

Abstract
In established obesity, inflammation and macrophage recruitment likely contribute to the development of insulin resistance. In the current study, we set out to explore whether adipose tissue infiltration by neutrophils that occurs early (3 days) after initiating a high-fat diet (HFD) could contribute to the early occurrence of hepatic insulin resistance and to determine the role of cytosolic phospholipase A2α (cPLA2α) in this process. The 3-day HFD caused a significant upregulation of cPLA2α in periepididymal fat and in the liver. A specific antisense oligonucleotide (AS) effectively prevented cPLA2α induction, neutrophil infiltration into adipose tissue (likely involving MIP-2), and protected against 3-day HFD–induced impairment in hepatic insulin signaling and glucose over-production from pyruvate. To sort out the role of adipose neutrophil infiltration independent of cPLA2α induction in the liver, mice were injected intraperitoneally with anti–intracellular adhesion molecule-1 (ICAM-1) antibodies. This effectively prevented neutrophil infiltration without affecting cPLA2α or MIP-2, but like AS, prevented impairment in hepatic insulin signaling, the enhanced pyruvate-to-glucose flux, and the impaired insulin-mediated suppression of hepatic glucose production (assessed by clamp), which were induced by the 3-day HFD. Adipose tissue secretion of tumor necrosis factor-α (TNF-α) was increased by the 3-day HFD, but not if mice were treated with AS or ICAM-1 antibodies. Moreover, systemic TNF-α neutralization prevented 3-day HFD–induced hepatic insulin resistance, suggesting its mediatory role. We propose that an acute, cPLA2α-dependent, neutrophil-dominated inflammatory response of adipose tissue contributes to hepatic insulin resistance and glucose overproduction in the early adaptation to high-fat feeding.
Established obesity, particularly if associated with insulin resistance–related morbidities, is characterized by systemic and adipose tissue inflammation (1–3). The complexity of the adipocytokines and inflammatory cell types involved in adipose inflammation is constantly increasing, and today, most myeloid cell types have been implicated in the process, including macrophages, B cells, various T-cell classes, and even eosinophils and mast cells (4–6). In contrast, much less is known about adipose tissue and liver adaptation to a short-term high-fat diet (HFD) before overt obesity is present. Metabolically, it appears that hepatic insulin resistance may be a front-line response to a short-term (3 days) HFD (7,8), representing “physiological adaptation” and/or an early maladaptive response on the causal pathway to obesity-induced whole-body insulin resistance. It currently remains unclear to what degree this early response to an HFD involves immune cells in general and, specifically, in adipose tissue.
In a previous study, we demonstrated that during the first week of initiating an HFD, adipose tissue is infiltrated by neutrophils (9). Adipose tissue protein levels of the neutrophil-specific myeloperoxidase (MPO) were increased, and correspondingly, histology detected an increased number of neutrophils within the parenchyma of adipose tissue (i.e., not restricted to blood vessels). This early appearance of neutrophils in adipose tissue was recently confirmed (10), suggesting that adipose tissue inflammation in obesity largely follows the classical inflammation paradigms of acute versus chronic inflammatory cell infiltrates, predominated first by neutrophils, then lymphocytes in the subacute period, and finally, by mononuclear macrophages when inflammation becomes chronic. Yet, the co-occurrence of increased adipose neutrophil infiltration (9,10) with the early hepatic insulin resistance (7,8) prompts the question of whether the former phenomenon is causative for the latter.
Cytosolic phospholipase A2α (cPLA2α) has received much attention as a key regulator of inflammation. It plays a major role in the stimulus-initiated production of eicosanoids (prostaglandins and the chemoattractant leukotrienes) and platelet activating factor (11). In a previous study, we demonstrated that cPLA2α is upregulated in vascular endothelial cells in adipose tissue of mice in response to the 3-day HFD and that it mediates the elevated expression of the endothelial intracellular adhesion molecule (ICAM-1) (12) that is used for adhesion by neutrophils and monocytes. In addition, cPLA2α has been demonstrated to regulate superoxide generation by NADPH oxidase activation (13), thus promoting phagocyte-induced oxidative stress. Intriguingly, in humans, even a single exposure to a high-fat meal induced NADPH activation and inflammatory cascades in circulating leukocytes (14–16). These findings suggest that cPLA2α could participate in priming/activation of circulating cells upstream in inflammatory cascades that ultimately lead to adipose tissue infiltration by neutrophils, way before obesity has developed. In the current study, we set out to reveal the role of cPLA2α and adipose tissue neutrophil infiltration in the acute adaptation to a 3-day HFD, with emphasis on whether these early inflammatory responses could underlie the development of hepatic insulin resistance.
RESEARCH DESIGN AND METHODS
Animals and diets.
The study was approved by Ben-Gurion University Institutional Animal Care and Use Committee (IL-35–2006) and conducted according to the Israeli Animal Welfare Act, following the guidelines of the Guide for Care and Use of Laboratory Animals (National Research Council, 1996). Male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) at 6 weeks of age were fed a low-fat diet (LFD; 6% calories from fat; Harlan Teklad 2018sc) or an HFD (60% calories from fat; Research Diets #12492), as previously performed (17).
Mice were killed by CO2 at the indicated times, and periepididymal fat was dissected out and immediately fixed in 4% formaldehyde or snap-frozen and stored in liquid nitrogen until further analyzed. A combination of two oligonucleotides antisenses (AS) (TCAAAGGTCTCATTCCACA, GCTGTCAGGGGTTGTAG) and their corresponding senses, with phosphorothioate modifications on the last three bases at both 5′ and 3′ ends, was used as described previously (18).
For insulin stimulation, insulin was administered intravenously 5 min before the mice were killed, and livers or epididymal fat pads were immediately obtained and flash-frozen until analysis. Insulin doses of 0.5 and 2 units/kg gave identical signaling responses. LEAF-purified anti-mouse Ly-6G antibodies, LEAF-purified anti-mouse tumor necrosis factor (TNF)-α antibodies (MP6-XT22), and LEAF-purified anti-mouse CD54 antibodies and their isotype control IgG antibodies (BioLegend, San Diego, CA) were used for neutrophil depletion, TNF-α neutralization, and ICAM-1 neutralization, respectively.
Immunoblot analysis.
Epididymal fat pads were lysed in ice-cold lysis buffer exactly as done earlier (9). Samples underwent 7.5% SDS-PAGE (for each 100-μg sample). The resolved proteins were electrophoretically transferred onto a nitrocellulose membrane, blocked in 5% milk in TBS-T (TBS containing 0.1% Tween-20), and incubated overnight at 4°C with goat anti-mouse MPO (1:100; Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-mouse β-actin (1:10,000), anti–phosphorylated (p)-Ser473 Akt (1:1000), anti-Akt (1:1000), anti–p-Ser9 glycogen synthase kinase (GSK; 1:1000), anti-GSK (1:1000), anti-cPLA2α (1:200; Cell Signaling, Beverly, MA), anti 5-lipoxygenase (5-LOX; 1:1000; Cayman Chemical, Ann Arbor, MI), or anti–cyclogenase-2 (COX-2; 1:1000; Abcam, Cambridge, U.K.). After being washed with TBS-T (four washes for 15 min each), the membranes were incubated with 1:10,000 dilutions of the respective horseradish peroxidase (HRP)–conjugated secondary antibodies for 1.5 h at room temperature. Proteins were quantified using video densitometry analysis (ImageGauge version 4.0, Fuji), as described previously (19,20).
