

Abstract
Glutathione reductase (Gsr)1 catalyzes the reduction of glutathione disulfide to glutathione, a major cellular antioxidant. We have recently shown that Gsr is essential for host defense against the Gram-negative bacteria Escherichia coli in a mouse model of sepsis. While we have demonstrated that Gsr is required for sustaining the oxidative burst and the development of neutrophil extracellular traps, the role of Gsr in other phagocytic functions remains unclear. It is also unclear whether Gsr-deficient mice exhibit host defense defects against Gram-positive bacteria. In the present study, we characterized the effects of Gsr deficiency on the innate immune responses to a Gram-positive bacterium, group B Streptococcus, and to the Gram-negative bacterial cell wall component lipopolysaccharide (LPS). We found that like, E. coli, group B Streptococcus resulted in a substantially more robust cytokine response and a markedly higher morbidity and mortality in Gsr-deficient mice than in wildtype mice. The increased morbidity and mortality were associated with greater bacterial burden in the Gsr-deficient mice. Interestingly, Gsr-deficient mice did not exhibit a greater sensitivity to LPS than did wildtype mice. Analysis of the neutrophils of Gsr-deficient mice revealed impaired phagocytosis. In response to thioglycollate stimulation, Gsr-deficient mice mobilized far fewer phagocytes, including neutrophils, macrophages, and eosinophils, into their peritoneal cavities than did wildtype mice. The defective phagocyte mobilization is associated with profound oxidation and aggregation of ascitic proteins, particularly albumin. Our results indicate that the oxidative defense mechanism mediated by Gsr is required for an effective innate immune response against bacteria, likely by preventing phagocyte dysfunction due to oxidative damage.
Keywords: Bacterial infection, phagocytes, redox regulation, oxidative burst, host defense, animal model, group B Streptococcus
Introduction
Reactive oxygen species (ROS) are pivotal in the host defense mediated by the innate immune cells, particularly neutrophils and macrophages, against bacterial and fungal pathogens. Upon encountering these pathogens, phagocytes engulf the microbial particles through phagocytosis, which activates NADPH oxidase, leading to a robust production of ROS, including H2O2. While ROS are critical for the killing of engulfed microbial pathogens, these ROS may oxidize biomolecules of the host and thereby inhibit phagocytic function. Glutathione, or GSH, is critical for the elimination of H2O2 in cells. GSH reacts with H2O2 through a chemical process catalyzed by glutathione peroxidase, resulting in glutathione disulfide (GSSG) [1, 2]. Glutathione reductase (Gsr) catalyzes the regeneration of GSH from GSSG, utilizing NADPH generated by the hexose monophosphate shunt. Therefore, by catalyzing the GSH/GSSG cycle to facilitate electron transfer from glucose to H2O2, Gsr functions to prevent the accumulation of excessive amounts of ROS and the resulting oxidative damage. Previously, it has been shown that Gsr is required for a sustained respiratory burst in neutrophils after activation [3, 4]. Loos et al. reported that neutrophils isolated from a family with minimal residual GSR activity exhibited a very brief respiratory burst and these neutrophils also displayed a marginal defect in bacterial killing assays in vitro [4]. Very recently, we have shown that Gsr-deficient mice are more sensitive to challenge by the Gram-negative bacteria, Escherichia coli [5]. Upon E. coli challenge, Gsr-null mice exhibited greater mortality, which was associated with a more robust cytokine storm, than did wildtype mice. Analyses of neutrophil function from Gsr-deficient mice revealed considerable defects in both phagocytic respiratory burst and the development of neutrophil extracellular traps. While the critical importance of Gsr in host defense against E. coli is clear, it is not clear whether Gsr also plays a significant role in host defense against other types of bacteria. It is also unclear whether Gsr-deficiency affects other aspects of the innate immune response besides the oxidative burst and the development of neutrophil extracellular traps.
In the present study, we investigated the effects of Gsr deficiency on host defense against a Gram-positive bacterium, group B Streptococcus (GBS), which is a major cause of meningitis, sepsis, and pneumonia in neonates. To understand the mechanisms underlying the substantially elevated mortality of Gsr-deficient mice following E. coli infection, we examined the effects of Gsr deficiency on cellular signaling and cytokine production during the response to LPS. We also studied the effects of Gsr deficiency on leukocyte mobilization and phagocytosis. Our studies revealed novel functions of Gsr in the innate immune system.
Materials and methods
Mice
Wildtype C3H/HeN mice (6–10 weeks old) were purchased from Harlan. The initial generation of the Gsr-deficient mice has been previously described [6]. The genetic mutation in these mice has been characterized as a 13 kb deletion that starts in intron 1 and ends in intron 5 of the Gsr gene [7]. This deletion eliminates a region in the open reading frame of Gsr and causes a frame shift in the resulting mutant Gsr mRNA, preventing its translation into a functional protein. The original Gsr mutant mice were crossbred to C3H/HeN mice for 10 generations in our laboratories and bred to homozygosity. The genotype of the offspring was determined by PCR, using the following primers: 5'-GCCTCAGTTCAGTTTCCAGC-3' and 5'-GTGCTCCTAAGCACCGAGTC-3' for detecting the wildtype allele (364 bp); 5'-TGGGAATTCCCTAAGGAGCTCCAT-3'and 5'-GTGCTCCTAAGCACCGAGTC-3' to detect the mutant allele (208 bp). Animals had free access to food and water in a specific pathogen-free vivarium at 25°C with humidity between 30% and 70% and with a 12-h alternating light-dark cycle. Animals were treated humanely according to the NIH guidelines and all animal experiments were approved by the Institutional Animal Care and Use Committees of the Research Institute at Nationwide Children's Hospital and Seattle Children's Research Institute.
Bacteria
The GBS serotype H1 strain (COH1), a virulent type III GBS originally isolated from a septic neonate, was purchased from ATCC. The bacteria were grown in tryptic soy broth until the OD600 value reached 0.6. Bacterial concentrations were verified by colony formation assays on tryptic soy agar plates, and bacteria were diluted to the appropriate working concentration in sterile PBS before inoculation. To obtain heat-killed GBS, live bacteria were placed in a 60°C water bath for 1 h, and death was verified by plating on tryptic soy agar plates.
Streptococcal infection, endotoxemia, bacterial burden
In the GBS infection model, mice were given varying doses of GBS, i.p. Control animals were given equal volumes of PBS or 107 colony forming unit (cfu) equivalents of heat-killed GBS, i.p. Mice were observed every 4 h for the first 24 h post infection, every 8 h between 24 and 48 h post infection, then every 24 h thereafter for 7 d for signs of morbidity and mortality. At every observation time-point, each mouse was assigned a score on a scale of one to six, based on clinical signs of sepsis or death (Table 1). Moribund mice were assigned a clinical score of 5 and were euthanized. Dead mice were assigned a clinical score of 6.
Table 1.
Grading system for clinical assessment of mice after GBS infectiona
| State | normal | moribund | dead | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Score | 1 | 2 | 3 | 4 | 5 | 6 |
| Activity | active | less active | slowed movement | slowed | inactive, wobbly, unable to walk | dead |
| Coat | smooth | rough | rough, starred | rough, starred | rough, starred | dead |
| Eyes | clear | clear | dull | some crusting | closed, crusted | dead |
| Breathing | normal | normal, slowed | slowed | labored | labored, shallow | none |
| Posture | good | some hunching | hunched | hunched | lying on side | none |
| Isolation | none | none | none | possible isolation | isolation | dead |
| Handling | resists | some resistance | less resistance | minimal resistance | little or no resistance | dead |
Mice were inoculated with GBS i.p., and scored for disease severity according to the score index given above. Moribund mice were given a score of 5 and dead mice were given a score of 6.
To determine bacterial burden, mice were dosed with 106 cfu of GBS, i.p., and euthanized by CO2 inhalation after 24 h. Blood was collected via cardiac puncture, and 100 μl of whole blood were combined with 900 μl tryptic soy broth. Serial dilutions were conducted and plated on tryptic soy agar to enumerate the bacterial counts, as previously described [8, 9].
In the endotoxin shock model, mice were challenged i.p. with LPS (E. coli O55:B5, Sigma-Aldrich) at various doses, and the survival of these animals was monitored.
