

Abstract
A current view of the inflammatory bowel diseases (IBD) includes the luminal triggering of innate immune disease in a genetically susceptible host. Given the unique anatomy and complex environment of the intestine, local microenvironmental cues likely contribute significantly to both disease progression and resolution in IBD. Compartmentalized tissue and microbe populations within the intestine result in significant metabolic shifts within these tissue microenvironments. During active inflammatory disease, metabolic demands often exceed supply, resulting in localized areas of metabolic stress and diminished oxygen delivery (hypoxia). There is much recent interest in harnessing these microenvironmental changes to the benefit of the tissue, including targeting these pathways for therapy of IBD. Here, we review the current understanding of metabolic microenvironments within the intestine in IBD, with discussion of the advantages and disadvantages of targeting these pathways to treat patients with IBD.
Keywords: mucosa, inflammation, colitis, neutrophil, epithelium, hypoxia, resolution
Introduction
Mucosal surfaces, such as the intestine, are lined by epithelial cells. The epithelium is uniquely positioned to serve as a conduit between the extensive mucosal immune system and the external environment. In their normal state, mucosal surfaces are exposed on the lumenal surface to high concentrations of foreign antigens, while at the same time, intimately associated with the immune system via subepithelial lymphoid tissue 1. Consequently, the epithelium forms an important barrier, preventing the free mixing of lumenal antigenic material with the lamina propria which houses the mucosal immune system 2. Like many aspects of biology, this view has changed over the past decade. The epithelium is now viewed as an active player in normal homeostatic mechanisms of mucosal immunity, and in some instances, the epithelium may be the central conductor in orchestrating mucosal immune responses in a variety of diseases, particularly inflammatory bowel disease (IBD).
It is widely understood that the gastrointestinal (GI) tract functions in a state of ‘low grade inflammation,’ wherein tissues of the GI tract remain “primed” for rapid responses to acute stresses. Such inflammatory priming results from the constant processing of luminal antigenic material during the development of oral tolerance and the activation of the mucosal immune system by antigens or microbes that may penetrate the barrier. Significant evidence indicates that effective mucosal responses are derived from microenvironmental cues generated by complex interactions at local tissue sites. Here, we review some of these microenvironmental signals and potential therapeutic implications therein.
Oxygen metabolism in the microenvironment as it relates to the immune response
There is significant recent interest in defining the contribution of oxygen metabolism to the microenvironment of the mucosa in IBD 3, 4. The intestine, in fact, provides a fascinating oxygenation profile. Under normal physiologic conditions, the intestinal mucosa experiences profound fluctuations in blood flow and metabolism. Less than 5% of total blood volume is present in the gut during fasting, however, following ingestion of a meal, approximately 30% of total blood volume is redirected to the gastrointestinal tract. Such changes in blood flow can result in marked shifts in local pO2. Notably, a steep oxygen gradient exists from the anaerobic lumen across the epithelium into a highly vascularized and metabolically active sub-epithelium. From this perspective, it is perhaps not surprising that the epithelium has evolved a number of features to cope with such metabolic shifts. Studies comparing functional responses between epithelial cells from different tissues have revealed that intestinal epithelial cells seem to be uniquely resistant to hypoxia and that an extremely low level of oxygenation within the normal intestinal epithelial barrier (so-called “physiologic hypoxia”) may be a regulatory adaptation mechanism to this steep oxygen gradient 5.
Sites of mucosal inflammation are characterized by profound changes in tissue metabolism, including local depletion of nutrients, imbalances in tissue oxygen supply and demand, and the generation of large quantities of reactive nitrogen and oxygen intermediates 6. In part, these changes can be attributed to recruitment of inflammatory cells, including myeloid cells such as neutrophils (polymorphonuclear cells; PMNs) and monocytes (Figure 1). PMNs are recruited by chemical signals, such as the chemokine interleukin 8, complement factor C5a, N-formylated peptides, platelet-activating factor and leukotriene B4, which are generated at sites of active inflammation as part of the innate host immune response to microorganisms. In transit, these cells expend tremendous amounts of energy. For instance, large amounts of adenosine 5′-triphosphate (ATP) are needed for the high actin turnover required for cell migration 7. Once at the sites of inflammation, the nutrient, energy and oxygen demands of the PMNs increase to accomplish the processes of phagocytosis and microbial killing. It has long been known that PMNs are primarily glycolytic cells, with few mitochondria and little energy produced from respiration 8. A predominantly glycolytic metabolism ensures that PMN can function at the low oxygen concentrations (even anoxia) associated with inflammatory lesions.