Preparation of neutrophils.
Heart blood was withdrawn before mice were killed. Neutrophils, at 95% purity, were obtained by Ficoll-Hypaque centrifugation, dextran sedimentation, and hypotonic lysis of erythrocytes. Cells were counted, and their viability was determined by trypan blue exclusion.
Immunohistochemistry for neutrophil detection.
Epididymal fat tissues were embedded in paraffin and sectioned using a Leica microtome (Leica Biosystem, Vienna, Austria). Neutrophil staining by rat anti-mouse monoclonal anti–NIMP-R14 antibody to identify neutrophils (Novus Biological Inc.) was analyzed as done before (9,13), based on an earlier study (21). Nonspecific reactivity was blocked by incubating the slides with 20% normal rabbit serum for 20 min with avidin-biotin VECTA-STAIN Kit Elite PK 6105 (Vector Laboratories, Burlingame, CA). Sections were then incubated in 1:10 dilution of anti–NIMP-R14 antibodies in PBS for 1 h at room temperature, washed, and further incubated with 1:200 dilution of biotinylated anti-rat IgG, followed by avidin-biotin complex/HRP-DAB, which resulted in brown staining of neutrophils. For each treatment, a negative control was prepared without the primary antibody. The sections were counterstained with hematoxylin and analyzed in a blinded fashion using an Olympus BX-60 microscope.
Superoxide generation.
The production of the superoxide anion by intact neutrophils was measured as the superoxide dismutase inhibitable reduction of acetyl ferricytochrome c by the microtiter plate technique, as previously described (22).
Cytokines and eicosanoids release measurements.
Epididymal fat tissue from mice was removed, weighed, sliced, and incubated for 3 h in 1 mL medium containing 0.1% BSA. The collected media were measured using commercial ELISA kits for TNF-α (Biolegend, San Diego, CA), 15 μg tissue for each sample, MIP-2, KC, leukotriene B4 (LTB4), IL-6 (all from R&D Systems, Inc., Minneapolis, MN), 150 μg tissue for each sample. Prostaglandin E2 (PGE2) levels were determined according to a dextran-coated charcoal radioimmunoassay protocol, using PGE2 standard, anti-PGE2 anti-serum (Sigma Israel, Rehovot, Israel), and [3H]PGE2 (Amersham Biosciences Corp., Piscataway, NJ), exactly as previously described (23).
Pyruvate tolerance test.
Pyruvate tolerance test was performed by intraperitoneal injection of pyruvate (2 g/kg body weight) in saline after an overnight fast. Blood glucose concentrations were measured in tail blood before and after injection at the times indicated (24). All glucose measurements were made using a glucometer (Abbott, Alameda, CA).
Homeostasis model assessment of insulin resistance.
Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated by multiplying fasting plasma insulin concentration (mU/L) with fasting plasma glucose (mmol/L) divided by 22.5 (25–27). Plasma insulin levels were determined by ELISA (ALPCO Diagnostics, Salem, NH).
Glucose clamp studies.
Glucose clamp studies in freely moving mice were performed as described (28). Steady-state glucose infusion rate was calculated once glucose infusion reached a near-constant rate to maintain glucose at 5 mmol/L for 20 min. The glucose disposal rate was calculated by dividing the rate of [3-3H]glucose infusion by the plasma [3-3H]glucose-specific activity (29,30). Endogenous glucose production during the clamp was calculated by subtracting the glucose infusion rate from the glucose disposal rate (29,30). Insulin-stimulated glucose disposal rate was calculated by subtracting basal endogenous glucose production (equal to basal glucose disposal rate) from the glucose disposal rate during the clamp (31).
Statistical analysis.
Data are presented as the mean ± SEM. Statistical significance for comparison between two groups was determined using the Student paired two-tailed t test, and by ANOVA for more than two treatment groups, followed by Bonferroni correction or by Newman-Keuls post hoc test, using GraphPad Prism 5.03 software.
RESULTS
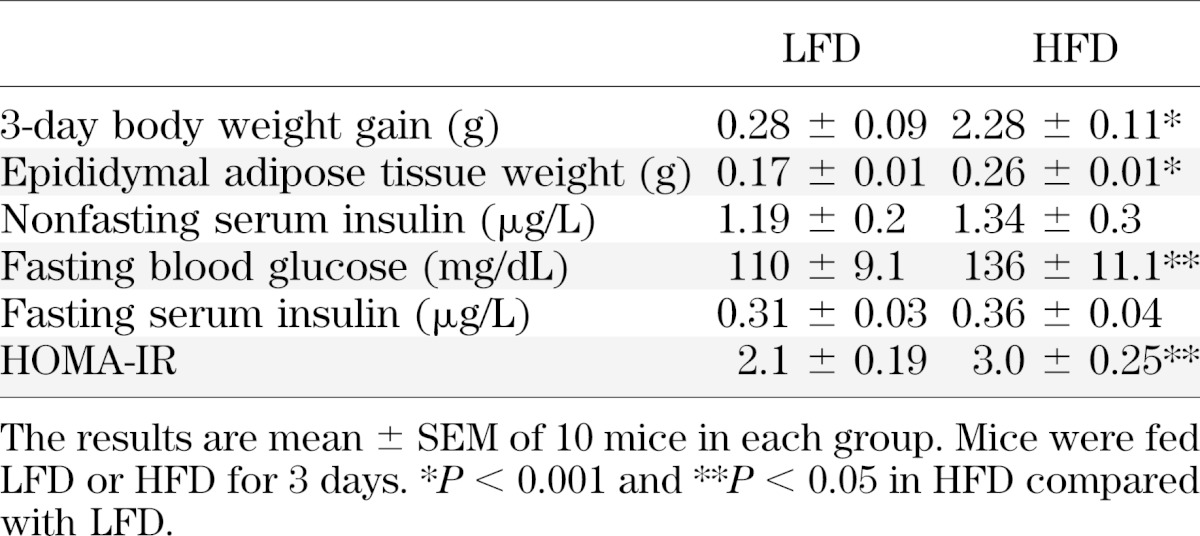
The time-course of development of insulin resistance in response to the HFD was determined in liver and in periepididymal fat pads by assessing insulin-stimulated phosphorylation of Akt. At different times after initiating the HFD, mice received an intravenous insulin injection 5 min before livers were harvested. Three days after initiating the HFD, hepatic insulin-stimulated Ser473-Akt phosphorylation was markedly attenuated (Fig. 1A). At this time point of marked diminution of insulin signaling in the liver, adipose tissue exhibited normal insulin-stimulated Ser473-Akt phosphorylation (Fig. 1B). Insulin resistance in this tissue required a longer duration of HFD to manifest (data not shown). Nevertheless, the 3-day HFD was already sufficient to induce a statistically significant increase in adipose tissue weight (Fig. 1C) and a notable increase in adipocyte cell size (Fig. 1D). As reported in a previous study (9), multiple neutrophils were visible between adipocytes, indicating infiltration of the tissue parenchyma. (Fig. 1D). Concomitantly, histologic analysis showed hepatic lipid content was notably mildly increased in mice fed the 3-day HFD (Fig. 1E), as were fasting glucose levels (Table 1). Calculated HOMA-IR values revealed systemic insulin resistance induced by the 3-day HFD (Table 1).