Determination of cytokine levels
To assess cytokine and chemokine levels in the blood, mice were euthanized and blood was harvested. Cytokines and chemokines in the sera were either determined by ELISA as previously described [10–12], or determined in plasma by using multiplex cytokine analysis kits according to the manufacturer's protocol (Millipore, Billerica, MA). In the multiplex assays, data were collected using the Luminex-200 system Version 1.7 (Luminex, Austin, TX). Data analysis was performed using the MasterPlex QT 1.0 system (MiraiBio, Alameda, CA).
Thioglycollate stimulation, peritoneal lavage, and ex vivo macrophage stimulation
Approximately 2 ml of thioglycollate medium (3%, Invitrogen) was injected into the peritonea of female mice. Peritoneal cells were harvested at various time-points following thioglycollate injection, by lavage with approximately 10 ml of phenol red-free DMEM. Cells were sedimented by centrifugation at 300×g for 5 min. These cells were then stained with leukocyte cell markers and analyzed by flow cytometry. Peritoneal macrophages were enriched by adherence to tissue culture dishes, and treated with LPS at a concentration of 100 ng/ml as previously described [13]. To generate bone marrow-derived macrophages (BMDM), bone marrow cells were harvested from wildtype and Gsr-null mice, and cultured in DMEM containing 10% FBS, 100 units/ml penicillin, 100 g/ml /streptomycin, and 25 ng/ml M-CSF (PeproTech) for 7 days.
Antibodies and fluorophores
For flow cytometry, the following antibodies or fluorophores were used. F4/80-PE-Cy7 (BM8) and CD11b-eFluor 605NC (M1170) were purchased from eBioscience. Ly-6G-Pacific Blue (1A8) and Gr-1-Pacific Blue (RB6-8C5) were purchased from BioLegend. Annexin V-FITC and PI (51-65874x), Siglec-F-PE (E50-2440), and rat anti-mouse CCR3 (CD193)-Alexa Fluor 647 (557974) were purchased from BD Bioscience. Goat anti-mouse CCR2 (CD192) (AAM72) was purchased from Serotec. Qdot 655-conjugated rabbit anti-goat IgG (H+L) (Q11821MP, Invitrogen) was used as a second antibody to detect CCR2. Alexa Fluor 488-conjugated phalloidin (A12379) was also from Invitrogen.
Flow cytometry
Thioglycollate-elicited peritoneal cells were harvested and analyzed by flow cytometry (LSR II, BD Biosciences). The populations of polymorphonuclear leukocytes (PMN) (CD11b+Ly-6G+F4/80−), eosinophils (CD11b−F4/80low), resident macrophages (CD11b+ F4/80high CCR2−), inflammatory macrophages (CD11b+ F4/80highCCR2+), and their cell viability parameters were compared between wildtype and Gsr-deficient mice.
Phagocytosis
Blood sample were used to assess the phagocytotic activity of wildtype and Gsr-null monocytes, as previously described [5]. Briefly, pH-sensitive pHrodo-conjugated E. coli bioparticles (Invitrogen) were opsonized using serum according to manufacturer's instruction. Heparinized blood samples (100 l) harvested from wildtype or Gsr-null mice were incubated with opsonized pHrodo-conjugated E. coli bioparticles at 37qC for 1 h. As controls, blood samples were incubated with pHrodo-conjugated E. coli bioparticles on ice. Cells were then stained with specific leukocyte makers (CD11b, Gr1, and F4/80), and subjected to flow cytometry analysis. Phagocytosis by monocytes (CD11b+Gr-1lowF4/80+) was quantified.
To assess F-actin formation, neutrophils were purified from mouse bone marrow by Percoll gradient centrifugation, as previously described [5, 14]. Bone marrow neutrophils were stimulated with 100 nM phorbol myristate acetate (PMA) for different lengths of time. Cells were then fixed, permeabilized, and stained with Alexa Fluor 488-labeled phalloidin (Molecular Probes). Cells were either examined by confocal microscope (LSM-710, Zeiss), or analyzed by flow cytometry.
Western blotting
Western blot analysis was performed as previously described (42) using antibodies against mitogen-activated protein kinase phosphatase (Mkp)-1 (Santa Cruz Biotechnology), phosphorylated and total c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK) (Cell Signaling Technology).
Mass spectrometry
Mice were injected with 2 ml thioglycollate medium i.p., and euthanized 48 h later. The gelatinous coating on the liver of Gsr-null mice was collected with clean and sterile forceps, and mixed with 1× NuPAGE lithium dodecyl sulfate (LDS) sample buffer (2.5% glycerol, 0.5% LDS, 1% Ficoll-400, 62 mM Tris-HCl pH8.5, 0.064 mM phenol red, 0.055 mM Serva blue G250, 0.13 mM EDTA, and 50 mM DTT). Similarly, sera and PBS lavage supernatants collected from healthy mice were mixed with 4× NuPAGE LDS sample buffer (Invitrogen). The mixtures were heated at 65°C for 10 min, and samples were centrifuged at room temperature for 5 min. The samples were resolved on 10% NuPAGE gels (Invitrogen), and the gels were stained with Coomassie G250. The major band at ~65 kDa was excised from the gel, incubated with 25 mM DTT to reduce cystine residues to cysteines, then incubated with 55 mM iodoacetamide to alkylate the cysteine residues. Finally, the protein was digested with trypsin, and the tryptic peptides analyzed by liquid chromatography coupled tandem mass spectrometry (LC-MS/MS). Further LC-MS/MS analysis coupled with peptide sequencing was used to quantify differentially oxidized albumin peptides. Peak areas of serum albumin peptides that only contained carbamidomethylated cysteine (or cystine) and peptides that were heavily oxidized (containing oxidized methionine and/or cysteic acid) were measured from the selected ion chromatogram. The degree of abnormal albumin oxidation was estimated by calculating the percentages of the peptide containing only carbamidomethylated cysteine (or cystine) and the peptide that contains oxidized methionine and/or cysteic acid(s), using the formula: [peak area of a given peptide/(peak area of carbamidomethylated peptide + peak area of heavily oxidized peptide)]×100.
Statistics
Differences in survival between wildtype and Gsr-deficient mice after GBS infection were determined by Kaplan-Meier analyses. Bacterial burdens were compared between wildtype and Gsr-deficient mice using χ2 analyses. Differences between the two groups in clinical scores were identified using two-way ANOVA. Differences between wildtype and Gsr-deficient mice in other parameters were identified using unpaired Student's t-tests. A p value of less than 0.05 was considered significant. The LD50 values and 95% confidence intervals of wildtype and Gsr-deficient mice following LPS challenge were calculated using the R package [15].
Results
Gsr-deficient mice exhibit increased morbidity and mortality as well as a more robust cytokine storm following group B Streptococcus infection
To assess the functional importance of Gsr in host defense against GBS, wildtype and Gsr-deficient mice were infected i.p. with GBS serotype H1 strain (COH1), a clinical isolate from a case of fatal infant septicemia. Animal survival was monitored over 7 days. Figure 1A depicts the survival curves of wildtype and Gsr-deficient mice after challenge with 1×107 cfu of GBS. Gsr-deficient mice not only exhibited a lower overall rate of survival but also succumbed to GBS earlier than did wildtype mice. To quantify the susceptibility of these two strains of mice to GBS, we infected wildtype and Gsr-deficient mice with a wide range of doses of bacteria, and documented the survival (Table 2). The LD50 values for wildtype and Gsr-deficient mice were estimated to be 3.01×107 and 1.36×106 cfu of GBS, respectively.
Figure 1.
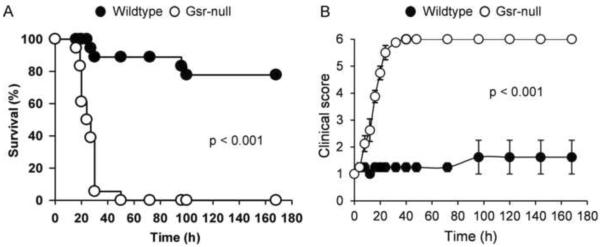
Morbidity and mortality of wildtype and Gsr-deficient mice following group B streptococcal challenge. A. Survival curves for wildtype and Gsr-deficient mice after GBS challenge. GBS (107 cfu) were introduced i.p. into wildtype and Gsr-deficient mice (n=18 for each group). Mortality was documented over 7 days. Differences between the two groups were determined by Kaplan-Meier analysis. B. Disease severity in wildtype and Gsr-deficient mice after GBS infection. GBS (107 cfu) were introduced i.p. into wildtype and Gsr-deficient mice (n=8 for each group, 4 male and 4 female). Morbidity was evaluated using the scoring system described in Table 1. Data in the graphs represent mean ± SEM of 8 animals. Differences between the two groups were identified using two-way ANOVA.