Figure 1. Signaling within the microenvironment of the crypt abscess.
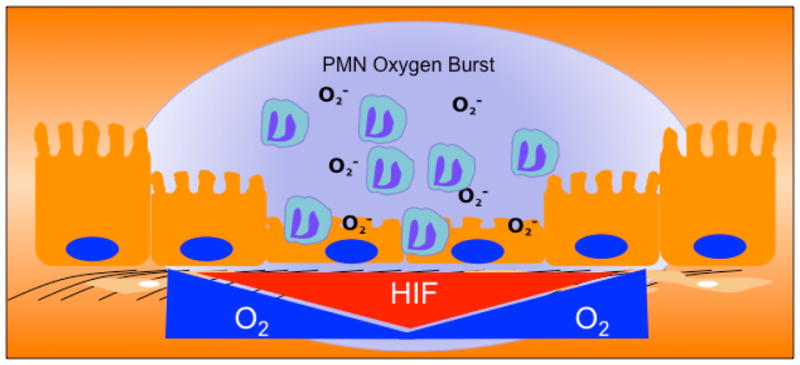
Transmigrating PMN become activated to consume large amounts of oxygen in the formation of superoxide anions (termed the PMN oxygen burst). As a result, the localized microenvironment becomes depleted of molecular O2, and culminates in the stabilization of HIF. The activation of multiple HIF target genes (see text) promotes the active resolution of inflammation within the mucosa.
Activated PMNs recognize and engulf pathogens and activate the release of antibacterial peptides, proteases and reactive oxygen species (ROS; superoxide anion, hydrogen peroxide, hydroxyl radical and hypochlorous acid) into the vacuole, which together kill the invading microbes (Figure 1) 9. ROS are produced by phagocytes in a powerful oxidative burst, driven by a rapid increase in oxygen uptake and glucose consumption, which in turn triggers further generation of ROS. When activated, it is estimated that PMN can consume up to 10 times more O2 than any other cell in the body. Notably, the PMN oxidative burst is not hindered by even relatively low O2 (as low as 4.5% O2) 10, which is important, since it means that ROS can be generated in the relatively low O2 environments of inflamed intestinal mucosa 6.
In stark contrast to the innate immune response, adaptive immune responses are characterized by a combination of recruitment and high rates of local T and B cell proliferation. As a result, adaptive immune responses have markedly different demands for glucose, oxygen and ATP within this microenvironment 11, 12. In the past decade, we have begun to understand the nature of interactions between microenvironmental metabolic changes and the generation of recruitment signals and molecular mechanisms of leukocyte migration into these areas. These metabolic changes that occur as a result of the recruitment and activation of leukocytes during inflammation provide information about the potential sources of hypoxia at the intestinal epithelial barrier (Figure 1).
IBD is a particularly interesting disease for studying the metabolic changes associated with inflammation, principally the development of hypoxia within inflammatory lesions 6. Some microvascular abnormalities that occur in patients with IBD have been associated with abnormal blood flow to the intestine, including increased production of tissue vasoconstrictor molecules and a reduced generation of nitric oxide by endothelial cells 13, as well as vascular endothelial growth factor-dependent angiogenesis 14. In addition, studies of active inflammation in mouse models of IBD have shown the intestinal epithelial cell to be a primary target for hypoxia 15.