FIG. 1.

Short-term HFD induces liver, but not adipose tissue insulin resistance. A: Liver insulin resistance was determined by impaired insulin-stimulated p-Akt on serine 473 after intravenous injection of insulin. Representative immunoblot of p-Akt in liver lysates from mice at day 0 up to day 28 on HFD compared with liver lysates of a mouse that was not treated with insulin (–). After densitometry analysis, the ratio of the intensity of the p-Akt band to total Akt (t-Akt) was calculated and presented, as arbitrary units, by the mean ± SEM of 10 mice studied in three independent experiments. +, treated with insulin. B: Normal insulin signaling in epididymal fat of 3-day HFD mice. A representative immunoblot of p-Akt in epididymal fat tissue lysates from mice at day 0, and HFD and control mice on LFD at day 3. Densitometry analysis was performed as in A. Results are mean ± SEM of 15 mice studied in five independent experiments. C: Epididymal fat mass of 10 mice fed the HFD and 10 mice fed the LFD at day 3 of the diet show a significant difference. *P < 0.001 between the groups. Results are mean ± SEM of two different experiments. D: Histological sections of epididymal adipose tissues and immunolocalization of neutrophils (shown by arrows) using anti-NIMP-R14 antibodies and hematoxylin counterstaining (original magnification ×400). E: Histological sections of liver stained by hematoxylin and eosin (original magnification ×200). Shown are representative images of 10 mice in each group.
TABLE 1.
Metabolic changes induced by the 3-day HFD
We hypothesized that adipose tissue neutrophil infiltration induced by the 3-day HFD (9) could participate in the early induction of hepatic insulin resistance. To test this hypothesis, adipose neutrophil recruitment was first prevented using oligo-AS against cPLA2α. This approach was based on previous findings demonstrating 1) the effectiveness of AS administration in inhibiting neutrophil recruitment to the site of inflammation (13) and 2) that the 3-day HFD induces cPLA2α upregulation in adipose tissue (12). Mice received an intravenous tail vein injection of 1 mg/kg AS, 1 mg/kg corresponding (control) sense oligonucleotide, or saline 1 day before and then daily during the 3-day HFD. Weight gain was not affected by the AS or sense treatments (Fig. 2A), but AS treatment significantly prevented the elevated cPLA2α protein expression detected in lysates of adipose tissue of the 3-day HFD mice (Fig. 2B). The AS treatment also significantly prevented the recruitment of neutrophils to the adipose tissue of the HFD mice, as detected by adipose protein levels of MPO (Fig. 2C) and by quantitation of neutrophils by NIMP-R14 immunostaining (Fig. 2D).
FIG. 2.

Inhibition of neutrophil recruitment to epididymal adipose tissue by cPLA2α-specific AS oligonucleotide prevents hepatic insulin resistance. Mice were intravenously injected 1 day before the initiation of the HFD and every day during the diet with 2 mg/kg AS against cPLA2α. The HFD control mice were administrated with 100 μL saline or 2 mg/kg sense (SE) oligonucleotide, and because there was no difference between the two groups, they were combined (SE). Control LFD mice were fed a normal diet for 3 days. Half of the control LFD mice were administrated 100 μL saline, which did not cause any effect compared with the noninjected mice. The weight and the densitometry of the immunoblot of the individual mice in each group performed in three independent experiments are presented. A: *P < 0.001 between the two groups of mice on the HFD and the LFD mice at day 3 of the diet. B: A representative immunoblot of cPLA2α and the corresponding β-actin protein expression in epididymal adipose tissue lysates of the different groups of mice. The intensity of each cPLA2α band was divided by the intensity of the respective β-actin band after quantitation by densitometry and expressed as arbitrary units. *P < 0.001 for control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. C: A representative immunoblot and densitometry analyses of MPO (a specific neutrophil marker) and β-actin in epididymal fat from LFD mice, AS-treated HFD mice, and control mice (SE). The intensity of each MPO band was divided by the intensity of the respective β-actin band after quantitation by densitometry and expressed as arbitrary units. *P < 0.001 in control HFD mice (SE) compared with AS-treated HFD mice or LFD mice. D: The presence of stained neutrophils using mouse anti-neutrophils antibody (NIMP-R14) in sections of epididymal adipose tissues. Results are the mean ± SEM number per field, derived from 20 individual fields for each mouse from two independent experiments. *P < 0.001 in control HFD mice (SE) compared with AS-treated HFD mice or LFD mice. E and F: For each group of mice, liver lysate of a mouse that was not treated with insulin (–) and lysates from two different mice treated with insulin (+) are presented. Densitometry analysis for p-Akt and p-GSK were performed as in Fig. 1A. *P < 0.001, significant decrease of control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. G: Pyruvate tolerance test (PTT) in 3-day HFD mice after overnight fast. The results are the mean ± SEM of six mice in each group performed in two independent experiments. The inset describes the area under the curve (AUC). *P < 0.05, **P < 0.01 compared with SE-treated HFD mice (SE) by two-way ANOVA and Bonferroni post-test analysis. t, total.
To analyze whether AS protected against hepatic insulin resistance, insulin induction of liver p-Ser473–Akt or p-Ser9–GSK-3β was assessed (32,33). AS, but not sense treatment, prevented HFD-induced impairment of insulin-stimulated Akt and GSK-3β phosphorylation (Fig. 2E and F), without affecting liver histology (data not shown). AS treatment did not affect the normal insulin signaling in the adipose tissue (data not shown). At the whole-body metabolic level, the 3-day HFD was associated with an exaggerated glucose production during a pyruvate tolerance test compared with LFD mice, and this enhanced pyruvate-to-glucose flux was prevented by AS compared with the sense-treated mice (Fig. 2G).
