

Abstract
The endogenous hormone relaxin increases vascular reactivity and angiogenesis. We demonstrate that acute relaxin infusion in lean C57BL/6J mice enhances skeletal muscle perfusion and augments muscle glucose uptake during a hyperinsulinemic-euglycemic clamp. However, an acute effect was absent in mice fed a high-fat (HF) diet for 13 weeks. In contrast, mice fed an HF diet for 13 weeks and continuously treated with relaxin for the final 3 weeks of the diet exhibited decreased fasting blood glucose. Insulin-stimulated whole-body glucose disappearance and percent suppression of hepatic glucose production are corrected by chronic relaxin. The increase in peripheral glucose utilization is a result of augmented in vivo skeletal muscle glucose uptake. Relaxin intervention improves endothelial-dependent vascular reactivity and induces a two-fold proliferation in skeletal muscle capillarity. The metabolic effects of the treatment are not attributed to changes in myocellular insulin signaling. Relaxin intervention reverses the accumulation of collagen III in the liver and collagen III and collagen IV in the heart; this is induced by HF feeding. These studies show the potential of relaxin in the treatment of diet-induced insulin resistance and vascular dysfunction. Relaxin provides a novel therapeutic approach targeting the extramyocellular barriers to insulin action, which are critical to the pathogenesis of insulin resistance.
Insulin resistance precedes the development of type 2 diabetes, and it is associated with cardiovascular disease. Recent evidence suggests that muscle insulin resistance coincides with extramyocellular adaptations, including extracellular matrix (ECM) remodeling and capillary rarefaction (1–4). Kang et al. (1) established that the accumulation of ECM proteins and lower capillary number correspond to muscle insulin resistance in mice fed a high-fat (HF) diet (1,2). The vascular and ECM abnormalities associated with obesity provide novel therapeutic targets to simultaneously treat insulin resistance and its coaggregates.
The hemodynamic action of insulin is fundamental to skeletal muscle metabolism during insulin stimulation (5,6). Hyperinsulinemia increases skeletal muscle microvascular blood volume, thus enhancing nutrient and hormone flux to this tissue (7–9). Previous studies estimated that 40% of insulin-stimulated muscle glucose uptake (MGU) was a result of increased muscle perfusion and that this hemodynamic response is diminished in insulin-resistant individuals (10,11). Studies applying metabolic control analysis demonstrated that the vascular delivery of glucose to the muscle is a major limitation to insulin-stimulated MGU (12,13). Mice fed an HF diet have attenuated vascular insulin signaling that precedes the impairment in insulin-responsive tissues such as skeletal muscle, liver, and adipose tissue (14). Kubota et al. showed that impaired endothelial insulin signaling in mice lacking endothelial insulin receptor substrate 2 prevented endothelial nitric oxide synthase activation and resulted in decreased muscle perfusion, substrate delivery, and MGU during steady-state hyperinsulinemia (15).
Relaxin (Rlx), a 6-kDa protein hormone, has potent vasodilatory and antifibrotic actions (16–20). Rlx augments circulating vascular endothelial growth factor (VEGF)-A concentrations, which have been shown to be essential to the vasodilatory response (21). Furthermore, Rlx-induced VEGF expression stimulates the integration of bone marrow–derived endothelial cells into sites of vasculogenesis to enhance vessel growth (22,23). In experimental models of type 1 diabetes and hypertension, Rlx attenuated the fibrotic response in cardiac and renal tissues, respectively (18,19). An important mechanism for the antifibrotic actions of Rlx is blunted transforming growth factor-β signaling, which can reduce collagen deposition (24–27). Rlx regulation of matrix metalloproteinase (MMP), such as MMP-2 and MMP-9, activities has been shown to be important to the ECM remodeling mechanism of Rlx and the acute vasodilatory response (24,27,28). The pleiotropic actions of Rlx provide an intriguing therapeutic candidate for insulin resistance.