Hypoxia in murine models of IBD was revealed using 2-nitroimidazole dyes, a class of compounds known to undergo intracellular metabolism depending on tissue oxygenation16. Tissue staining with these nitroimidazole dyes revealed two profound observations. First, in the normal intestinal epithelial cells, especially in the colon, physiologic hypoxia predominates. Whether such low oxygen levels function to regulate basal gene expression in intestinal epithelial cells is not known. Second, the inflammatory lesions seen in these mouse models are profoundly hypoxic or even anoxic, similar to that seen in some large tumors, and penetrate deep into the mucosal tissue. It is likely that there are multiple contributing factors, such as vasculitis, vasoconstriction, edema and increased O2 consumption (Figure 1), which predispose the inflamed intestinal epithelia to decreased oxygen delivery and hypoxia 15. While these 2-niroimidazole compounds have not been used to image inflammatory lesions, they have shown significant clinical utility in tumor imaging and in the identification of stroke regions within the brain of patients 17. As opposed to other imaging techniques, these compounds have the advantages that they image only viable tissue and are not active within apoptotic or necrotic regions 18. Likewise, studies are underway to use these compounds as adjunct radiosensitizers for enhancing chemotherapy targeting 19.
Hypoxia-inducible factor
The discovery of hypoxia-inducible factor (HIF) in 1993 20 laid the ground work for decades of fascinating work in far reaching areas of cancer biology, metabolism and development. Our work demonstrating inflammation-related hypoxia also revealed the expression of HIF-1 in inflammatory lesions 15. [Short summary of HIF pathway] Many cell types, including intestinal epithelial cells 21, express both HIF-1 and HIF-2 isoforms of this transcriptional regulator and murine knockout studies suggest that these proteins have non-redundant roles 22. Some have suggested that distinct transcriptional responses mediated by HIF-1 and HIF-2 may be integrated in ways that support particular adaptations to hypoxia. For example, the transcriptional responses that coordinate the glycolytic pathways include more than 11 target genes and seem to be more selective for the HIF-1α than for the HIF-2α isoform 22. Studies addressing the selectivity of the two isoforms for erythropoietin induction have suggested a more important role for the HIF-2α isoform 22. Currently, this specificity is not well understood. Some have suggested that binding of HIF-1α or HIF-2α to other transcription factors at the site of DNA binding could determine such specificity 22, but these studies have not been conclusive.
Several studies have shown that HIF-1 triggers the transcription of many genes that enable intestinal epithelial cells to act as an effective barrier 5, 23–25. Originally shown by microarray analysis of hypoxic intestinal epithelial cells 24, these studies have been validated in animal models of intestinal inflammation 15, 26–30 and in human intestinal inflammation tissues 31–33. The functional proteins encoded by hypoxia-induced, HIF-dependent mRNAs localize primarily to the most luminal aspect of polarized epithelia. Molecular studies of these hypoxia-elicited pathway(s) have shown a dependence on HIF-mediated transcriptional responses. Notably, epithelial barrier protective pathways driven by HIF tend not to be the classical regulators of barrier function, such as the tight junction proteins occludin or claudin. Rather, the HIF-dependent pathways are predisposed to regulate overall tissue integrity, ranging from increased mucin production 34, including molecules that modify mucins, such as, intestinal trefoil factor 5, to xenobiotic clearance by P-glycoprotein23, to nucleotide metabolism (by ecto-5′-nucleotidase and CD73) 24, 25 and nucleotide signaling through the adenosine A2B receptor 25.
As an extension of the original studies identifying HIF induction within the intestinal mucosa, Karhausen et al. generated transgenic mice expressing either mutant Hif1a (causing constitutive repression of Hif1-α) or mutant von Hippel-Lindau (causing constitutive overexpression of HIF) targeted to the intestinal epithelial cells15. Loss of epithelial HIF-1α resulted in a more severe colitic phenotype compared with wild-type animals, with increased weight loss, decreased colon length and increased epithelial permeability, whereas constitutively active intestinal epithelial HIF was protective for these parameters. These findings may be somewhat model-dependent, since epithelial HIF-based signaling has also been shown to promote inflammation in another study 30. However, the findings confirmed that intestinal epithelial cells can adapt to hypoxia and that HIF may contribute to this adaptation.