Liver cPLA2α was also upregulated by the 3-day HFD, and as in adipose tissue, this increased expression was prevented by AS treatment (Fig. 3A). However, neutrophil infiltration to adipose tissue seemed to constitute a unique response of this tissue to the 3-day HFD, because no similar elevation occurred in the levels of the neutrophil-selective protein MPO in the liver (Fig. 3B). Thus, AS directed against cPLA2α decreased the elevated expression of this phospholipase in adipose tissue and in the liver and concomitantly prevented the tissue-selective neutrophil infiltration into adipose tissue that was induced by the 3-day HFD. These effects were associated with prevention of impaired hepatic insulin signaling and enhanced pyruvate-to-glucose flux that developed during early adaptation to the HFD.
FIG. 3.

The effect of cPLA2α AS treatment in livers of HFD mice. A: A representative immunoblot analysis of cPLA2α and the corresponding β-actin protein expression in liver lysates of the different groups of mice. Densitometry analysis for cPLA2α was performed as in Fig. 2B. *P < 0.001 significant increase of control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. B: A representative immunoblot of MPO in liver of LFD mice, control HFD mice (SE), and AS-treated HFD mice. Densitometry analysis for MPO was performed as in Fig. 2C. There are no differences among the various groups of mice. d, day.
To further support the role of neutrophil recruitment to adipose tissue in inducing liver insulin resistance and to differentiate its effect from the possible consequences of increased expression of cPLA2α in the liver, anti–ICAM-1 antibodies were used to prevent neutrophil recruitment. Mice were injected intraperitoneally with 100 μg anti-mouse ICAM-1 antibodies or isotype control IgG antibodies every other day, beginning 2 days before the HFD was initiated. Treatment with anti–ICAM-1 antibodies, but not with control IgG antibodies, reduced MPO expression in adipose tissue (Fig. 4A) and the number of neutrophils, as analyzed histologically by NIMP-R14, from 11.56 ± 1.2 to 3.49 ± 0.9 neutrophils per high-power field (P < 0.001), indicating the prevention of adipose neutrophil recruitment, without affecting body weight (Fig. 4B). Importantly, this approach to prevent HFD-induced adipose neutrophil infiltration did not attenuate the diet-induced elevation in cPLA2α protein expression in the liver (Fig. 4C). Inhibition of neutrophil infiltration to adipose tissue significantly improved hepatic insulin signaling, as detected by insulin-stimulated p-Akt and p-GSK (Fig. 4D and E), without affecting liver histology (data not shown) or adipose insulin signaling (not shown) and, consistently, alleviated the augmented gluconeogenic flux from pyruvate induced by the 3-day HFD (Fig. 4F).
FIG. 4.

Prevention of neutrophil recruitment to epididymal adipose tissue by anti–ICAM-1 antibodies prevented hepatic insulin resistance in response to 3-day HFD. Mice were intraperitoneally injected (100 μg/kg) with anti–ICAM-1 antibodies 2 days before the initiation of the HFD and every other day thereafter. The HFD control mice were administrated 100 μL saline or rat IgG2b, isotype control. The weight and the densitometry of the immunoblot of the individual mice in each group performed in three different experiments are presented. A: A representative immunoblot of MPO in epididymal fat. Densitometry analysis for MPO was performed as described in Fig. 2C. *P < 0.001, significant increase of control HFD mice (open circles) and anti-IgG–treated HFD mice (down gray triangles) compared with HFD mice treated with anti–ICAM-1 antibodies (up gray triangles) and LFD mice (filled circles). B: Effect of treatment on body weight. *P < 0.001 between the groups of mice fed the HFD and the group fed the LFD at day 3 of the diet. C: A representative immunoblot analysis of cPLA2α and the corresponding β-actin protein expression in adipose tissue lysates of the different groups of mice. Densitometry analysis for cPLA2α was performed as in Fig. 2B. There is a significant difference (*P < 0.001) among the groups of mice fed the HFD compared with the LFD at day 3 of the diet. D and E: Representative immunoblots and densitometry analyses of p-Akt (serine 473) and p-GSK (serine 9). For each group of mice, liver lysate of a mouse that was not treated with insulin (–) and lysates from two different mice treated with insulin (+) are presented. Densitometry analyses for p-Akt and p-GSK were performed as in Fig. 1A. *P < 0.001, significant decrease of control HFD mice and anti-IgG–treated HFD mice compared with HFD mice treated with anti–ICAM-1 antibodies and LFD mice. F: Pyruvate tolerance test (PTT) after an overnight fast. The inset describes the area under the curve (AUC). The results are the mean ± SEM of six mice in each group performed in two independent experiments. **P < 0.01, *P < 0.05 compared with ICAM-1 antibody-treated HFD mice by two-way ANOVA and Bonferroni post-test analysis. Hyperinsulinemic-euglycemic clamp studies with glucose infusion rate (GIR) (G), insulin-inhibited endogenous glucose production (EGP) (H), and insulin-stimulated glucose disposal rate (IS-GDR) (I) were conducted in chow-fed (open), IgG-treated HFD-fed (gray), and ICAM-treated HFD-fed (black) mice. (Further clamp results are in Supplementary Fig. 1.) Results are the means of four (chow-fed) or four (HFD-fed) animals. All error bars represent SEM. *P < 0.05, **P < 0.01 ANOVA, followed by Newman-Keuls post hoc test. J and K: Neutrophil depletion prevented insulin resistance in the 3-day HFD liver. Representative immunoblots and densitometry analyses are shown for p-Akt (serine 473) and p-GSK (serine 9). For each group of mice, liver lysate of a mouse that was not treated with insulin (–) and lysates from two different mice treated with insulin (+) are presented. Densitometry analyses for p-Akt and p-GSK were performed as in Fig. 1A. *P < 0.001, significant decrease of control HFD mice compared with neutrophil-depleted HFD mice and LFD mice. t, total.
To further confirm that prevention of adipose infiltration by neutrophils improved hepatic insulin sensitivity, hyperinsulinemic-euglycemic clamp studies were conducted. The glucose infusion rate was significantly decreased in HFD-fed mice (Fig. 4G and Supplementary Fig. 1). However, no difference was observed between IgG- and ICAM-treated mice (Fig. 4G and Supplementary Fig. 1). Insulin-mediated inhibition of endogenous (mainly reflecting hepatic) glucose production was significantly reduced in IgG-treated HFD-fed mice (Fig. 4H). In contrast, ICAM-treated HFD-fed mice showed a conserved response to insulin, suggesting improved hepatic insulin sensitivity in ICAM-treated compared with IgG-treated HFD-fed mice (Fig. 4H). Of note, the 3-day HFD did not significantly affect the insulin-stimulated glucose disposal rate (mainly reflecting skeletal muscle insulin sensitivity), although a slight decrease was apparent in the group treated with anti–ICAM-1 (Fig. 4I). High variability in the insulin-stimulated glucose disposal rate may have prevented a significant difference in the glucose infusion rate between the two HFD-fed groups (Fig. 4G). These hyperinsulinemic-euglycemic clamp results are consistent with a state of selective hepatic insulin resistance induced by the 3-day HFD, which can be alleviated by anti–ICAM-1 antibodies that prevent adipose tissue neutrophil infiltration.