The goal of the current investigation was to determine the viability of Rlx intervention in rescuing muscle insulin resistance. The hypotheses tested herein are that acute Rlx infusion will enhance skeletal muscle perfusion and insulin action in lean mice but not in mice fed an HF diet, and chronic Rlx intervention in mice fed an HF diet will reverse muscle insulin resistance, enhance endothelial reactivity, and augment skeletal muscle capillarity.
RESEARCH DESIGN AND METHODS
Mouse models.
The Vanderbilt University Animal Care and Use Committee approved all animal protocols. Mice were housed with a 12:12-h light:dark cycle in a temperature-controlled and humidity-controlled environment. Male 6-week-old C57BL/6J mice (The Jackson Laboratory) were placed on either chow diet (5001 Laboratory Rodent Diet) or HF diet (F3282 Bioserv) containing 5.5% or 60% calories as fat, respectively. In protocol 1, mice fed chow diet or HF diet for 13 weeks had a primed (10 µg) continuous infusion (15 µg/h) with recombinant H2 Rlx (Corthera) or vehicle (20 mmol/L sodium acetate; pH 5.0) for a total of 6.5 h. The infusion of Rlx or vehicle began 1 h after the onset of the fast and lasted for the duration of the hyperinsulinemic-euglycemic clamp. In protocol 2, mice were fed an HF diet for 13 weeks. At week 10 of the HF diet, osmotic minipumps (Alzet models 2001 and 2002; replaced after 2 weeks) were implanted subcutaneously to deliver Rlx at a rate of 1 mg · kg−1 · day−1 or vehicle.
Assessment of body composition and cardiac function.
Body composition was determined in protocol 2 using an mq10 nuclear magnetic resonance analyzer (Bruker Optics). Echocardiogram (Sonos 5500 system; Agilent) and blood pressure were measured with a blood pressure transducer via a carotid arterial catheter with the assistance of the Cardiovascular Pathophysiology and Complications Core of the Vanderbilt Mouse Metabolic Phenotyping Center.
Hyperinsulinemic-euglycemic clamps (insulin clamp).
One week before insulin clamps, mice had carotid artery and jugular vein catheters surgically implanted for sampling and infusions, respectively (29). In the chronic Rlx and vehicle protocols, osmotic minipumps were replaced at the time of catheter implantation to avoid volume depletion. Mice were fasted for 5 h before the start of the insulin clamp. The insulin clamps were performed as described previously (1,29–31). The method used by our laboratory does not require that mice are handled (29,32). Erythrocytes were replaced to prevent a decline in hematocrit that occurs with repeated blood sampling. Basal arterial glucose–specific activity was measured at −15 min and −5 min, and arterial insulin was measured at −5 min. The clamp was initiated at 0 min with a continuous insulin infusion (4 mU · kg−1 · min−1) that was maintained for 155 min. Arterial glucose was determined at 10-min intervals to provide feedback to adjust the rate of exogenous glucose (glucose infusion rate [GIR]) as needed to clamp glucose. [3-3H]glucose kinetics were determined at 10-min intervals between 80 and 120 min because insulin action is in a steady state by this interval. Plasma insulin during the clamp was measured at 100 min and 120 min. A 13-µCi intravenous bolus of 2[14C]deoxyglucose (2[14C]2DG) was administered at 120 min. 2[14C]2DG was used to determine the glucose metabolic index (Rg), an indication of tissue-specific glucose uptake. Blood samples were collected at 2, 15, 25, and 35 min after the injection to measure the disappearance of 2[14C]DG from the plasma. After the last sample of the insulin clamp, 50 µL yellow DYE-TRAK 15-μm microspheres were injected into the carotid artery in some studies to determine microsphere content in skeletal muscle and the left and right kidneys.
Processing of plasma and tissue samples.