Table 2.
Survival of mice after GBS infectiona
| Dose of infection (cfu) | Survivedb (total infected) |
|
|---|---|---|
| Wildtype | Gsr-deficient | |
| 1 × 105 | 8 (8) | 6 (8) |
| 1 × 106 | 13 (14) | 9 (14) |
| 1 × 107 | 14 (18) | 0 (18) |
| 1 × 108 | 2 (12) | 0 (12) |
| 1 × 109 | 0 (4) | 0 (4) |
Mice were inoculated with GBS i.p.
Number represents animals survived after 7 days.
A clinical scoring system was developed to evaluate disease severity in wildtype and Gsr-deficient mice following GBS challenge (Table 1). Gsr-null mice not only exhibited more severe disease, but also developed disease earlier than did wildtype mice (Figure 1B). At a dose of 107 cfu of GBS per animal, Gsr mice developed significantly more severe signs of disease, including rough coat and slow or labored breathing within 8 h, while wildtype mice had few signs of disease at this time-point. By 16 h, all Gsr-deficient mice developed signs of severe sepsis, whereas the wildtype mice remained relatively asymptomatic. All 8 Gsr-deficient mice reached a clinical score of 6 by 40 h post bacterial challenge. In contrast, with the same dose of GBS only one out of 8 wildtype mice had reached a clinical score of 6 at 96 h post inoculation, while all others survived the 7 day observation period.
To understand the mechanisms underlying the high morbidity and mortality of Gsr-deficient mice after GBS infection, we measured the levels of cytokines and chemokines in the plasmas of animals. Mice were given PBS, 106, or 107 cfu of live GBS per animal. As an additional control, mice were also given 107 cfu equivalents of heat-killed GBS per animal. Mice were euthanized 24 h post bacterial administration, and cytokines and chemokines in the plasma were measured using multiplex assays (Figure 2). The levels of cytokines and chemokines in the PBS-treated wildtype and Gsr-deficient mice were very low. Infection of wildtype mice with 106 cfu of live GBS resulted in little changes in blood cytokine and chemokine levels, suggesting that inflammation was minimal in these mice at 24 h. Compared to wildtype mice, Gsr-deficient mice infected with 106 cfu of live GBS exhibited a much more robust inflammatory response, as indicated by dramatic increases in blood cytokine and chemokine levels. Specifically, Gsr-deficient mice had substantially higher concentrations of TNF-. IL-6, IL-10, IFN-, KC, MCP-1, MIP-1, MIP-1, MIP-2, RANTES, IP-10, G-CSF, GM-CSF, and M-CSF, than did wildtype micẽ Infection of wildtype mice with a higher dose of GBS (107 cfu) substantially increased the levels of TNF-. IFN-, KC, MCP-1, MIP-1, RANTES, IP-10, G-CSF, and M-CSF, suggesting significant inflammation in these mice. Furthermore, the concentrations of TNF-. IL-6, IL-10, KC, MCP-1, MIP-1, MIP-1, MIP-2, RANTES, G-CSF, GM-CSF, and M-CSF in Gsr-deficient mice infected with 107 cfu of GBS were higher than those in similarly infected wildtype mice. The differences in plasma cytokines and chemokines between the wildtype and Gsr-deficient mice were dependent on the viability of the administered GBS, since wildtype and Gsr-deficient mice given 107 cfu of heat-killed GBS had levels of blood cytokines and chemokines comparable to mice given PBS.
Figure 2.

Serum cytokine and chemokine levels in wildtype and Gsr-deficient mice after GBS challenge. Wildtype and Gsr-deficient mice were infected i.p. with GBS (either 106 or 107 cfu per animal), or given heat-killed GBS (107 cfu equivalent, marked as GBS HK in the graph), or given PBS. Animals were euthanized 24 h after bacterial challenge, and blood was harvested by cardiac puncture. The concentrations of cytokines and chemokines were determined using cytokine multiplex kits. Data in the graphs represent mean ± SEM, n = 4–6. *, p < 0.05, compared to similarly treated wildtype mice. †, p < 0.05, compared to mice given heat-killed GBS (Student's t-test).
Gsr-deficient mice display higher bacterial burdens following group B Streptococcus infection
To understand the effects of Gsr deficiency on bactericidal function against GBS, we assessed bacterial burden in infected mice. As shown in Table 3, 9 out of 10 wildtype mice dosed with 106 cfu of GBS cleared their bacteria by 24 h, with only one animal showing low but detectable levels of bacteria (<104 cfu/ml blood). In contrast, out of 10 Gsr-deficient mice only one mouse cleared GBS and another mouse had bacterial counts in the low but detectable range of bacterial burden (<104 cfu/ml blood), while the remaining 8 mice exhibited high bacterial counts (>2×105 cfu/ml blood). χ2 analysis revealed a significant difference in the bacterial loads between wildtype and Gsr-deficient mice (P<0.0005).
Table 3.
Bacterial burden in the blood of GBS-infected micea
| Strain | animal number | # of mice with blood bacterial counts in the range of |
|||
|---|---|---|---|---|---|
| 0 cfu/ml | 0 < cfu/ml < 104 | > 2 × 105 cfu/ml | |||
| Wildtype | Male | 5 | 4 | 1 | 0 |
| Female | 5 | 5 | 0 | 0 | |
| Total | 10 | 9 | 1 | 0 | |
|
| |||||
| Gsr-deficient | Male | 5 | 1 | 1 | 3 |
| Female | 5 | 0 | 0 | 5 | |
| Total | 10 | 1 | 1 | 8 | |
Mice were infected with 1 × 106 cfu GBS per animal, and euthanized 24 h later. Blood was harvested by cardiac puncture aseptically, and bacterial burden in the blood was determined by culture.
Phagocytotic defects of Gsr-null phagocytes
Phagocytes play a critical role in host defense against extracellular bacteria [16], and Gsr-deficient mice displayed higher bacterial burdens. We have shown previously that neutrophils deficient in Gsr exhibit marked impairment in phagocytic activity [5]. However, it is unclear whether other types of phagocytic cells also exhibit similar impairment. We investigated the effects of Gsr deficiency on phagocytic activity of monocytes, since they play an important role in the detection of invading pathogens. Heparinized blood was harvested from uninfected wildtype or Gsr-deficient mice, and blood leukocytes were incubated with opsonized pHrodo-conjugated E. coli bioparticles at 37°C or 0°C for 1 h. The leukocytes were then stained with specific leukocyte makers (CD11b, Gr1, and F4/80), and subjected to flow cytometry analysis. pHrodo Green-conjugated E. coli bioparticles are non-fluorescent outside the leukocytes at neutral pH, but fluoresce brightly green at acidic pH such as in phagosomes. In combination with monocyte markers (CD11b+Gr-1lowF4/80+), phagocytosis by mature monocytes was quantified (Figure 3A, Left panel). At 0°C, very few E. coli bioparticles were engulfed by monocytes from either wildtype or Gsr-null mice, and no differences in phagocytotic activities were detected between the two groups (Top and 2nd histograms in left panel of Figure 3A). However, at 37°C, there was a clear difference in the phagocytotic properties between these two monocyte groups. Compared to wildtype monocytes, Gsr-deficient monocytes engulfed fewer E. coli particles, indicated by the leftward shift of the fluorescent spectrum (3rd and bottom histograms in left panel of Figure 3A) and decreased mean fluorescence intensity (MFI) in these cells (Figure 3A, Bar graph).
Figure 3.