Non-epithelial cell types within the gastrointestinal mucosa have also been studied for HIF expression and response to hypoxia. Activated T cells increased expression of HIF-1α, which prevents them from undergoing activation-induced cell death in hypoxic settings. T-cell survival in hypoxia is, at least in part, mediated by the vasoactive peptide adrenomedullin 35. Other studies using chimeric mice bearing HIF-1α-deficient T and B cells have revealed lineage-specific defects that result in increased autoimmunity, including autoantibodies, increased rheumatoid factor expression and kidney damage 12. Most recently, it was shown that diminished O2 in the colitic microenvironment triggers HIF-dependent regulation of T cell differentiation 36. Indeed, Clambey et al. showed that one of the key transcriptional differentiation factors, FoxP3, is a HIF target gene and that HIF-1 was required for optimal regulatory T cell (Treg) abundance and function. HIF-1α-deficient Tregs failed to control T-cell mediated colitis, providing an important role for microenvironmental O2 in productive mucosal immune responses.
HIF function has also been studied in some detail in myeloid cells. Cre-LoxP-based elimination of HIF-1α in cells of the myeloid lineage (lysozyme M promoter) have revealed multiple features which importantly implicate metabolic control of myeloid function 37. In particular, these studies have shown that PMN and macrophage bacterial killing capacities are severely limited in the absence of HIF-1α, as HIF-1α is central to production of antimicrobial peptides and granule proteases. These findings are explained, at least in part, by the inability of myeloid cells to mount appropriate metabolic responses to the diminished O2 characteristic of infectious sites 37. Finally, compelling evidence has revealed that HIF-1α transcriptionally controls the critical integrin important in all myeloid cell adhesion and transmigration, namely the β2 integrin (CD18) 38, 39. Such findings are important for our current understanding of the role of functional PMN in IBD. A recent study, for example, used PMN depletion to document a central role for PMN in the resolution of inflammation in several murine IBD models 40.
Microenvironmental control of NF-κB in IBD
The oxygen-dependent regulation of the microenvironment may not be restricted to the HIF pathway and may provide a means to better understand how hypoxia contributes to other aspects of inflammation. For instance, NF-κB may interact with the HIF pathway and is activated during inflammation. NF-κB consists of either homo- or hetero-dimers, which, upon activation, translocate to the nucleus and bind with the transcriptional coactivator CBP/p300 to initiate transcription or repression of various genes. The activity of NF-κB is regulated by the inhibitory IκB proteins 41. The best-studied complex is IκBα bound to the NF-κB p50-p65 dimer41. The interaction with IκBα inhibits NF-κB from binding to DNA and maintains the complex in the cytoplasm. Upon activation by various extracellular signals, IκB kinase (IKK) is activated, resulting in phosphorylation 42 and polyubiquitylation of IκBα43. The polyubiquitinated IκBα is then selectively degraded by the S26 proteasome. Once dissociated from IκBα, NF-κB rapidly enters the nucleus and activates gene expression.
It is now appreciated that NF-κB-dependent pathways are also regulated by the enzymes which promote the degradation of HIF-α subunits, namely HIF prolyl hydroxylases (PHDs) and that the usefulness of PHD inhibitors in murine colitis models also target the NF-κB pathway 26. Indeed, hypoxia has been shown to activate NF-κB and this activation seems, at least in part, to be dependent on PHD-mediated hydroxylation 44, 45 of IKKβ.38 In normoxia, IKKβ activity is held in check through LXXLAP-dependent hydroxylation by PHD1 and PHD2 45. It is notable that conditional deletion of the NF-κB pathway in intestinal epithelial cells in mice leads to an increased susceptibility to colitis 46, similar to that of the mice expressing homozygous mutant HIF-1α 15. This implicates epithelial NF-κB in a prominently protective role in colitis, probably through the expression of anti-apoptotic genes in intestinal epithelial cells and through enhanced epithelial barrier function. Some studies have suggested that both the HIF and NF-κB pathways may also be influenced by mediators found within inflammatory sites, including microbial products, cytokines and even intact bacteria 37. NF-κB is a classic transcriptional regulator activated by a spectrum of agonists, the activation of which drives a complex series of receptor-mediated signaling pathways. Recent studies indicate that transcription of HIF-1α is activated by NF-κB-mediated signaling 47. Inflammation-associated upregulation of HIF-1α mRNA occurs in an NF-κB-dependent manner 47. It also seems that increased NF-κB activity in hypoxia can be regulated by HIF-1 48, and thus a cross-regulatory loop may exist between these two pathways and may involve other transcriptional regulators that bear non-redundant PHD sensitivity, including Activating Transcription Factor-4 and Notch 49, 50, both critical regulators of cell fate. Given that intestinal epithelial cells are situated in an environment with constant exposure to potentially inflammatory stimuli, the cross regulation of HIF and NF-κB may have profound implications for intestinal epithelial cell function and survival under both homeostatic and disease conditions.