Arterial insulin was determined by ELISA (Alpco). Radioactivity of [3-3H]glucose, 2[14C]DG, and 2[14C]DG-6-phosphate were assessed by liquid scintillation counting (31). Whole-body glucose appearance (Ra) and glucose disappearance (Rd) were calculated using non–steady-state equations (33). Endogenous glucose production (endoRa) was calculated by subtraction of the GIR from total glucose Ra. Muscle Rg was calculated as previously described (31). Free fatty acids were assessed spectrometrically by an enzymatic calorimetric assay (NEFA C Kit; Wako Chemicals). Basal free fatty acids were an average of samples taken at −15 min and −5 min, and the free fatty acid levels during the insulin clamp were the average at 80 min and 120 min. After overnight tissue digestion, microspheres were resuspended and the fluorescent dye was eluted as previously described (34). Absorbance of the eluent was read at 450 nm. Microspheres with absorbance at 670 nm were added to monitor the assay recovery. The plasma and tissue VEGF were assayed by the manufacturer’s specifications (VEGF ELISA Kit, Mouse No. QIA52; Calbiochem) to detect VEGF120 and VEGF164 isoforms.
Ex vivo muscle glucose uptake.
Isolated muscle (soleus and extensor digitorum longus) 2-deoxyglucose uptake was measured as previously described (35). After a 15-min basal incubation period, muscles were transferred to fresh media and incubated for 30 min in the absence or presence of insulin (10 mU/mL). After stimulation, 2-deoxy-d-glucose uptake was measured for 10 min in fresh media in the absence or presence of insulin by adding cold 2-deoxy-d-glucose (1 mmol/L) and tracers 2-[1,2-3H]deoxy-d-glucose (0.25 μCi/mL) and D-[1-14C]mannitol (0.16 Ci/mL). Muscles were then lysed, and radioactivity in the supernatant was measured.
Aortic ring reactivity.
After the insulin clamps in protocols 1 and 2, mouse aortas were excised and placed directly in HEPES buffer (140 mmol/L NaCl, 4.7 mmol/L KCl, 1.0 mmol/L MgSO4, 1.0 mmol/L NaH2PO4, 1.5 mmol/L CaCl2, 10 mmol/L glucose, and 10 mmol/L HEPES; pH 7.4) and immediately suspended in a muscle bath apparatus. Subcutaneous fat and adventitial tissues were removed, after which the aorta was sectioned to create rings. Smooth muscle relaxation and contraction were determined with sodium nitroprusside and phenylephrine after maximal contraction with KCl. Endothelial-dependent relaxation was determined with carbachol as described (36,37).
Gelatin zymography.
The activation of MMP-2 and of MMP-9 was determined using the gelatin zymograph technique (38). Briefly, gastrocnemius was homogenized in buffer containing 0.5% Triton X-100, 100 mmol/L EDTA, and 10 µL/mL protease inhibitor (pH 7.5). Homogenates were centrifuged at 13,000 rpm for 20 min. Supernatants were incubated at 4°C for 2 h with 40 µL gelatin-Sepharose (Pharmacia). The gelatin-Sepharose beads were resuspended in nonreducing sodium dodecyl sulfate sample buffer and loaded on 10% zymogram gels (Invitrogen). The gel was developed according to the manufacturer’s instructions.
Immunohistochemical analyses of collagen and capillary density.
Collagen III, collagen IV, and CD31 were assessed by immunohistochemistry in paraffin-embedded tissue sections as described previously (1). Five-micron sections were placed on charged slides, and paraffin was removed. The sections were then incubated with the following primary antibodies for 60 min: anticollagen III (CosmoBio), anticollagen IV (Abcam), or anti-CD31 (BD Biosciences). Staining was quantified with ImageJ software.
Immunoblotting.
Cardiac, liver, and gastrocnemius samples were homogenized in buffer containing 50 mmol/L Tris-HCl (pH 7.5), 1 mmol/L EDTA, 1 mmol/L EGTA, 10% glycerol, 1% Triton X-100, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 5 µg/mL protease inhibitor, 50 mmol/L NaF, and 5 mmol/L sodium pyrophosphate. Samples were then centrifuged at 13,000 rpm for 20 min at 4°C; 30 µg supernatant was loaded onto 4–12% SDS-PAGE gel. The gel was transferred to polyvinylidene fluoride membrane and incubated overnight with phosphorylated (Ser 473) and total Akt antibodies (Cell Signal) or glyceraldehyde-3-phosphate dehydrogenase (Abcam) in liver and gastrocnemius samples. Cardiac samples were incubated with phosphorylated SMAD2 (Ser 465/467) and total SMAD2 (cell signal), and glyceraldehyde-3-phosphate dehydrogenase was used as the loading control.