Compromised phagocytosis and actin polymerization in Gsr-deficient phagocytes. A. Compromised phagocytosis in Gsr-deficient monocytes. Heparinized whole blood from uninfected wildtype or Gsr-deficient mice was incubated with pHrodo-conjugated E. coli Bioparticles (200 μg/ml) at 0°C (on ice) or at 37 °C for 1 h, and then stained with leukocyte surface proteins. The cells were then analyzed by flow cytometry and the leukocytes were gated to assess engulfment of pHrodo-conjugated E. coli Bioparticles by monocytes (CD11b+Gr-1lowF4/80+). Left panel: Representative histogram of phagocytosis by blood monocytes. Right panel: Graphic depiction of phagocytosis by monocytes. Data represent the mean fluorescence intensity (MFI) of pHrodo™ particles engulfed by blood monocytes (CD11b+Gr-1lowF4/80+). B. Representative images of F-actin in control and PMA-stimulated neutrophils. Neutrophils were isolated from bone marrow and seeded on uncoated glass coverslips. After 1 h, neutrophils were stimulated with PMA (100 nM) for the indicated time, or left untreated (control). F-actin (green) was stained with Alexa Fluor 488-conjugated phalloidin and DNA (blue) was stained with DAPI. Cells were examined by confocal microscopy. Scale bar represents 5 μm. C. Flow cytometry-based quantification of F-actin assembly (measured as MFI) in PMA-stimulated wildtype and Gsr-deficient neutrophils. Bone marrow neutrophils were stimulated with PMA (100 nM) for different periods of time, stained with Alexa Fluor 488-conjugated phalloidin, and analyzed by flow cytometry. For normalization, MFI of untreated neutrophils were set as 100%. Values in the graphs in the right panel of A and C represent mean ± SEM from at least 3 independent experiments. *, p < 0.05; **, p < 0.01 (Student's t-test), compared between the wildtype and Gsr-deficient groups.
Phagocytosis is mediated by rapid actin polymerization and depolymerization [17, 18]. The most striking feature of phagocytosis is the formation of the phagocytic cup characterized by focal accumulation of actin filaments (F-actin), which can be visualized using fluorescent-labeled phalloidin [19]. In addition to phagocytosis of foreign particles, actin assembly in neutrophils can also be invoked by phorbol myristate ester (PMA) stimulation [20]. To examine the role of Gsr in actin assembly in phagocytes, we investigated F-actin formation in both wildtype and Gsr−/− neutrophils by confocal fluorescent microscopy. Without PMA stimulation, F-actin was primarily located near the cell surface (Figure 3B). Within 5 min after PMA stimulation, increased F-actin was detected on the cell membrane in both wildtype and Gsr-deficient neutrophils. However, while F-actin was relatively stable and lasted over 60 min on the cell membrane in wildtype neutrophils, F-actin rapidly disappeared from the cell membrane in Gsr-deficient neutrophils. By 15 min post PMA stimulation, F-actin was only barely detectable on the membrane in a small fraction of the Gsr-deficient neutrophils. In the majority of the Gsr-deficient neutrophils, F-actin was either undetectable or faintly detectable near the segmented nuclear lobe, suggesting rapid F-actin depolymerization in these cells.
F-actin remodeling in neutrophils upon exposure to PMA was also quantified by flow cytometry after staining with fluorescent phalloidin (Figure 3C). PMA stimulated a significant actin polymerization in wildtype neutrophils, which lasted for at least 30 min. While PMA also tended to increase actin polymerization in Gsr-deficient neutrophils, similar to the pattern of the confocal results F-actin detected by flow cytometry was short-lived such that by 15 min after PMA stimulation F-actin was significantly lower in Gsr-deficient neutrophils than in wildtype neutrophils (Figure 3C). These results suggest that Gsr-deficient neutrophils have impaired cytoskeleton remodeling during phagocytosis.
Gsr-deficient mice do not exhibit increased sensitivity to endotoxin
To understand whether Gsr-deficient mice have defects in the innate immune response to bacterial components, we challenged wildtype and Gsr-deficient mice with lipopolysaccharide (LPS), and documented mouse survival. Although Gsr-deficient mice exhibited an increased susceptibility to E. coli infection [5], these animals did not exhibit an increased sensitivity to LPS (Table 4). In fact, Gsr−/− mice were slightly more resistant to LPS than were wildtype mice. The LD50 value for wildtype mice, representing the dose of LPS that caused a mortality of 50%, was estimated to be 14.9 mg/kg body weight (95% confidence interval: 13.9 – 19.0 mg/kg). On the other hand, the LD50 value for Gsr−/− mice was estimated to be 20.9 mg/kg body weight (95% confidence interval: 17.6 – 24.4 mg/kg).
Table 4.
Survival of WT and Gsr−/− mice at 72 h after LPS challengea
| LPS (mg/kg) | Survived (total challenged) |
|
|---|---|---|
| WT | Gsr−/− | |
| 7.5 | 5 (5) | 5 (5) |
| 10 | 5 (5) | 5 (5) |
| 15 | 8 (15) | 13 (15) |
| 20 | 2 (7) | 2 (6) |
| 25 | 1 (2) | 0 (2) |
| 30 | 0 (12) | 3 (12) |
| 35 | 0 (2) | 0 (2) |
Mice were challenged i.p. with the indicated doses of LPS.
LD50 was calculated by logistic regression.
LD50 wt= 16.2 mg/kg body weight; LD50 Gsr= 20.7 mg/kg body weight.
Consistent with the overall comparable endotoxin sensitivities of the two strains of mice, serum TNF- levels were similar in wildtype and Gsr-deficient mice at both early and late time-points (Figure 4A). Similarly, no marked differences between wildtype and Gsr−/− groups were observed in the serum levels of KC and MCP-1, two important chemokines. Nonetheless, modest differences were found in the serum levels of IL-6 and IL-10. Serum IL-10 levels were higher in wildtype than in Gsr-null mice 2 h after LPS challenge. At 18 h after LPS challenge, IL-6 and IL-10 levels in wildtype mice were significantly higher than in Gsr-deficient mice. These results suggest that the inflammatory response to LPS at this time-point in wildtype mice could be more robust than in Gsr-deficient mice.
Figure 4.

LPS responses of Gsr-deficient mice and macrophages. A. Serum cytokine and chemokine levels in wildtype and Gsr-deficient mice. Mice were challenged with LPS i.p. (15 mg/g body weight) and euthanized at the indicated time-points. Blood was collected by cardiac puncture, and serum cytokines were measured by ELISA. Values in the graphs represent mean ± SEM of 5–10 independent experiments. B. Cytokines produced by thioglycollate-elicited peritoneal macrophages. Macrophages were stimulated with LPS (100 ng/ml) for 8 or 24 h, and cytokines in the medium were assessed by ELISA. *, p < 0.05 (Student's t-test), compared between the wildtype and Gsr-deficient groups. Values in the graphs represent mean ± SEM of at least 3 independent experiments. C. The activation of MAPKs and Mkp-1 induction in LPS-stimulated peritoneal macrophages. Macrophages were harvested from thioglycollate-stimulated mice, and stimulated with LPS (100 ng/ml). The activation of MAPKs and the induction of Mkp-1 were assessed by Western blot analysis using antibodies against phospho-MAPKs and Mkp-1. The blots were stripped and blotted with antibodies against total MAPKs. Data shown are representative results.
To examine the effect of Gsr deficiency on the macrophage response to LPS, we elicited peritoneal macrophages with thioglycollate and assessed cytokine production of isolated peritoneal macrophages. Thioglycollate-elicited peritoneal macrophages from both wildtype and Gsr-deficient mice produced comparable amounts of cytokines after LPS stimulation (Figure 4B). Likewise, BMDM generated from both wildtype and Gsr-deficient mice also produced comparable amounts of TNF-, IL-6, IL-10, and IL-1 after LPS stimulation (data not shown). Similarly, Gsr deficiency did not noticeably affect the kinetics of LPS-induced mitogen-activated protein kinase (MAPK) activation in thioglycollate-elicited peritoneal macrophages. Activation of ERK, JNK, and p38, as well as induction of Mkp-1, occurred similarly in both wildtype and Gsr-deficient macrophages (Figure 4C). These results indicate that Gsr deficiency in macrophages neither markedly alters the cellular signaling events nor substantially affects cytokine production during the response to LPS.