To gain further support for the role of adipose neutrophil infiltration in the induction of hepatic insulin resistance, neutrophil infiltration was prevented by an alternative approach (i.e., by neutrophil depletion using Ly6G-specific monoclonal antibody), as described (34,35). Mice were injected intraperitoneally with 150 μg anti-mouse Ly-6G antibodies or isotype control IgG antibodies 1 day before the 3-day HFD was initiated. Neutrophil depletion was validated by fluorescence-activated cell sorter analysis using rat anti-mouse neutrophils antibodies (MCA771F, Serotec), as performed in a previous study (13). Neutrophil depletion significantly improved hepatic insulin responsiveness, as detected by insulin-stimulated p-Akt (Fig. 4J) and p-GSK (Fig. 4K). Collectively, inhibiting adipose neutrophil infiltration, independent of attenuating cPLA2α induction early after initiating the HFD, prevented the early diet-induced hepatic insulin resistance and glucose overproduction.
We next assessed possible mechanisms related to 1) neutrophil adaptation to HFD and recruitment to adipose tissue and 2) the induction of hepatic insulin resistance in this specific model of short-term hypernutrition. Neutrophil tissue infiltration is a process associated with cellular activation and with specific chemoattractants that direct these cells to the site of inflammation. Indeed, peripheral blood neutrophils from mice fed the 3-day HFD released significantly higher levels of superoxides (Fig. 5A), suggesting that neutrophil activation is induced by the diet. Consistent with our previous report (13), the AS treatment attenuated the release of superoxides.
FIG. 5.
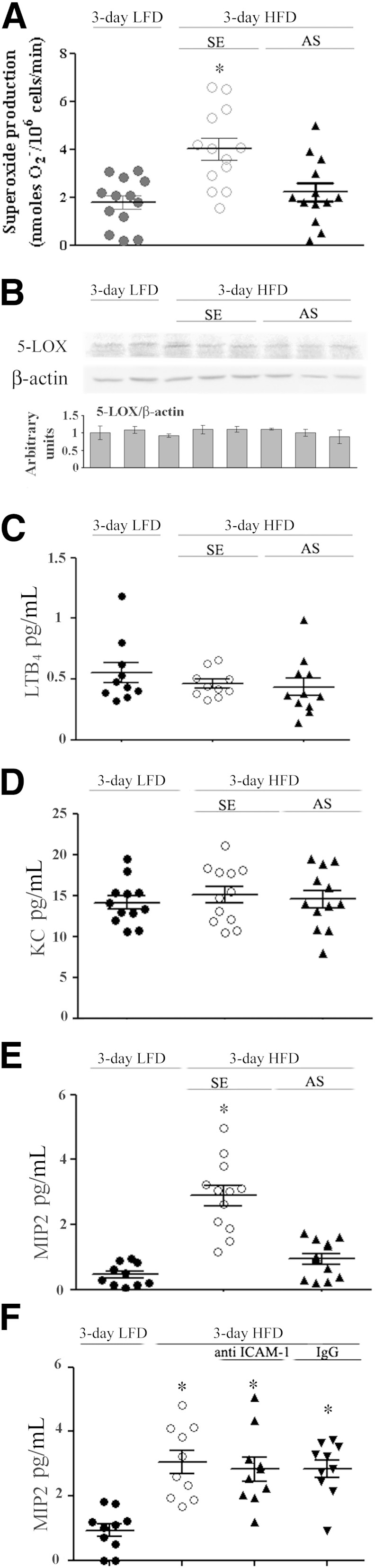
Peripheral blood neutrophil and adipose tissue release of MIP-2 after the 3-day HFD. A: Spontaneous superoxide production by isolated peripheral blood neutrophils from individual HFD mice and LFD mice at day 3 of the diet performed in three different experiments is presented. *P < 0.001, significant increase of control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. There are no differences between HFD mice with AS treatment and LFD mice. B: A representative immunoblot of 5-LOX in epididymal fat lysates of LFD mice, control HFD mice (SE), and AS-treated HFD mice. Densitometry analyses were performed as for cPLA2α in Fig. 2A. Results are mean ± SEM of 12 mice. Release of LTB4 (C) and KC (D) from adipose tissue explants of the different groups. There are no significant differences among the groups. E: MIP-2 release from adipose tissue explants was significantly increased (*P < 0.001) in control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. F: MIP-2 release from adipose tissue explants was significantly higher (*P < 0.001) in the groups of mice fed the HFD compared with mice fed the LFD at day 3 of the diet.
Several putative adipose tissue–derived neutrophil chemoattractants are known as potent activators. These factors include the cPLA2-dependent metabolite LTB4, previously shown by us to recruit neutrophils to the peritoneal cavity of a mouse model of sterile peritonitis (13), and the neutrophil-selective chemoattractant IL-8. The IL-8 ortholog in mice is keratinocyte chemoattractant (KC or CXCL1), and MIP-2 (CXCL2), both ligands for the chemokine receptor CXCR2 (36). Epididymal fat tissue from 3-day HFD–fed mice and control (3-day LFD) mice expressed similar levels of 5-LOX protein and released similar levels of LTB4, with no effect on these parameters by AS treatment (Fig. 5B and C). Similarly, secretion of the neutrophil chemoattractant KC was not affect by 3-day HFD or by the AS treatment (Fig. 5D). Yet, a significantly increased secretion of MIP-2 from adipose tissue of the 3-day HFD mice was detected (Fig. 5E and F). AS-treated 3-day HFD mice secreted significantly lower levels of MIP-2, similar to those of the control mice (Fig. 5E), suggesting that cPLA2α is involved in regulation of MIP-2 production. In contrast, treatment of the 3-day HFD mice with anti–ICAM-1 antibodies did not attenuate MIP-2 secretion (Fig. 5F), suggesting that cPLA2α upregulation, but not the consequential neutrophil infiltration, is involved in adipose tissue MIP-2 production. Jointly, these findings suggest a putative role for adipose MIP-2 as a chemoattractant involved in neutrophil recruitment to the epididymal fat tissue early after initiating the HFD.