Adenosine metabolism in the inflammatory microenvironment
Nucleosides such as adenosine (Ado) influence nearly every aspect of physiology and pathophysiology. Extracellular nucleotides liberated at local sites of inflammation are metabolized through regulated phosphohydrolysis by a series of ecto-nucleotidases. The formation of extracellular Ado within the microenvironment from ATP is accomplished primarily through ectonucleoside triphosphate diphosphohydrolase-1 (CD39) and ecto-5′-nucleotidase (CD73), found on the surface of a variety of cell types (Figure 2). Once generated, Ado is available to bind and activate one of four G-protein-coupled Ado receptors (A1AR, A2AAR, A2BAR, or A3AR).
Figure 2. Nucleotide and lipid mediator metabolism as pro-resolving signals generated in the microenvironment.
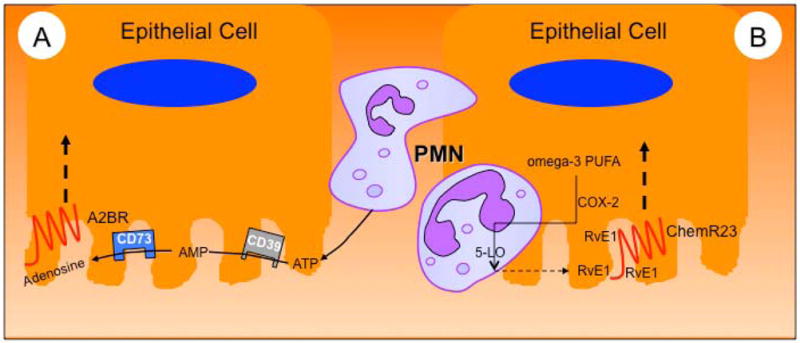
In model A, activated PMN release large amounts of ATP during transmigration. This ATP is subsequently metabolized to adenosine by a two-step enzymatic reaction involving ecto-apyrase (CD39) and ecto-nucleotidase (CD73). Adenosine binding to apical adenosine A2B receptors promotes the resolution of inflammation. In model B, RvE1 production is amplified by transcellular biosynthesis via the interactions of PMN with epithelial cells during transmigration, each contributing an enzymatic product. In the example shown here, epithelial cell COX-2 generates 18-HEPE from dietary omega-3 PUFA and PMN-expressed 5-LO then generates RvE1. Such locally generated RvE1 is then made available to activate apically expressed ChemR23 which in turn promotes resolution through a number of mechanisms (see text).
Individual Ado receptors are widely expressed within the GI tract and their function has been studied recently. While less is known about A1AR in GI disease models, the A2AAR and A2BAR receptors have been studied in some detail 51–53. The A2AAR is critical for A2AAR signaling in T cell-mediated regulation of colitis. Moreover, treatment with a specific A2AAR agonist attenuated the production of pro-inflammatory cytokines and attenuation of colitis 54. Recent studies have performed “head-on” comparisons between A2AAR−/− and A2BAR−/− in murine colitis models and revealed a relative importance for the A2BAR in murine colitis 55. Studies in A3AR−/− mice have proven quite interesting and potentially revealed a specific role for A3AR in the resolution of GI inflammation. For example, Butler et al. and Ren et al. have recently shown that A3AR−/− mice show overall decreased pathology in acute DSS colitis 56, but these animals failed to resolve inflammation associated with chronic inflammation in this model 56. Likewise, the adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, has proven to be protective in at least two murine models of colitis57. In humans, A3AR expression negatively correlates with acute inflammatory score, Crohn’s Disease Activity Index (CDAI) and disease chronicity 58, strongly implicating a need for further investigation of this receptor subtype.