Abnormal leukocyte mobilization after thioglycollate stimulation in Gsr-deficient mice
When we prepared thioglycollate-elicited peritoneal macrophages, we noticed that there were far fewer peritoneal cells recovered from Gsr-deficient mice after thioglycollate stimulation than from wildtype mice (Figure 5A). However, in the absence of any stimulation, peritoneal lavage cell numbers were not different between the two strains of mice. To understand the mechanism underlying the dramatic difference in the population of thioglycollate-elicited peritoneal macrophages between the two genotypes, we characterized the time-course of appearance of peritoneal lavage cells in wildtype and Gsr-deficient mice after thioglycollate stimulation. Several differences were observed between wildtype and Gsr-deficient mice in these cells. First, the total number of peritoneal lavage cells started to differ significantly between the two groups of mice by 24 h after thioglycollate stimulation, and the difference increased through 96 h (Figure 5A). Initially (6–24 h), the majority of the peritoneal lavage cells were CD11b+Ly-6GhighF4/80− neutrophils in both wildtype and Gsr-deficient mice (Figure 5B). However, the numbers of neutrophils that migrated to the peritoneal cavity in wildtype mice were significantly higher than those in Gsr-deficient mice. The differences in neutrophil numbers were initially modest (6 h), but became substantial by 24 h. After 24 h, both the absolute numbers and percentages of neutrophils among peritoneal lavage cells declined in both groups. Second, far fewer monocytes/macrophages were recruited to the peritoneal cavity of Gsr-deficient mice following thioglycollate stimulation (Figure 5C). After thioglycollate stimulation, monocytes/macrophages (CD11b+F4/80high) arrived to the peritoneal cavity later than did neutrophils (Figure 5C, enclosed in ovals). In wildtype mice, macrophage numbers were increased by 24 h and reached maximal levels at about 48 h, which persisted through 96 h. In contrast, macrophage numbers in the Gsr-deficient mice did not increase substantially following thioglycollate stimulation. Third, Gsr-deficient mice failed to accumulate eosinophils in the peritoneal cavity after thioglycollate stimulation (Figure 5C). Similar to macrophages, a group of CD11b−F4/80low cells also migrated to the peritoneal cavity after thioglycollate stimulation (Figure 5C, outlined in the squares). Flow cytometry analysis indicated that this group of CD11b−F4/80low cells expressed both Siglec-F and CCR3 (Figure 5C, column on the far right), two markers characteristic of eosinophils [21]. The eosinophil populations were dramatically lower in Gsr-deficient exudates than in the wildtype exudates. Finally, among the monocytes/macrophages, the inflammatory macrophage populations (CD11b+F4/80highCCR2+) [22] were substantially lower in the Gsr-deficient exudates than in the wildtype exudates (Figure 5D). In the wildtype mice, thioglycollate stimulation resulted in a substantial increase in the inflammatory macrophage population. In contrast, accumulation of inflammatory macrophages in the peritonea of the Gsr-deficient mice was minimal.
Figure 5.

Abnormal leukocyte mobilization in Gsr-deficient mice after thioglycollate stimulation. Resident and thioglycollate-elicited peritoneal cells were harvested from the peritoneal cavity of wildtype and Gsr-deficient mice either prior to or after thioglycollate stimulation at various time-points. Numbers of total leukocytes in peritoneal lavage fluid were counted using a hemocytometer. The peritoneal lavage cells were sedimented by centrifugation, stained with leukocyte markers, and analyzed by flow cytometry. Peritoneal lavage cells were first gated on forward scatter (FSC) and side scatter (SSC) to exclude cell debris, and viable cells were then gated for CD11b+Ly-6GhighF4/80− neutrophils, and CD11b+F4/80high monocytes/macrophages. The CD11b−F4/80low cells outlined in the squares of were further gated for CCR3 and Siglec-F expression (right column). Since the CD11b−F4/80low cells were positive for Siglec-F and CCR3, these cells are considered to be eosinophils. A. Time-dependent changes in the numbers of total peritoneal elicited leukocyte cells (PEC). B. Time-dependent changes in the numbers (bar graph on the left) and flow cytometry plots (on the right) of peritoneal neutrophils. Dots in the rectangle regions within the scatter plots are neutrophils (CD11b+Ly-6GhighF4/80−), and numbers given on the right side of the rectangles indicate the percentage of neutrophils among the total PEC. C. Time-dependent changes in the numbers (bar graph on the left) and flow cytometry plots (on the right) of peritoneal monocytes/macrophages and eosinophils. Dots in the ovals and squares within the scatter plots are macrophages/monocytes (CD11b+F4/80high) and eosinophils (CD11b−F4/80lowSiglec-F+CCR3+), respectively. The percentages of the monocytes/macrophages (on the right side of the ovals) and the eosinophils (above the squares) among the total PEC are indicated in the plots. Cells in the squares were further gated and plotted for CCR3 and Siglec-F expression (right columns). Since the CD11b−F4/80low cells were positive for Siglec-F and CCR3, these cells are considered as eosinophils. D. Time-dependent changes in the numbers (bar graph on the left) and flow cytometry plots (on the right) of inflammatory monocytes/macrophages. The CD11b+F4/80high monocytes/macrophages outlined in the ovals of cytometry plots in panel B were further gated for CCR2 expression to identify inflammatory monocytes/macrophages (CD11b+F4/80highCCR2+). The numbers given in the scatter plots in D indicate the percentage of inflammatory monocytes/macrophages within the monocyte/macrophage population. Absolute numbers of each leukocyte subset were calculated according to the total PEC numbers and the percentages of each leukocyte groups determined by flow cytometry. Values in the graphs represent mean ± SEM of at least 3 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (Student's t-test), compared between wildtype and Gsr-deficient groups at the same time-points. The scatter plots shown are results from representative experiments.
The abnormalities in leukocyte mobilization were not due to decreases in chemokine production. The levels of the neutrophil chemokine KC and the monocyte chemokine MCP-1 in the blood and peritoneal lavage fluids of Gsr-deficient mice after thioglycollate stimulation were similar to those in the wildtype mice (data not shown). However, thioglycollate-stimulated Gsr-deficient mice contained greater numbers of apoptotic cells in their peritoneal cavities than did similarly treated wildtype mice (Figure 6). Apoptosis was more prevalent in the Gsr-deficient neutrophils. Additionally, more apoptotic eosinophils were also found in the Gsr-deficient mice at 24 h (Figure 6). Interestingly, Gsr-deficient macrophages did not exhibit enhanced apoptosis. These results indicate that Gsr plays an important role in the mobilization of inflammatory leukocytes after inflammatory stimulation, perhaps in part by facilitating cell survival in the face of cellular stress.
Figure 6.

Increased phagocyte apoptosis in the peritoneal cavity of Gsr-deficient mice after thioglycollate stimulation. Mice were treated as described for Figure 5, and peritoneal leukocytes were harvested by lavage. The percentages of apoptotic cells in total PEC and in distinct leukocyte populations were quantified by flow cytometry after staining with annexin V and PI. Cells that were positive for annexin V were counted as apoptotic cells. Data represent mean ± SEM from at least 3 independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (Student's t-test), compared between the two different groups at the same time-points.
To investigate the mechanism underlying the attenuated leukocyte mobilization we performed necropsy. To our surprise, a thick layer of gelatinous material was found on internal organs, particularly on the liver, in the thioglycollate-stimulated Gsr-deficient mice, but not on the internal organs of wildtype mice (Figure 7A). The lobes of the livers in the thioglycollate-stimulated wildtype mice could be separated easily. In contrast, the lobes of the livers in the Gsr-deficient mice were difficult to separate, as if the gelatinous material acted as an adhesive between the hepatic lobes. It is worth noting that in the absence of thioglycollate stimulation, no gelatinous material was found in Gsr-deficient mice (data not shown). The gelatinous material on the liver of Gsr-deficient mice was clearly seen in the histology sections (Figure 7B). The gelatinous material on the livers of thioglycollate-stimulated Gsr-deficient mice was collected and placed in buffer containing 1% Triton X-100. Because the material was largely insoluble in this buffer, we mixed the material with 1× NuPAGE sample buffer containing 50 mM DTT and 0.5% LDS (a strong detergent similar to SDS). After heating the samples to 65 C for 10 min, the samples were resolved by electrophoresis using 10% NuPAGE gel (Figure 7C). To characterize the composition of the gelatinous material, we also separated serum and lavage supernatants isolated from wildtype and Gsr-deficient mice. Proteins in these samples were stained by Coomassie blue. The overall protein patterns in these different samples were very similar, suggesting that the gelatinous material covering the internal organs in the thioglycollate-stimulated Gsr-deficient mice was ascitic protein aggregates. The most abundant protein in the gelatinous material recovered on the livers of thioglycollate-stimulated Gsr-deficient mice exhibited an apparent molecular mass of approximately 65 kDa.
Figure 7.