To shed light on the mechanisms by which neutrophil infiltration to adipose tissue might induce hepatic insulin resistance, we determined if secretion/expression of PGE2, COX2, and the proinflammatory cytokines IL-6 and TNF-α are increased already 3 days after initiating the HFD. cPLA2α and COX2 are both robustly increased in various tissues during active inflammatory processes, and PGE2, the metabolite of arachidonic acid, has been recently implicated in insulin resistance (37). Yet, no increase in COX2 expression or PGE2 secretion from adipose tissue was observed in 3-day HFD mice (Fig. 6A) compared with control mice (Fig. 6B). There was also no increase in the secretion of IL-6 (Fig. 6C), but TNF-α (Fig. 6D and E) was significantly elevated in adipose tissue of the HFD-fed mice, an effect of the diet, prevented by AS or anti–ICAM-1 antibodies. To further establish the mediatory role of TNF-α in hepatic insulin resistance after the 3-day HFD, TNF-α was neutralized, using anti-mouse IgG1 monoclonal antibody, as in a previous study (38). Mice were injected intraperitoneally with 150 μg anti-mouse TNF-α antibodies or isotype control IgG1 antibodies 1 day before initiation of the HFD and at day 2 of the HFD. The elevated serum TNF-α levels in HFD mice (8.7 ± 0.9 compared with 5.5 ± 0.7 pg/mL in LFD mice) was significantly (P < 0.001) reduced by anti–TNF-α antibodies treatment (4.5 ± 0.6 pg/mL) but not by control IgG1 antibodies (8.1 ± 0.7 pg/mL). Serum TNF-α neutralization significantly improved hepatic insulin responsiveness, as detected by insulin-stimulated p-Akt and p-GSK (Fig. 6F and G).
FIG. 6.

TNF-α is the possible adipose mediator for the induction of hepatic insulin resistance in response to short-term HFD. A: A representative immunoblot of COX-2 in adipose lysates of LFD mice, control HFD sense (SE)-treated mice, and AS-treated HFD mice. Densitometry analyses were performed as for cPLA2α in Fig. 2A. Release of PGE2 (B) and IL-6 (C) from adipose tissue explants. No statistically significant differences among the groups were noted. D: TNF-α release from adipose tissue explants was significantly increased (*P < 0.001) in control HFD mice (SE) compared with HFD mice with AS treatment or LFD mice. E: TNF-α release from adipose tissue explants was significantly increased (*P < 0.001) in control HFD mice (open circles) and HFD mice with IgG treatment (down gray triangles) compared with HFD mice with anti–ICAM-1 treatment (up gray triangles) or LFD mice (filled circles). F and G: TNF-α neutralization prevented insulin resistance in the 3-day HFD liver. Representative immunoblots and densitometry analyses are shown for p-Akt (serine 473) and p-GSK (serine 9). For each group of mice, liver lysate of a mouse that was not treated with insulin (–) and lysates from two different mice treated with insulin (+) are presented. Densitometry analyses for p-Akt and p-GSK were performed as in Fig. 1A. *P < 0.001, significant decrease in control and IgG-treated HFD mice compared with TNF-α–depleted HFD mice or LFD mice. 3-day LFD (filled circles), 3-day HFD (open circles), 3-day HFD treated with anti-TNF antibodies (up gray triangles), and 3-day HFD treated with control IgG (down gray triangles). t, total.
DISCUSSION
The current study shows that hepatic insulin resistance induced by a short-term HFD reflects a dysregulated adipose-to-liver axis that is initiated by a cPLA2α-mediated, neutrophil-predominated, adipose tissue inflammation. Our studies reveal that:
1) Hepatic insulin resistance develops as early as 3 days after initiating an HFD, consistent with recently published studies (7,8).
2) Recruitment of neutrophils to adipose tissue is critical for the induction of hepatic insulin resistance in response to the 3-day HFD. Increased adipose tissue TNF-α secretion may constitute a mediator in this response, because increased TNFα secretion and hepatic insulin resistance were both prevented when neutrophil infiltration was attenuated, and neutralizing TNF-α significantly prevented hepatic insulin resistance.
3) The recruitment of neutrophils to adipose tissue likely involves cPLA2α-dependent MIP-2 secretion.
Consistent with our previous (9) and present studies, recruitment of neutrophils to adipose tissue after 3 days of the HFD was recently confirmed by others (10), and the role of adipose tissue neutrophils in insulin resistance was also demonstrated. Adipose tissue recruitment of neutrophils is also consistent with a study demonstrating increased leukocyte–endothelial cells–platelet interaction in the microcirculation of visceral adipose tissue in obesity (39). Moreover, in humans, pregnant obese women with preeclampsia exhibited extensive vascular inflammation characterized predominantly by neutrophils, demonstrable in adipose tissue blood vessels (40). The current study, however, offers several novel observations and mechanistic insights: the 3-day HFD constitutes a unique state predominantly affecting hepatic insulin responsiveness, independent of skeletal muscle, and/or adipose tissue insulin resistance that apparently takes longer to develop. Under such conditions of “isolated hepatic insulin resistance,” we were able to demonstrate that neutrophil recruitment, specifically to adipose tissue (and not to the liver), is necessary for hepatic insulin resistance to develop. Moreover, induction of adipose cPLA2α upregulation is deemed necessary for adipose tissue neutrophil infiltration and is upstream of an inflammatory pathway involving the upregulation of the neutrophil chemoattractant MIP-2, the upregulation of the endothelial adhesion molecule ICAM-1 (12), and the adipose tissue neutrophil–dependent increased production of TNF-α. Importantly, although cPLA2α is upregulated also in the liver, as was neutrophil elastase in a recent study,(10) our results assign a role specifically for adipose, and not hepatic, cPLA2α upregulation in the induction of hepatic insulin resistance and glucose overproduction: By antagonizing ICAM-1, 3-day HFD–induced increase in cPLA2α was still present, but adipose tissue neutrophil infiltration was prevented, and hepatic insulin resistance was still averted. We thus provide compelling evidence that adipose cPLA2α-mediated neutrophil infiltration, specifically to adipose tissue, contributes to hepatic insulin resistance in response to acute short-term high-fat feeding. It may be speculated that this early inflammatory response to a HFD, consistent with studies in human circulating leukocytes (14–16), represents, at least in the short-term, a physiological adaptation: isolated hepatic insulin resistance with preserved adipose tissue insulin sensitivity would assist in directing more storage of excess calories in adipose tissue and less in ectopic sites.
Recruitment of different immune cell types to adipose tissue and consequential changes in secretion of cytokines have been documented in recent years, mainly at much later time points of diet-induced obesity. These include CD11c+, M1-like macrophages (2,4,41,42), and B and CD8+ T cells (43–45). However, the recruitment of lymphocytes to adipose tissue was not directly linked with induction of insulin resistance (5). Although Kupffer cells were proposed to mediate the induction of hepatic insulin resistance by 3-day HFD (7), another study reported opposite results (8), suggesting that whereas macrophage-mediated tissue inflammation is a key component of chronic obesity-associated insulin resistance, it is not critical for the decrease in insulin sensitivity at the early adaptation to HFD feeding. We therefore propose that most likely, in the acute response to HFD, neutrophils may well constitute the major driver of adipose tissue inflammation and hepatic insulin resistance.