CD73 is the terminal enzyme in the generation of extracellular Ado and is therefore considered the pacemaker for Ado actions within the microenvironment (Figure 2) 59. Original characterization the Cd73−/− mouse revealed that of the tissues surveyed, the colon showed the highest level of enzyme activity, a somewhat surprising result, as a number of previous studies suggested that the kidney likely carried the highest activity of any tissue. Instead, it was found that the colon expresses nearly twice as much activity as the kidney, with the rank order of tissue activity as follows: colon > kidney = brain > liver > lung > heart ≫ muscle 60. During experimental colitis induced by the hapten trinitrobenzene sulfonic acid (TNBS), Cd73−/− mice develop a more severe phenotype 61. Cytokine profiling revealed similar increases in both IFN-gamma and TNF-alpha mRNA in colitic animals, independent of genotype. However, IL-10 mRNA increased in wild-type mice on day 3 after TNBS administration, whereas Cd73−/− mice mounted no IL-10 response. This IL-10 response was restored in the Cd73−/− mice by exogenous IFN-αA 61. More recently it was shown that CD73 is central to both the magnitude and the resolution of murine DSS colitis62.
As CD39 hydrolyzes nucleotides to generate AMP (Figure 2), it has become a point of interest in IBD. In one study, the authors hypothesized that CD39 might protect against IBD 63. They studied these possibilities in a mouse model of colitis using mice with global CD39 deletion and also tested whether human genetic polymorphisms in the CD39 gene might influence susceptibility to Crohn’s disease. Mice deficient for CD39 were highly susceptible to chemically induced colitis, with heterozygote mice showing an intermediate phenotype. Moreover, they identified a common SNP that tags CD39 mRNA expression levels in man. The SNP tagging low levels of CD39 expression was associated with increased susceptibility to Crohn’s disease in a case-control cohort comprised of 1,748 patients and 2,936 controls. These data indicate that CD39 deficiency exacerbates murine colitis and suggest that CD39 polymorphisms are associated with IBD in humans 63. Other studies have identified CD39 as a specific marker for Tregs, and implicate CD39-dependent ATP/ADP breakdown in autocrine enhancement of the anti-inflammatory functions of this group of T-cells 64.
Pro-resolving Lipids in the Microenvironment of IBD
Resolution of inflammation and return to tissue homeostasis is an exceptionally well-coordinated process. There is much recent interest in the identification of pro-resolving molecules generated within the inflammatory microenvironment. Significant progress has been made in this regard, especially pro-resolving lipids generated during the resolution phase of ongoing inflammation that actively stimulate restoration of tissue homeostasis 65.
Particular attention has been paid to the resolvins (Figure 2) 66. Resolvins are omega-3 fatty acid-derived mediators that are agonist-dependent, temporally distinct and carry novel mucosal-directed signals 67. The first resolvin, known today as resolvin E1 (RvE1), was identified in 1999 as a potent and active initiator of inflammatory resolution 68. Inordinate, unrestricted acute inflammation is now acknowledged as an instigating factor, which when unchecked, contributes to numerous chronic disease states, including IBD. As such, an understanding of the pharmacology of anti-inflammation and endogenous pro-resolution has been a significant venture 67.
A recent microarray screen to identify RvE1-regulated genes in intestinal epithelial cells revealed two important findings. First, these studies revealed the previously unappreciated native expression of the RvE1 receptor ChemR23 on epithelial cells. A screen of various epithelial cell lines revealed prominent expression of ChemR23 on human intestinal epithelial cell lines (T84 and Caco-2). Unique was the pattern of expression on polarized epithelia as this analysis revealed that ChemR23 localizes predominantly to the apical membrane surface, which was somewhat unexpected given that most other G-protein-coupled receptors exhibit basolateral expression in polarized epithelia 69. Such membrane distribution of ChemR23 suggested that the localized generation of RvE1 during PMN-epithelial interactions could occur at the apical (lumenal) aspect of the tissue. This is an intriguing possibility given that the other known function for RvE1 on mucosal epithelia is to promote the termination and clearance of PMN following transmigration 70, through well-characterized CD55-dependent mechanisms 71, 72. Thus, the PMN-epithelial interactions that occur within the lumen of the intestine may initiate a pro-resolving signature for the epithelium during PMN transit through the mucosa.