Statistical analysis.
Student t test or two-way ANOVA, followed by Tukey post hoc tests when appropriate, was used to determine statistical significance. Data are expressed as mean ± SE. The significance level was P ≤ 0.05.
RESULTS
Protocol 1: Acute Rlx infusion enhances insulin-stimulated MGU in chow-fed but not in HF-fed mice
Glucoregulatory.
To test the hypothesis that a 6.5-h Rlx infusion enhances insulin-stimulated MGU in chow-fed mice, insulin clamps were performed in 5-h fasted mice. There was no difference in body weight between treatment groups (Table 1). The acute Rlx group had greater fasting arterial insulin; however, no change in fasting arterial glucose was present (Table 1). Insulin clamps were performed to determine in vivo muscle insulin action in conscious unrestrained mice (29). During the steady-state period of the insulin clamp (80–120 min), Rlx-treated mice required a higher GIR to maintain euglycemia compared with vehicle-infused mice (P ≤ 0.05; Fig. 1A). The increase in GIR was independent of changes in the suppression of endoRa (Fig. 1B). The enhanced GIR in the Rlx group was attributable to an augmented whole-body glucose disappearance (Rd) during the clamp steady-state period (P ≤ 0.05; Fig. 1B). The Rg in the Rlx infusion group was greater in the gastrocnemius, superior vastus lateralis (SVL), and heart (P ≤ 0.05; Fig. 1C). The in vivo muscle glucose flux data coincided with augmentation of the ratio of phosphorylated Akt to total Akt in the gastrocnemius (P ≤ 0.05; Supplementary Fig. 1B).
TABLE 1.
Insulin clamp characteristics in protocol 1
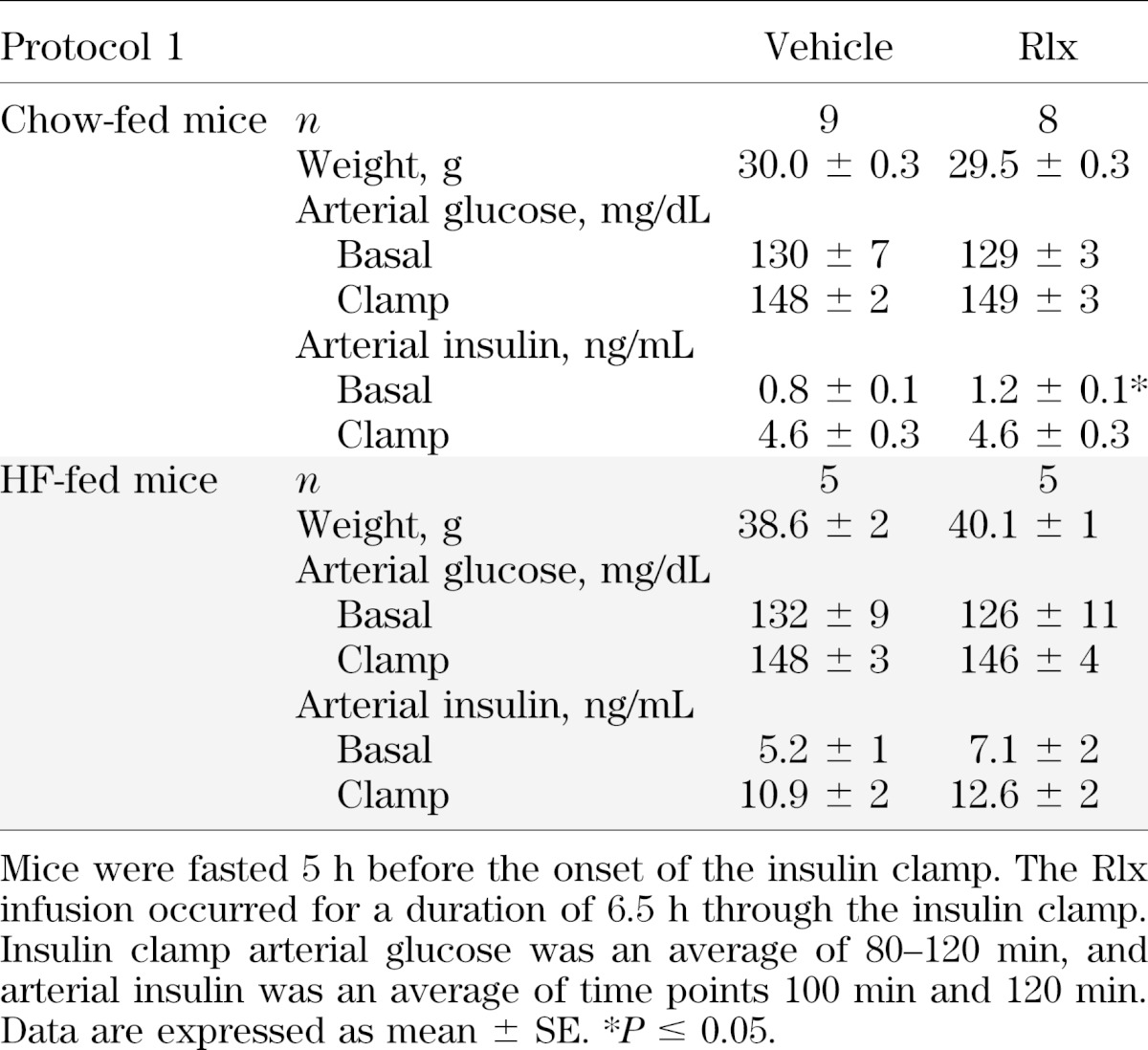
