs were incubated for 48 hours in DMEM supplemented with 0.5% fetal bovine serum, penicillin and streptomycin. The conditioned media containing soluble SmoCRD protein was harvested, subjected to centrifugation to remove cellular debris, and concentrated 10-fold by ultrafiltration through a 10 kDa cutoff concentration device (Amicon). The media was then supplemented with dodecyl-β-maltoside (0.5% final concentration) and protease inhibitors, and was used in ligand affinity assays as described above for detergent extracts of cells expressing full-length Smo proteins.
Supplementary Material
Acknowledgements
We thank Yoshito Kishi and members of his laboratory for help with chiral chromatography, and Rajat Rohatgi for the initial gift of 20-OHC beads. A.S. is supported in part by NIH grant RO1 GM092924.
Footnotes
Competing financial interests The authors declare no competing financial interests.
Author contributions D.N. and A.S. performed cellular and biochemical experiments. J.L., C.Y.J. and A.S. designed and synthesized reported compounds. J.L. purified and characterized the compounds. Y.X. and D.N. developed automated image analysis software, and D.N. analyzed imaging data. All authors contributed data to the manuscript. A.S. wrote the manuscript, with input from all other authors.
References
- 1.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 2.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes & development. 2001;15:3059–3087. doi: 10.1101/gad.938601. doi:10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 3.Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 4.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes & development. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frank-Kamenetsky M, et al. Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J Biol. 2002;1:10. doi: 10.1186/1475-4924-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robarge KD, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorganic & medicinal chemistry letters. 2009;19:5576–5581. doi: 10.1016/j.bmcl.2009.08.049. doi:10.1016/j.bmcl.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 7.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nature chemical biology. 2006;2:29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 9.Dwyer JR, et al. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. The Journal of biological chemistry. 2007;282:8959–8968. doi: 10.1074/jbc.M611741200. [DOI] [PubMed] [Google Scholar]
- 10.Corcoran RB, Scott MP. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8408–8413. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim WK, Meliton V, Amantea CM, Hahn TJ, Parhami F. 20(S)-hydroxycholesterol inhibits PPARgamma expression and adipogenic differentiation of bone marrow stromal cells through a hedgehog-dependent mechanism. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22:1711–1719. doi: 10.1359/jbmr.070710. doi:10.1359/jbmr.070710. [DOI] [PubMed] [Google Scholar]
- 12.Nachtergaele S, et al. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nature chemical biology. 2012;8:211–220. doi: 10.1038/nchembio.765. doi:10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 14.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 15.Svard J, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Developmental cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Cooper MK, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 17.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Zhou Z, Walsh CT, McMahon AP. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2623–2628. doi: 10.1073/pnas.0812110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3196–3201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer cell. 2010;17:388–399. doi: 10.1016/j.ccr.2010.02.027. doi:10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazan JF, de Sauvage FJ. Structural ties between cholesterol transport and morphogen signaling. Cell. 2009;138:1055–1056. doi: 10.1016/j.cell.2009.09.006. doi:10.1016/j.cell.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, et al. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chemistry & biology. 2012;19:972–982. doi: 10.1016/j.chembiol.2012.06.012. doi:10.1016/j.chembiol.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. doi:10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Developmental cell. 2012;23:823–835. doi: 10.1016/j.devcel.2012.07.004. doi:10.1016/j.devcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Infante RE, et al. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. The Journal of biological chemistry. 2008;283:1064–1075. doi: 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- 26.Motamed M, et al. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. The Journal of biological chemistry. 2011;286:18002–18012. doi: 10.1074/jbc.M111.238311. doi:10.1074/jbc.M111.238311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano Y, et al. Functional domains and sub-cellular distribution of the Hedgehog transducing protein Smoothened in Drosophila. Mechanisms of development. 2004;121:507–518. doi: 10.1016/j.mod.2004.04.015. doi:10.1016/j.mod.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Aanstad P, et al. The extracellular domain of Smoothened regulates ciliary localization and is required for high-level Hh signaling. Current biology : CB. 2009;19:1034–1039. doi: 10.1016/j.cub.2009.04.053. doi:10.1016/j.cub.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tukachinsky H, Lopez L, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. The Journal of cell biology. 2010;191 doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, Salic A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell reports. 2012;2:308–320. doi: 10.1016/j.celrep.2012.07.010. doi:10.1016/j.celrep.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.