Second, these microarray studies identified a prominent RvE1-dependent antimicrobial signature within the epithelium, including the induction of BPI and the BPI-like molecule PLUNC (palate, lung, nasal epithelium clone)73. Also notable was the induction of epithelial ALPI by RvE1. Surface expressed ALPI was shown to retard Gram negative bacterial growth and to potently neutralize LPS through a mechanism involving dephosphorylation of 1,4′-bisphosphorylated glucosamine disaccharide of LPS lipid A 74, 75. This observation was translated to the murine DSS colitis model and revealed that induction of ALPI by RvE1 in vivo strongly correlated with the resolution phase of inflammation (Figure 2). Moreover, inhibition of ALPI activity was shown to increase the severity of colitic disease and abrogated the protective influences of RvE1 73. Like those defining epithelial expression of BPI 76, these studies provide an example of the critical interface between inflammatory resolution and the importance of antimicrobial mechanisms.
Conclusions
The gastrointestinal mucosa is an interesting tissue in which to investigate the microenvironment and tissue-based metabolism. In this review, we have outlined the evidence for mediators generated in the local microenvironment as important signaling mechanisms within the intestinal mucosa. Studies from cultured cell systems, animal models and patient-derived materials have documented that the metabolism of nucleotides and lipids provide a significant component of the inflammatory microenvironment. Likewise, studies to date in animal models of intestinal inflammation have demonstrated an almost uniformly beneficial influence of oxygen metabolism and HIF stabilization on disease outcomes. Ongoing studies to define the differences and similarities between innate and adaptive immune responses will continue to teach us important lessons about the complexity of the gastrointestinal tract. Such information will provide new insight into the pathogenesis of the disease and importantly, will provide new targets as templates for the development of therapies for human disease.
Acknowledgments
This work was supported by National Institutes of Health grants DK50189, HL60569, DK95491 and by grants from the Crohn’s and Colitis Foundation of America.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Beagley KW, Husband AJ. Intraepithelial lymphocytes: origins, distribution, and function. Crit Rev Immunol. 1998;18:237–54. doi: 10.1615/critrevimmunol.v18.i3.40. [DOI] [PubMed] [Google Scholar]
- 2.McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Curr Opin Gastroenterol. 2007;23:647–54. doi: 10.1097/MOG.0b013e3282f0153b. [DOI] [PubMed] [Google Scholar]
- 3.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology. 2011;140:1748–55. doi: 10.1053/j.gastro.2011.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuta GT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Ex Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2008;85:1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 7.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 8.Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. 1982;70:550–7. doi: 10.1172/JCI110647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30:279–89. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 10.Gabig TG, Bearman SI, Babior BM. Effects of oxygen tension and pH on the respiratory burst of human neutrophils. Blood. 1979;53:1133–9. [PubMed] [Google Scholar]
- 11.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 12.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–21. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 13.Hatoum OA, Heidemann J, Binion DG. The intestinal microvasculature as a therapeutic target in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:78–97. doi: 10.1196/annals.1326.003. [DOI] [PubMed] [Google Scholar]
- 14.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol. 2007;178:6017–22. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 15.Karhausen JO, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans SM, et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000;60:2018–24. [PubMed] [Google Scholar]
- 17.Takasawa M, Moustafa RR, Baron JC. Applications of nitroimidazole in vivo hypoxia imaging in ischemic stroke. Stroke. 2008;39:1629–37. doi: 10.1161/STROKEAHA.107.485938. [DOI] [PubMed] [Google Scholar]
- 18.Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci. 2009;100:1366–73. doi: 10.1111/j.1349-7006.2009.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25:4066–74. doi: 10.1200/JCO.2007.12.7878. [DOI] [PubMed] [Google Scholar]
- 20.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Nat Acad Sci (USA) 1993;90:4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastrogiannaki M, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009 doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–5. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comerford KM, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–94. [PubMed] [Google Scholar]
- 24.Synnestvedt K, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 (HIF-1) mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cummins EP, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–65. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Han IO, Kim HS, Kim HC, Joe EH, Kim WK. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73:659–69. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 28.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–18. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 29.Robinson A, et al. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–55. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah YM, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–48. 2048 e1–3. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giatromanolaki A, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–13. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mariani F, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–9. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Matthijsen RA, et al. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4:e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louis NA, et al. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–27. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 35.Makino Y, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol. 2003;171:6534–40. doi: 10.4049/jimmunol.171.12.6534. [DOI] [PubMed] [Google Scholar]