FIG. 1.

Protocol 1: chow-fed mice. Hyperinsulinemic-euglycemic clamps, glucose flux analysis, and vascular reactivity after a 6.5-h Rlx or vehicle infusion were performed in lean mice. A: Glucose infusion rate (top) and arterial glucose (bottom) during the insulin clamp. Mice were fasted 5 h before the onset of the clamp. Blood glucose was maintained at ∼150 mg/dL during steady state (80–120 min), and the time course is displayed to demonstrate quality of the clamp; 50% glucose was infused to maintain euglycemia. B: EndoRa and Rd during the insulin clamp. Basal values are an average of −15 min and −5 min, and the insulin clamp values are an average of 80–120 min (steady state). C: Rg after the insulin clamp in the gastrocnemius, SVL, and heart. D: Endothelial and smooth muscle–dependent relaxation in aortas excised from mice after the insulin clamp in response to carbachol (Cch) and sodium nitroprusside (SNP), respectively. Data are expressed as mean ± SE. n = 8–9. *P ≤ 0.05.
The acute Rlx infusion of protocol 1 was repeated in HF-fed mice. In contrast to the chow-fed mice, there was no difference in insulin action and glucose fluxes between the Rlx and vehicle-infused groups. There was no difference in body weight or arterial glucose and insulin levels in the fasted and clamp states (Table 1). The GIR during the insulin clamps were equal (Fig. 2A). The endoRa and Rd were comparable between groups in the basal and insulin clamp states (Fig. 2B). The Rg data corresponded to the flux analysis with no difference in the gastrocnemius, SVL, or heart (Fig. 2C). HF-fed mice are resistant to the acute effects of Rlx.
FIG. 2.