cPLA2α was recently suggested to be involved in the storage of lipids in the adipose tissue and liver during prolonged obesity (46) and in 3T3-L1 cells (47). In the current study, prevention of cPLA2α upregulation by AS treatment in the 3-day HFD mice did not interfere with weight gain. Thus, cPLA2α regulates the adaptation to the 3-day HFD independent of weight gain or adipocyte hypertrophy. The results of the current study suggest that MIP-2, a mouse ortholog of human IL-8, signals neutrophils recruitment downstream of cPLA2α (Fig. 6). This observation may also be highly relevant for humans in whom adipose tissue IL-8 levels were elevated (48–50). MIP-2 has been shown to be one of the major inducible chemokines with the ability to attract neutrophils to the site of inflammation (51). KC and MIP-2 have temporally distinct patterns of expression and cell type-specificity (52). MIP-2 was shown to be produced by resident macrophages and by neutrophils (36,51). Therefore, in response to the 3-day HFD, MIP-2 is likely produced by resident macrophages and not by neutrophils, because MIP-2 production was reduced only with the AS treatment and not with anti–ICAM-1 antibodies treatment, whereas in both treatments, the recruitment of neutrophils to the adipose fat was prevented. Because MIP-2 synthesis is Toll-like receptor (TLR)–dependent (36) and saturated free fatty acids act as a naturally occurring ligands of macrophage TLR4 (53), it is possible that elevation of free fatty acid in the intra-abdominal adipose tissue by the 3-day HFD triggers the production of cPLA2α-dependent MIP-2 in resident macrophages, leading to neutrophil recruitment. Thus, cPLA2α facilitates neutrophil recruitment by two important steps of the process: the secretion of the chemoattractant MIP-2 and the production of the adhesion molecule ICAM-1 (12).
We show here that circulating neutrophils are activated in mice fed the 3-day HFD, as shown by the release of superoxides (Fig. 5A). This is consistent with the immediate activation of circulating neutrophils, the release of superoxides, increased expression of NADPH oxidase, and the increased proinflammatory cytokines demonstrated in healthy human subjects exposed to a high-fat meal or a high-fat-carbohydrate meal or during a glucose challenge (14–16,54–57). The activated adipose neutrophil-produced factors that mediate hepatic insulin resistance have yet to be identified. Although neutrophil elastase is a likely candidate (10), it should be remembered that this was demonstrated at 12 weeks of the HFD, when its expression was also elevated in the liver. LTB4 or PGE2 were shown to impair insulin signaling (37,58), and adipose tissue LTB4 secretion was recently reported to contribute to elevation of adipose macrophages and T cells at 15 weeks of an HFD (59). Yet, they are unlikely to be involved in the response to the 3-day HFD. Elevated local and circulating levels of TNF-α and IL-6 are known to be associated with obesity in humans and rodent models (60–62), and both cytokines have been shown to induce hepatic insulin resistance (63,64). Although there was no increase in adipose secretion of IL-6 at the 3-day HFD, here we show that adipose tissue released significantly higher levels of TNF-α and that prevention of neutrophil infiltration to adipose tissue significantly attenuated this response. TNF-α blood levels were significantly elevated by the short-term HFD, consistent with a recent study (8), and neutralizing these elevated TNF-α levels prevented the development of hepatic insulin resistance by the 3-day HFD, further supporting its causal role in the process. In accordance with our results, a much higher increase in TNF-α than in IL-6 gene expression in adipose tissue at day 3 of the HFD was reported (8). We thus propose TNF-α as a potential mediator of primed adipose tissue neutrophils in causing hepatic insulin resistance induced by the 3-day HFD.
In conclusion, we show here that adipose cPLA2α has a profound role in initiating hepatic insulin resistance during the early response to hypernutrition. Adipose cPLA2α has a major role in promoting infiltration of activated neutrophils to adipose tissue in a short-term HFD, probably by increasing the production of the neutrophil chemoattractant MIP-2 and upregulating ICAM-1. The presence of neutrophils in the adipose tissue induces elevated secretion of TNF-α, which likely contributes to the early development of isolated hepatic insulin resistance.
Acknowledgments
N.H. was supported by a grant from Goldman Faculty Fund for Medical Research in Community Health of the Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel. A part of the work was supported by research grants from the Swiss National Science Foundation (No. 310030-141238, to D.K.).
No potential conflicts of interest relevant to this article were reported.
N.H. researched the data and reviewed the manuscript. O.B., V.E.-C., and Y.S. researched the data. S.W., F.I., and D.K. conducted and interpreted the clamp studies. A.R. contributed to study conceptualization, results interpretation, and participated in writing and reviewing the manuscript. R.L. designed the study and wrote the manuscript. R.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1300/-/DC1.
REFERENCES
- 1.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 2003;112:1785–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007;132:2169–2180 [DOI] [PubMed] [Google Scholar]
- 4.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1304–1310 [DOI] [PubMed] [Google Scholar]
- 6.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–920 [DOI] [PubMed] [Google Scholar]
- 7.Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol 2010;298:G107–G116 [DOI] [PubMed] [Google Scholar]
- 8.Lee YS, Li P, Huh JY, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011;60:2474–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 2008;49:1894–1903 [DOI] [PubMed] [Google Scholar]
- 10.Talukdar S, Oh da Y, Bandyopadhyay G, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 2012;18:1407–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JD, Lin LL, Kriz RW, et al. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991;65:1043–1051 [DOI] [PubMed] [Google Scholar]
- 12.Hadad N, Tuval L, Elgazar-Carmom V, Levy R, Levy R. Endothelial ICAM-1 protein induction is regulated by cytosolic phospholipase A2α via both NF-κB and CREB transcription factors. J Immunol 2011;186:1816–1827 [DOI] [PubMed] [Google Scholar]
- 13.Raichel L, Berger S, Hadad N, et al. Reduction of cPLA2alpha overexpression: an efficient anti-inflammatory therapy for collagen-induced arthritis. Eur J Immunol 2008;38:2905–2915 [DOI] [PubMed] [Google Scholar]
- 14.Mohanty P, Ghanim H, Hamouda W, Aljada A, Garg R, Dandona P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am J Clin Nutr 2002;75:767–772 [DOI] [PubMed] [Google Scholar]
- 15.Aljada A, Mohanty P, Ghanim H, et al. Increase in intranuclear nuclear factor kappaB and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: evidence for a proinflammatory effect. Am J Clin Nutr 2004;79:682–690 [DOI] [PubMed] [Google Scholar]
- 16.Ghanim H, Abuaysheh S, Sia CL, et al. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 2009;32:2281–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strissel KJ, Stancheva Z, Miyoshi H, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007;56:2910–2918 [DOI] [PubMed] [Google Scholar]