Demonstration and characterization of protein aggregation coating the internal organs of Gsr-deficient mice after thioglycollate stimulation. Female wildtype and Gsr-deficient mice were stimulated with thioglycollate medium (3%, 2 ml, i.p.) and euthanized 48 h later. The peritoneal cavities were opened for photography, or collection of the coating on the livers. Livers were excised from the animals, fixed, and liver sections were stained with H&E. A. Photographs of the internal organs of wildtype and Gsr-deficient mice. Note the whitish gelatinous coating materials on the liver of Gsr-deficient mice (indicated by arrows). B. H&E staining of the liver sections. Note the layer of fibrous coating on the surface of the liver of the Gsr-deficient mice marked by an arrow. Scale bar represents 100 μm. C. Coomassie blue staining of proteins in the gelatinous material, serum, and lavage samples. Mice were euthanized to collect blood by cardiac puncture, or peritoneal fluids by lavage with PBS. Additional mice were euthanized to collect the gelatinous coating materials on the liver of Gsr-deficient mice. The lavage fluids were centrifuged and supernatants were separated on NuPAGE gel together with serum and the gelatinous coating collected from the surface of the livers of thioglycollate-stimulated Gsr-deficient mice. The band marked with an asterisk was subjected to mass spectrometry analysis.
The major band of ~65 kDa was excised from the gel, incubated with 25 mM DTT to reduce cystine residues to cysteines, and then incubated with 55 mM iodoacetamide to alkylate the cysteine residues. Finally, the protein was subjected to enzymatic digestion with trypsin, then to LC-MS/MS. LC-MS/MS identified 31 peptides from mouse serum albumin with high abundance, 12 peptides from murine hemopexin with medium abundance, as well as peptides derived from several other serum proteins in substantially lower quantities. The results were not surprising, since a single band was subjected to analysis. To understand the nature of protein aggregation in Gsr-deficient mice, we analyzed the albumin peptides for signs of oxidation. Further LC-MS/MS analysis coupled with peptide sequencing revealed 67 differentially oxidized albumin peptides (see partial list in Table 5). The cystine residues that normally exist as disulfide bonds in albumin were mostly oxidized to cysteic acids in the aggregated samples. A wide disparity in the percentage of oxidation was observed among the different cystine residues, ranging from 2% at Cys340 (data not shown) to 53% at Cys501 (Table 5). We estimated that on average at least 3–4 cystine residues per albumin molecule, which has a total of 36 cysteine/cystine residues, were oxidized to cysteic acids. In summary, the gelatinous material observed in Gsr-deficient mice following thioglycollate stimulation is largely composed of abnormally oxidized serum albumin.
Table 5.
Partial list of albumin-derived peptides containing oxidized cysteine residues
| Sequence | Modified residue | Peak Areaa | % |
|---|---|---|---|
| 76TC(CAM)VADESAANC86(CAM)DK88 | 68628062 | 90.10 | |
| 76TC77(SO3)VADESAANC86(CAM)DK88 | C77 | 7540211 | 9.90 |
|
| |||
| 106ENYGELADC114(CAM)C115(CAM)TK117 | 1081000552 | 93.30 | |
| 106ENYGELADC114(CAM)C115(SO3)TK117 | C115 | 36494575 | 3.15 |
| 106ENYGELADC114(SO3)C115(CAM)TK117 | C114 | 41261358 | 3.56 |
|
| |||
| 123NEC125(CAM)FLQHK130 | 189327692 | 85.50 | |
| 123NEC125(SO3)FLQHK130 | C125 | 31989339 | 14.50 |
|
| |||
| 131DDNPSLPPFERPEAEMC148(CAM)TSFK152 | 55278063 | ||
| 131DDNPSLPPFERPEAEM(OX)C148(CAM)TSFK152 | 42665989 | 75.01 | |
| 131DDNPSLPPFERPEAEMC148(SO3)TSFK152 | 5174401 | ||
| 131DDNPSLPPFERPEAEM(OX)C148(SO3)TSFK152 | C148 | 27455790 | 24.99 |
|
| |||
| 470LPC472(CAM)VEDYLSAILNR483 | 415788330 | 87.76 | |
| 470LPC472(SO3)VEDYLSAILNR483 | C472 | 57983286 | 12.24 |
|
| |||
| 484VC485(CAM)LLHEKR490 | 1064877544 | 78.6 | |
| 484VC485(SO3)LLHEKR490 | C485 | 290225327 | 21.4 |
|
| |||
| 500C500(CAM)C501(CAM)SGSLVER508 | 19366640 | 47.0 | |
| 500C500(CAM)C501(SO3)SGSLVER508 | C501 | 21702104 | 53.0 |
|
| |||
| 509RPC511(CAM)FSALTVDETYVPK524 | 119614451 | 84.54 | |
| 509RPC511(SO3)FSALTVDETYVPK524 | C511 | 1124067375 | 15.46 |
|
| |||
| 528AETFTFHSDIC538(CAM)TLPEK543 | 148799481 | 79.20 | |
| 528AETFTFHSDIC538(CAM)TLPEKEK454 | 154526450 | ||
| 528AETFTFHSDIC538(SO3)TLPEK543 | C538 | 64693802 | 20.80 |
| 528AETFTFHSDIC538(SO3)TLPEKEK454 | 15091409 | ||
Peak area is measured from the selected ion chromatogram generated for a given peptide(s). The last column (%) represents the percentages of peptides whose cysteine(s) was carboamidomethylated and peptides where at least one cysteine was oxidized into cysteic acid or a methionine is oxidized. For example, in the top raw 90.1% of C77 is not oxidized while 9.9% of C77 is oxidized into cysteic acid. The percentage of a given peptide is estimated based on the area of the peptide peak relative to the total area of all the peaks derived from the same peptide.
CAM: carbamidomethyl group.
Carbamidomethylated cysteine residues are derived from either cysteine or cystine.
Discussion
Our studies reveal a number of previously under-appreciated functions of Gsr in innate host immunity against bacterial infection. Our findings presented here indicate that Gsr-deficient mice are defective in their bactericidal mechanism against GBS (Table 3). In the absence of Gsr, mice more readily developed cytokine storm (Figure 2), and exhibited greater susceptibility to GBS infection (Figure 1, Table 2). Studies of the phagocytotic function of the neutrophils and monocytes of Gsr-deficient mice revealed defects in phagocytosis (Figure 3). Moreover, Gsr-deficient mice exhibit a marked defect in the mobilization of inflammatory leukocytes (Figure 5), which likely contributes to the antimicrobial defects in these mice. Analysis of the peritoneal lavage cells from thioglycollate-stimulated mice indicate that Gsr deficiency renders neutrophils, and to a lesser degree eosinophils, more susceptible to apoptosis (Figure 6). The defects in the leukocyte mobilization of Gsr-deficient mice were associated with accumulation of profoundly oxidized serum albumin, one of the most abundant proteins in ascites. Taken together, our studies indicate that the redox regulation mediated by Gsr is crucial for optimal host defense against bacterial infection.
Gsr catalyzes the reduction of GSSG to GSH, a major cellular antioxidant. During infection, neutrophils engulf bacteria, and undergo a robust respiratory burst to produce ROS, including H2O2 and O2•−, in the phagolysosome. Although H2O2 and O2•− created through the respiratory burst are critical for bactericidal activity in the phagolysosome, these ROS can cause oxidation and inactivation of a variety of host biomolecules when they diffuse out from the phagolysosome. In the cytosol, H2O2 is detoxified by GSH, through the reaction (H2O2+2GSH→2H2O+GSSG) mediated by glutathione peroxidase [1, 2]. The cellular pool of GSH is replenished by two mechanisms: 1) GSH regeneration from GSSG mediated by Gsr and 2) GSH synthesis mediated by both -glutamate-cysteine ligase and glutathione synthetase. Gsr converts GSSG back to GSH via the reaction (GSSG+NADPH+H+→2GSH+NADP+), thus supplying GSH for cytosolic H2O2 detoxification to protect phagocytes from the collateral damage of the respiratory burst [1, 2]. NADPH is produced by the hexose monophosphate shunt. Thus, by catalyzing the reduction of GSSG to GSH, Gsr facilitates the NADPH-mediated electron transfer from glucose to H2O2. As a result of the GSH recycling process, phagocytes can sustain the respiratory burst and kill engulfed bacteria. In the absence of Gsr, this GSSG/GSH cycle is non-functional. It is conceivable that during massive bacterial infection, GSH consumption for cytosolic H2O2 detoxification could overwhelm GSH synthesis. Once this occurs, ROS will spill into the cytosol resulting in oxidation of biologically active host molecules, likely leading to compromised phagocytic activity. This is consistent with the higher bacterial burden in GBS-infected Gsr-null mice, and the greater morbidity and mortality in these mice (Figure 1). The dramatic increases in serum cytokine and chemokine levels observed in Gsr-deficient mice after GBS infection are likely the consequence of greater bacterial growth and spread (Figure 2).