Protocol 1: HF-fed mice. Hyperinuslinemic-euglycemic clamps, glucose flux analysis, and vascular reactivity after a 6.5-h Rlx or vehicle infusion were performed in HF-fed mice. A: Glucose infusion rate (top) and arterial glucose (bottom) during the insulin clamp. Mice were fasted 5 h before the onset of the clamp. Blood glucose was maintained at ∼150 mg/dL during steady state (80–120 min), and the time course is displayed to demonstrate quality of the clamp; 50% glucose was infused to maintain euglycemia. B: EndoRa and Rd during the insulin clamp. Basal values are an average of −15 min and −5 min, and the insulin clamp values are an average of 80–120 min (steady state). C: Rg after the insulin clamp in the gastrocnemius, SVL, and heart. D: Endothelial and smooth muscle–dependent relaxation in aortas excised from mice after the insulin clamp in response to carbachol (Cch) and sodium nitroprusside (SNP), respectively. Data are expressed as mean ± SE. n = 5.
Ex vivo glucose uptake was determined in isolated muscle from chow-fed mice that underwent 6.5-h Rlx infusion identical to the insulin clamp cohorts. Isolated glucose uptake was performed only in the chow-fed mice because of the enhanced insulin clamp Rg. Glucose uptake in isolated muscle removes the vascular delivery barrier of MGU during hyperinsulinemia. There was no difference in basal or insulin-stimulated glucose uptake in isolated soleus or extensor digitorum longus muscles between groups (Supplementary Fig. 1A).
Vascular.
The 6.5-h Rlx infusion enhanced the hemodynamic response to insulin in chow-fed mice. Endothelial-dependent aortic ring relaxation in the Rlx group was amplified, with no difference in smooth muscle–dependent relaxation (P ≤ 0.05; Fig. 1D). The acute enhancement in endothelial-specific vascular reactivity was absent in the HF-fed cohort (Fig. 2D). Muscle blood flow was increased in the Rlx group as indicated by a 2.5-fold increase in gastrocnemius microsphere content (P ≤ 0.05; Supplementary Fig. 1C). The 6.5-h Rlx infusion resulted in a 2.5-fold increase in MMP-9 and pro-MMP-2 activities (P ≤ 0.05; Supplementary Fig. 1D).
Protocol 2: Rlx intervention reverses diet-induced insulin resistance and the associated extramyocellular adaptations
Glucoregulatory.
To test the hypothesis that intervention with Rlx can reverse muscle insulin resistance and the extramyocellular adaptations to a HF diet, mice were treated with Rlx or vehicle for the final 3 weeks of a 13-week HF diet. Rlx intervention did not alter body weight or composition (Table 2). Furthermore, the 3-week Rlx treatment did not result in differences in mean arterial blood pressure or cardiac morphology (Table 2). Insulin clamps were performed after the 3-week intervention. Fasting (5 h) arterial glucose was lower in the Rlx-treated mice (P ≤ 0.05; Table 2). During the steady-state period of the insulin clamp, the Rlx group required a higher GIR to maintain euglycemia at ∼150 mg/dL (P ≤ 0.05; Fig. 3A). Rlx did not affect fasting glucose fluxes. Insulin suppressed endoRa during the steady-state period to a greater extent with Rlx intervention (P ≤ 0.05; Fig. 3B), suggesting improved hepatic insulin action. The greater GIR in the Rlx-treated group corresponded to enhanced whole-body Rd during the steady-state period of the insulin clamp (P ≤ 0.05; Fig. 3B). Rg was elevated in the gastrocnemius and SVL in the Rlx group compared with vehicle during hyperinsulinemia (P ≤ 0.05), and cardiac muscle Rg tended to be higher (P = 0.1; Fig. 3C). Skeletal muscle and ratio of hepatic phosphorylated Akt to total Akt tended to be higher after the Rlx intervention, but differences were not significant (Fig. 4A).
TABLE 2.