- 36.Clambey ET, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109:E2784–93. doi: 10.1073/pnas.1202366109. Epub 2012 Sep 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–17. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101:10440–5. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong T, Scully M, Shelley CS, Colgan SP. Identification of Pur alpha as a new hypoxia response factor responsible for coordinated induction of the beta 2 integrin family. J Immunol. 2007;179:1934–41. doi: 10.4049/jimmunol.179.3.1934. [DOI] [PubMed] [Google Scholar]
- 40.Kuhl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–92. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 42.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cockman ME, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc Natl Acad Sci U S A. 2006;103:14767–72. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaph C, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–6. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 47.Rius J, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coleman ML, et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282:24027–38. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 50.Koditz J, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–7. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 51.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–20. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberger P, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 53.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naganuma M, et al. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177:2765–9. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 55.Frick JS, et al. Contribution of Adenosine A2B Receptors to Inflammatory Parameters of Experimental Colitis. J Immunol. 2009 doi: 10.4049/jimmunol.0801324. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butler M, et al. Impairment of adenosine a3 receptor activity disrupts neutrophil migratory capacity and impacts innate immune function in vivo. Eur J Immunol. 2012;2:201242655. doi: 10.1002/eji.201242655. [DOI] [PubMed] [Google Scholar]
- 57.Mabley J, et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–9. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 58.Rybaczyk L, et al. New bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm Bowel Dis. 2009;15:971–84. doi: 10.1002/ibd.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–75. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson LF, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Louis NA, et al. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180:4246–55. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- 62.Bynoe MS, et al. CD73 is critical for the resolution of murine colonic inflammation. J Biomed Biotechnol. 2012:260983. doi: 10.1155/2012/260983. Epub 2012 Oct 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friedman DJ, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A. 2009;106:16788–93. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campbell EL, Serhan CN, Colgan SP. Antimicrobial aspects of inflammatory resolution in the mucosa: a role for proresolving mediators. J Immunol. 2011;187:3475–81. doi: 10.4049/jimmunol.1100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. 2008;153 (Suppl 1):S200–15. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Serhan CN, et al. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wozniak M, Keefer JR, Saunders C, Limbird LE. Differential targeting and retention of G protein-coupled receptors in polarized epithelial cells. J Recept Signal Transduct Res. 1997;17:373–83. doi: 10.3109/10799899709036615. [DOI] [PubMed] [Google Scholar]
- 70.Campbell EL, et al. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 2007;21:3162–70. doi: 10.1096/fj.07-8473com. [DOI] [PubMed] [Google Scholar]
- 71.Lawrence DW, et al. Antiadhesive role of apical decay-accelerating factor (CD55) in human neutrophil transmigration across mucosal epithelia. J Exp Med. 2003;198:999–1010. doi: 10.1084/jem.20030380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Louis NA, Hamilton KE, Kong T, Colgan SP. HIF-dependent induction of apical CD55 coordinates epithelial clearance of neutrophils. FASEB J. 2005;19:950–9. doi: 10.1096/fj.04-3251com. [DOI] [PubMed] [Google Scholar]
- 73.Campbell EL, et al. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc Natl Acad Sci U S A. 2010;107:14298–303. doi: 10.1073/pnas.0914730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mata-Haro V, et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–32. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 75.Moyle PM, Toth I. Self-adjuvanting lipopeptide vaccines. Curr Med Chem. 2008;15:506–16. doi: 10.2174/092986708783503249. [DOI] [PubMed] [Google Scholar]
- 76.Canny G, et al. Lipid mediator-induced expression of bactericidal/ permeability- increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci U S A. 2002;99:3902–7. doi: 10.1073/pnas.052533799. [DOI] [PMC free article] [PubMed] [Google Scholar]