The contrast in the susceptibilities of Gsr-deficient mice to E. coli and LPS
Previously, we have demonstrated that Gsr-deficient mice are dramatically more sensitive to infection by bacteria, including E. coli [5]. In the present study, we found that compared to wildtype mice Gsr-deficient animals were not more sensitive to LPS, a cell wall component of Gram-negative bacteria. In fact, Gsr-deficient mice are actually slightly more resistant to LPS, indicated by a small increase in LD50 value (Table 4). Perhaps reflecting this small increase in LPS resistance, we found that serum IL-6 and IL-10 levels in the Gsr-deficient mice 18 h post LPS challenge were lower than that in the wildtype mice (Figure 4A). Analyses of cytokine production and cellular signaling events of both wildtype and Gsr-deficient macrophages following LPS stimulation revealed no substantial differences between these two groups (Figures 4B & C). The overall comparable macrophage responses indicate that Gsr does not play an essential role in the detection of LPS as well as in the initiation of the inflammatory response. These results suggest that the increased susceptibility of Gsr-deficient mice to bacterial infection is probably not due to a hyper-responsiveness of these animals to bacterial products, but rather due to their inability to contain and eliminate the invading and proliferating micro-organisms. This is consistent with the observation that Gsr-deficient neutrophils fail to mount a robust and persistent oxidative burst and to generate neutrophil extracellular traps [5].
The role of Gsr in phagocytosis
Depletion of GSH stores in the neutrophils of the Gsr-deficient mice may also be a contributing factor to their phagocytotic defects. Phagocytosis is a highly dynamic process mediated by actin polymerization and depolymerization controlled by intricate signal transduction pathways [17, 18]. It is conceivable that the engulfment of bacterial particles initiates the respiratory burst, and that the oxidation of host proteins, as a result of GSH depletion, vastly changes the dynamics of the cellular events within the phagocytes, eventually compromising the subsequent phagocytotic process. This idea is consistent with the observation that Gsr-deficient neutrophils were defective in reorganizing F-actin (Figures 3B &C). Previously, we have shown that Gsr-deficient neutrophils exhibit marked impairment in phagocytic activity. Here we demonstrated that in addition to neutrophils, Gsr-deficient monocytes also display a significant defect in phagocytic activity (Figure 3A). We speculate that the effect of Gsr deficiency on F-actin represents only the “tip of the iceberg” of the phagocytic impairments of the Gsr−/− phagocytes. For example, it has been demonstrated that GSH depletion prevents microtubule assembly and causes breakdown of preassembled cytoplasmic microtubules in human neutrophils [23, 24]. Additionally, it has been shown that growth factor-mediated actin polymerization into filaments, translocation to the cell periphery, and membrane ruffling are regulated by S-glutathionylation [25–27]. Actin S-glutathionylation induced by ROS during cell adhesion is essential for cell spreading and cytoskeleton remodeling [26]. It is plausible that depletion of GSH in Gsr-deficient phagocytes would substantially alter the dynamics of protein S-glutathionylation, leading to impaired cytoskeleton remodeling during phagocytosis. Very recently, Sakai et al. have shown that actin undergoes glutathionylation in a ROS-dependent manner [28]. They further demonstrated that actin glutathionylation attenuates actin polymerization and impairs phagocytosis. Since Gsr deficiency shifts the GSH/GSSG balance and Gsr-deficient phagocytes exhibit compromised actin polymerization (Figure 3) and impaired phagocytosis [5], we speculate that actin glutathionylation may be significantly enhanced in Gsr-deficient phagocytes.
The effects of Gsr deficiency on leukocyte mobilization
Leukocyte mobilization plays a critical role in organizing an effective innate immune response [22, 29]. Thioglycollate stimulation recruits neutrophils, monocytes, and eosinophils to the peritoneal cavity to clear the irritant substances through chemokine-mediated leukocyte trafficking [30–33]. We found that leukocyte populations, including neutrophils, inflammatory macrophages, and eosinophils, in thioglycollate-stimulated Gsr-deficient mice are substantially smaller than those in the wildtype mice (Figure 5). While the exact mechanisms underlying such dramatic differences are not entirely clear, we can offer some speculations. Leukocyte recruitment in response to infection is dependent on the dynamic interaction among the various phagocytic cell types [22]. The resident macrophages produce chemokines such as KC and MCP-1 to induce the mobilization of neutrophils and inflammatory monocytes/macrophages into the peritoneal cavity [30–34]. The concentrations of KC (critical for neutrophil trafficking) and MCP-1 (crucial for monocyte chemotaxis) in the blood and peritoneal lavage after thioglycollate stimulation were either similar in the two groups of mice or actually higher in Gsr-deficient mice (data not shown). Therefore, abnormalities in leukocyte mobilization are unlikely to be caused by defects in chemokine production.
We think that the failure of Gsr-deficient mice to mobilize leukocytes to the peritoneum after thioglycollate stimulation may be the consequence of defective redox regulation in leukocytes. It has been shown that neutrophils play a pivotal role in recruiting inflammatory monocytes by secreting chemotactic substances through degranulation [35, 35]. Since neutrophils in Gsr-deficient mice exhibit a dramatic abnormality, we speculate that defects in neutrophils are likely the major contributing factor to defects in leukocyte mobilization in Gsr-deficient mice after thioglycollate stimulation. The inflammatory stimuli might cause activation of blood neutrophils, leading to a production of small amounts of ROS. In the absence of Gsr, ROS produced may overwhelm the antioxidant mechanism and cause oxidative damage to neutrophils, compromising their trafficking to the peritoneal cavity and the resultant recruitment of inflammatory macrophages. On the other hand, Gsr deficiency may also alter the intrinsic chemotactic properties of leukocytes. Oxidation of proteins as the result of altered redox state in Gsr-deficient phagocytes may inhibit the chemotactic process through inactivating critical signaling events [36], thereby compromising phagocyte mobilization. Sakai et al. have shown that actin glutathionylation plays a critical role in actin polymerization and cytoskeletal remodeling and defects in the regulation of protein glutathionylation impairs chemotaxis [28]. As discussed earlier, Gsr deficiency may alter the glutathionylation of actin, compromising actin polymerization and cytoskeletal remodeling. Since chemotaxis depends on a very dynamic cytoskeletal remodeling [28], dysregulation of actin glutathionylation may explain the defects of Gsr-null mice in neutrophil trafficking and consequently monocyte mobilization to peritoneal cavity following thioglycollate injection. Alternatively, due to the absence of Gsr-mediated GSH regeneration ROS may directly inactivate chemokines or other chemoattractants in the proximal extracellular environment surrounding he activated neutrophils. This concept is supported by the highly abnormal albumin oxidation (Table 5) in the thioglycollate-stimulated Gsr-deficient mice. In normal conditions, serum albumin exists as a highly folded polypeptide containing a large number of disulfide bonds, with only one cysteine residue, Cys34, in the reduced sulfhydryl state [37, 38]. We assume that the observed abnormal oxidation of a large numbers of cystines to cysteic acids (sulfonic acids) probably causes the denaturation of this protein, eventually resulting in the precipitation in the abdominal cavity. This highly abnormal albumin oxidation in the Gsr-deficient mice after thioglycollate stimulation raises the possibility that other proteins in the ascitic fluid, for example chemokines, may also be subject to a highly oxidative environment. Since intramolecular protein disulfide bonds are critical for chemokine activity [39–42], oxidation of the critical cystine residues on chemokine molecules to cysteic acids may provide a plausible postulate for the defects in leukocyte trafficking in the Gsr-deficient mice. Finally, accumulation of oxidants as the result of Gsr deficiency may also render phagocytes in the peritoneal cavities of the Gsr-deficient mice more susceptible to apoptosis [43, 44]. Regardless of the mechanisms leading to the abnormalities in leukocyte mobilization, the impairments in leukocyte recruitment are likely to contribute to the defective bactericidal activity of Gsr-deficient mice.
Conclusion
We have identified a critical function of Gsr in host defense against bacterial infection. Our studies indicate that Gsr is essential for host survival when challenged with large doses of bacteria. In the absence of Gsr, mice fail to contain invading bacterial pathogens, likely due to impairments in leukocyte mobilization, phagocytosis, and bactericidal activities. However, Gsr deficiency does not affect the detection of TLR ligands by macrophages and the subsequent cellular responses. Our studies illustrate an intricate relationship between bactericidal action and phagocyte protection. Our findings suggest that regeneration of GSH mediated by Gsr plays a critical role for the protection of the bactericidal functions of phagocytes, particularly those that produce large quantities of oxidants during massive bacterial infection. Thus, it appears that the function of Gsr is to protect the cytosolic organelles of phagocytes and the proximal extracellular environment surrounding the phagocytes from the collateral damage of the oxidative burst that occurs in the phagolysosomes, thus maximizing phagocytic bactericidal activity.