Treatment group characteristics in protocol 2
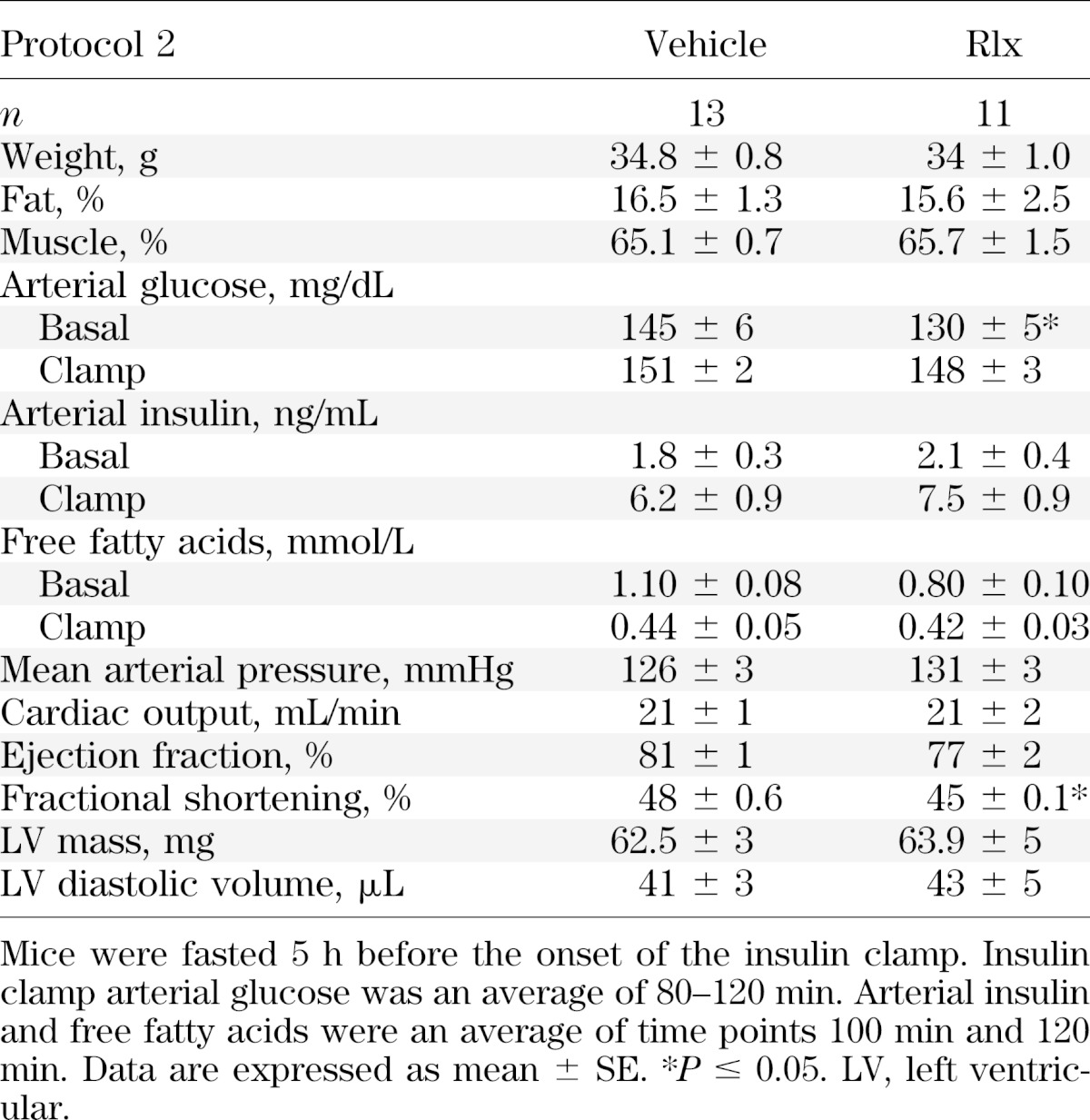
FIG. 3.