- 18.Liberty IF, Raichel L, Hazan-Eitan Z, et al. Cytosolic phospholipase A2 is responsible for prostaglandin E2 and leukotriene B4 formation in phagocyte-like PLB-985 cells: studies of differentiated cPLA2-deficient PLB-985 cells. J Leukoc Biol 2004;76:176–184 [DOI] [PubMed] [Google Scholar]
- 19.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998;47:1562–1569 [DOI] [PubMed] [Google Scholar]
- 20.Hazan-Eitan Z, Weinstein Y, Hadad N, Konforty A, Levy R. Induction of Fc gammaRIIA expression in myeloid PLB cells during differentiation depends on cytosolic phospholipase A2 activity and is regulated via activation of CREB by PGE2. Blood 2006;108:1758–1766 [DOI] [PubMed] [Google Scholar]
- 21.Lubberts E, Joosten LA, van Den Bersselaar L, et al. Adenoviral vector-mediated overexpression of IL-4 in the knee joint of mice with collagen-induced arthritis prevents cartilage destruction. J Immunol 1999;163:4546–4556 [PubMed] [Google Scholar]
- 22.Dana R, Leto TL, Malech HL, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J Biol Chem 1998;273:441–445 [DOI] [PubMed] [Google Scholar]
- 23.Szaingurten-Solodkin I, Hadad N, Levy R. Regulatory role of cytosolic phospholipase A2alpha in NADPH oxidase activity and in inducible nitric oxide synthase induction by aggregated Abeta1-42 in microglia. Glia 2009;57:1727–1740 [DOI] [PubMed] [Google Scholar]
- 24.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A 2007;104:12861–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 26.Mather K. Surrogate measures of insulin resistance: of rats, mice, and men. Am J Physiol Endocrinol Metab 2009;296:E398–E399 [DOI] [PubMed] [Google Scholar]
- 27.Saraswathi V, Morrow JD, Hasty AH. Dietary fish oil exerts hypolipidemic effects in lean and insulin sensitizing effects in obese LDLR-/- mice. J Nutr 2009;139:2380–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes 2011;60:56–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher SJ, Kahn CR. Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J Clin Invest 2003;111:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JK, Michael MD, Previs SF, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000;105:1791–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saberi M, Bjelica D, Schenk S, et al. Novel liver-specific TORC2 siRNA corrects hyperglycemia in rodent models of type 2 diabetes. Am J Physiol Endocrinol Metab 2009;297:E1137–E1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab 2002;13:444–451 [DOI] [PubMed] [Google Scholar]
- 33.Nov O, Kohl A, Lewis EC, et al. Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010;151:4247–4256 [DOI] [PubMed] [Google Scholar]
- 34.Czuprynski CJ, Brown JF, Maroushek N, Wagner RD, Steinberg H. Administration of anti-granulocyte mAb RB6-8C5 impairs the resistance of mice to Listeria monocytogenes infection. J Immunol 1994;152:1836–1846 [PubMed] [Google Scholar]
- 35.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 2008;83:64–70 [DOI] [PubMed] [Google Scholar]
- 36.De Filippo K, Henderson RB, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol 2008;180:4308–4315 [DOI] [PubMed] [Google Scholar]
- 37.Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen CH, Shih KC. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity (Silver Spring) 2009;17:1150–1157 [DOI] [PubMed] [Google Scholar]
- 38.Plessner HL, Lin PL, Kohno T, et al. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J Infect Dis 2007;195:1643–1650 [DOI] [PubMed] [Google Scholar]
- 39.Nishimura SM, Manabe I, Nagasaki M, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest 2008;118:710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah TJ, Walsh SW. Activation of NF-kappaB and expression of COX-2 in association with neutrophil infiltration in systemic vascular tissue of women with preeclampsia. Am J Obstet Gynecol 2007;196:48.e41–48.e48 [DOI] [PubMed] [Google Scholar]
- 41.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 2007;282:35279–35292 [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007;115:1029–1038 [DOI] [PubMed] [Google Scholar]
- 44.Duffaut C, Galitzky J, Lafontan M, Bouloumié A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 2009;384:482–485 [DOI] [PubMed] [Google Scholar]
- 45.Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol 2009;10:689–695 [DOI] [PubMed] [Google Scholar]
- 46.Ii H, Hatakeyama S, Tsutsumi K, Sato T, Akiba S. Group IVA phospholipase A2 is associated with the storage of lipids in adipose tissue and liver. Prostaglandins Other Lipid Mediat 2008;86:12–17 [DOI] [PubMed] [Google Scholar]
- 47.Maslowska M, Legakis H, Assadi F, Cianflone K. Targeting the signaling pathway of acylation stimulating protein. J Lipid Res 2006;47:643–652 [DOI] [PubMed] [Google Scholar]
- 48.Bruun JM, Pedersen SB, Richelsen B. Regulation of interleukin 8 production and gene expression in human adipose tissue in vitro. J Clin Endocrinol Metab 2001;86:1267–1273 [DOI] [PubMed] [Google Scholar]
- 49.Bruun JM, Lihn AS, Madan AK, et al. Higher production of IL-8 in visceral vs. subcutaneous adipose tissue. Implication of nonadipose cells in adipose tissue. Am J Physiol Endocrinol Metab 2004;286:E8–E13 [DOI] [PubMed] [Google Scholar]
- 50.Fain JN, Bahouth SW, Madan AK. Involvement of multiple signaling pathways in the post-bariatric induction of IL-6 and IL-8 mRNA and release in human visceral adipose tissue. Biochem Pharmacol 2005;69:1315–1324 [DOI] [PubMed] [Google Scholar]
- 51.Matzer SP, Baumann T, Lukacs NW, Röllinghoff M, Beuscher HU. Constitutive expression of macrophage-inflammatory protein 2 (MIP-2) mRNA in bone marrow gives rise to peripheral neutrophils with preformed MIP-2 protein. J Immunol 2001;167:4635–4643 [DOI] [PubMed] [Google Scholar]
- 52.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol 2004;75:641–648 [DOI] [PubMed] [Google Scholar]
- 53.de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett 2008;582:97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bae JH, Bassenge E, Kim KB, et al. Postprandial hypertriglyceridemia impairs endothelial function by enhanced oxidant stress. Atherosclerosis 2001;155:517–523 [DOI] [PubMed] [Google Scholar]
- 55.Alipour A, van Oostrom AJ, Izraeljan A, et al. Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler Thromb Vasc Biol 2008;28:792–797 [DOI] [PubMed] [Google Scholar]
- 56.Deopurkar R, Ghanim H, Friedman J, et al. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010;33:991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab 2000;85:2970–2973 [DOI] [PubMed] [Google Scholar]
- 58.Spite M, Hellmann J, Tang Y, et al. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol 2011;187:1942–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mothe-Satney I, Filloux C, Amghar H, et al. Adipocytes secrete leukotrienes: contribution to obesity-associated inflammation and insulin resistance in mice. Diabetes 2012;61:2311–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001;280:E745–E751 [DOI] [PubMed] [Google Scholar]
- 61.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997;389:610–614 [DOI] [PubMed] [Google Scholar]
- 62.Ventre J, Doebber T, Wu M, et al. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes 1997;46:1526–1531 [DOI] [PubMed] [Google Scholar]
- 63.Klover PJ, Clementi AH, Mooney RA. Interleukin-6 depletion selectively improves hepatic insulin action in obesity. Endocrinology 2005;146:3417–3427 [DOI] [PubMed] [Google Scholar]
- 64.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91 [DOI] [PubMed] [Google Scholar]