Highlights
Gsr−/− mice are highly susceptible to challenge by group B Streptococcus.
Gsr−/− phagocytes exhibit defects in phagocytosis.
Gsr−/− mice do not exhibit decreased resistance to LPS challenge.
Gsr−/− mice show defects in phagocyte trafficking upon thioglycollate elicitation.
Gsr−/− mice show abnormal ascites albumin oxidation upon thioglycollate injection.
Acknowledgments
We thank Walter Pretsch for providing the original Gsr-deficient mice. We are grateful to Dr. James Cooper for veterinary advice and to Drs. Emilio Flano and Santiago Partida-Sanchez for advice in flow cytometry and critical reading of the manuscript. We thank Katherine Lintner and Haiwa Wu for technical assistance, Wei Wang and Han Yin for their assistance with statistical analyses. This work was supported by the National Institutes of Health grants AI68956 and AI57798 (both to Y.L.), HL75261 (to L.D.N.), and GM44263 (to C.V.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: Gsr, glutathione reductase; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; Mkp, MAPK phosphatase; GSH, glutathione; GSSG, glutathione disulfide; ROS, reactive oxygen species; F-actin, actin filaments; BMDM, bone marrow-derived macrophages; PMN, polymorphonuclear leukocytes; PEC, peritoneal elicited leukocyte cells; LPS, lipopolysaccharides; PMA, phorbol myristate acetate; GBS, group B Streptococcus; FSC, forward scatter; SSC, side scatter; LC-MS/MS, liquid chromatography coupled tandem mass spectrometry; MFI, mean fluorescence intensity; MOI, multiplicity of infection; cfu, colony forming unit.
References
- [1].Reed PW. Glutathione and the hexose monophosphate shunt in phagocytizing and hydrogen peroxide-treated rat leukocytes. J. Biol. Chem. 1969;244:2459–2464. [PubMed] [Google Scholar]
- [2].Strauss RR, Paul BB, Jacobs AA, Sbarra AJ. The role of the phagocyte in host-parasite interactions. XIX. Leukocytic glutathione reductase and its involvement in phagocytosis. Arch. Biochem. Biophys. 1969;135:265–271. doi: 10.1016/0003-9861(69)90539-6. [DOI] [PubMed] [Google Scholar]
- [3].Loos H, Roos D, Weening R, Houwerzijl J. Familial deficiency of glutathione reductase in human blood cells. Blood. 1976;48:53–62. [PubMed] [Google Scholar]
- [4].Roos D, Weening RS, Voetman AA, van Schaik ML, Bot AA, Meerhof LJ, Loos JA. Protection of phagocytic leukocytes by endogenous glutathione: studies in a family with glutathione reductase deficiency. Blood. 1979;53:851–866. [PubMed] [Google Scholar]
- [5].Yan J, Meng X, Wancket LM, Lintner K, Nelin LD, Chen B, Francis KP, Smith CV, Rogers LK, Liu Y. Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J. Immunol. 2012;188:2316–2327. doi: 10.4049/jimmunol.1102683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pretsch W. Glutathione reductase activity deficiency in homozygous Gr1a1Neu mice does not cause haemolytic anaemia. Genet. Res. 1999;73:1–5. doi: 10.1017/s0016672398003590. [DOI] [PubMed] [Google Scholar]
- [7].Rogers LK, Tamura T, Rogers BJ, Welty SE, Hansen TN, Smith CV. Analyses of glutathione reductase hypomorphic mice indicate a genetic knockout. Toxicol. Sci. 2004;82:367–373. doi: 10.1093/toxsci/kfh268. [DOI] [PubMed] [Google Scholar]
- [8].Wang X, Meng X, Kuhlman JR, Nelin LD, Nicol KK, English BK, Liu Y. Knockout of Mkp-1 enhances the host inflammatory responses to Gram-positive bacteria. J. Immunol. 2007;178:5312–5320. doi: 10.4049/jimmunol.178.8.5312. [DOI] [PubMed] [Google Scholar]
- [9].Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, Cato AC, Liu Y. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J. Immunol. 2009;183:7411–7419. doi: 10.4049/jimmunol.0804343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J. Immunol. 2002;169:6408–6416. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- [11].Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J. Biol. Chem. 2004;279:54023–54031. doi: 10.1074/jbc.M408444200. [DOI] [PubMed] [Google Scholar]
- [12].Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: Attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 2005;280:8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]
- [13].Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nusse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- [15].Hojsgaard S, Halekoh U. [Accessing date: 12-17-2012. 11-25-2012];doBy: doBy - Groupwise summary statistics, general linear contrasts, population means (least-squares-means), and other utilities. Link: http://cran.r-project.org/web/packages/doBy/ Publication date: 11-25-2012;
- [16].Klebanoff SJ, Clark RA. The neutrophil: Function and clinical disorders. North-Holland Publishing Company; Amsterdam: 1978. [Google Scholar]
- [17].Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- [18].Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr. Opin. Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- [19].Wang K, Feramisco JR, Ash JF. Fluorescent localization of contractile proteins in tissue culture cells. Methods Enzymol. 1982;85(Pt B):514–562. doi: 10.1016/0076-6879(82)85050-7. [DOI] [PubMed] [Google Scholar]
- [20].Downey GP, Chan CK, Lea P, Takai A, Grinstein S. Phorbol ester-induced actin assembly in neutrophils: role of protein kinase C. J. Cell Biol. 1992;116:695–706. doi: 10.1083/jcb.116.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang JQ, Biedermann B, Nitschke L, Crocker PR. The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur. J. Immunol. 2004;34:1175–1184. doi: 10.1002/eji.200324723. [DOI] [PubMed] [Google Scholar]
- [22].Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- [23].Oliver JM, Albertini DF, Berlin RD. Effects of glutathione-oxidizing agents on microtubule assembly and microtubule-dependent surface properties of human neutrophils. J. Cell Biol. 1976;71:921–932. doi: 10.1083/jcb.71.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Burchill BR, Oliver JM, Pearson CB, Leinbach ED, Berlin RD. Microtubule dynamics and glutathione metabolism in phagocytizing human polymorphonuclear leukocytes. J. Cell Biol. 1978;76:439–447. doi: 10.1083/jcb.76.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid. Redox. Signal. 2011;15:233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J. Biol. Chem. 2006;281:22983–22991. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- [27].Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- [28].Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia Y, Dickinson BC, Zhong J, Ye K, Chang CJ, Ho YS, Zhou J, Luo HR. Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity. 2012;37:1037–1049. doi: 10.1016/j.immuni.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Imhof BA, urrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004;4:432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- [30].Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- [31].Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, Bidzhekov K, Rottenberg ME, Weber C, Lindbom L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008;112:1461–1471. doi: 10.1182/blood-2008-02-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Marty C, Ye RD. Heterotrimeric G protein signaling outside the realm of seven transmembrane domain receptors. Mol. Pharmacol. 2010;78:12–18. doi: 10.1124/mol.110.063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hayashi T, Shimoyama Y, Kuwata K, Era S. Investigation of slow dynamics of the sulfhydryl in the solution and gel states of bovine serum albumin: A vector electron paramagnetic resonance study. Jpn. J. Physiol. 1999;49:27–33. doi: 10.2170/jjphysiol.49.27. [DOI] [PubMed] [Google Scholar]
- [38].Candiano G, Petretto A, Bruschi M, Santucci L, Dimuccio V, Prunotto M, Gusmano R, Urbani A, Ghiggeri GM. The oxido-redox potential of albumin methodological approach and relevance to human diseases. J. Proteomics. 2009;73:188–195. doi: 10.1016/j.jprot.2009.06.006. [DOI] [PubMed] [Google Scholar]
- [39].Schall TJ, Bacon KB. Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- [40].Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol Regul. Integr. Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- [41].Miller MD, Krangel MS. Biology and biochemistry of the chemokines: a family of chemotactic and inflammatory cytokines. Crit Rev. Immunol. 1992;12:17–46. [PubMed] [Google Scholar]
- [42].Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [43].Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- [44].Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat. Rev. Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]