Protocol 2: hyperinsulinemic-euglycemic clamps, glucose flux analysis, and isolated muscle glucose uptake after the 3-week Rlx or vehicle intervention in HF-fed mice. A: Glucose infusion rate (top) and arterial glucose (bottom) during the insulin clamp. Mice were fasted 5 h before the onset of the clamp. Blood glucose was maintained at ∼150 mg/dL during steady state (80–120 min) and the time course is displayed to demonstrate quality of the clamp; 50% glucose was infused to maintain euglycemia. B: EndoRa and Rd during the insulin clamp. Basal values are an average of −15 min and −5 min, and the insulin clamp values are an average of 80–120 min (steady state). C: Rg after the insulin clamp in the gastrocnemius (gastroc), SVL, and heart. D: Isolated muscle glucose uptake on the soleus and extensor digitorum longus (EDL). Mice were fasted for 5 h before muscles were excised. Data are expressed as mean ± SE. n = 11–13. *P ≤ 0.05.
FIG. 4.

Protocol 2: insulin signaling and immunohistochemical stain of skeletal muscle and liver. Western blot analysis of the activation status of Akt (A) analyzed as the ratio of phosphorylated Akt (p-Akt) to total Akt in liver (top) and skeletal muscle (bottom) from protein extracted from mice after the insulin clamp (V, vehicle; R, relaxin). Immunohistochemical detection of skeletal muscle collagen III (B) and collagen IV (C) from the gastrocnemius muscle. Immunohistochemical detection of hepatic collagen III (D). Immunohistochemical analysis was performed on tissues fixed immediately after the insulin clamp. Data are expressed as mean ± SE. AU, arbitrary units. n = 5–6. *P ≤ 0.05.
Ex vivo glucose uptake in isolated soleus and extensor digitorum longus showed no difference between the vehicle or Rlx groups (Fig. 3D). This suggests that the extramyocellular effects of Rlx are key to the effectiveness of Rlx intervention in HF-fed mice.
ECM remodeling.
Collagen III and collagen IV levels were equivalent in skeletal muscle between treatment groups (Fig. 4B and C). The improvement in hepatic insulin action with Rlx treatment was associated with reduced collagen III (P ≤ 0.05; Fig. 4D). Cardiac collagen III and collagen IV were increased in response to the HF diet but were reduced by ∼50% and ∼65%, respectively, after the 3-week Rlx intervention (P ≤ 0.05; Fig. 5A and B). This effectively normalized these ECM proteins to levels of lean mice. The abatement in cardiac ECM protein associated with a reduction in SMAD2 phosphorylation (P ≤ 0.05; Fig. 5C), which is a marker of transforming growth factor-β receptor downstream activation.
FIG. 5.

Protocol 2: collagen protein levels and SMAD2 signaling in cardiac muscle. Immunohistochemical detection of collagen III (A) and collagen IV (B) from hearts fixed after the insulin clamp. Western blot analysis of phosphorylated SMAD2 (p-SMAD2), total SMAD2, and the ratio of p-SMAD2 to total SMAD2 (C) from cardiac protein extracts after the insulin clamp. Data are expressed as mean ± SE. AU, arbitrary units. n = 5–6. *P ≤ 0.05.
Vascular.
The enhanced muscle insulin action in Rlx-treated mice was associated with a two-fold expansion of skeletal muscle capillary density (P ≤ 0.05; Fig. 6A). Improved vascular reactivity was present after the Rlx intervention, as indicated by augmented endothelial-dependent aortic relaxation (P ≤ 0.05; Fig. 6C). There was no difference in smooth muscle–dependent relaxation in response to sodium nitroprusside and no significant change in the vasculature response to phenylephrine (PE) (Fig. 6C and D). A potential common mechanism for the changes in skeletal muscle capillary density and endothelial-dependent relaxation is the elevated arterial VEGF concentrations after 3 weeks of continuous Rlx administration (P ≤ 0.05; Fig. 6B).
FIG. 6.
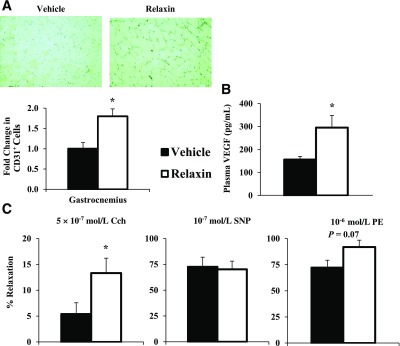
Protocol 2: capillarity density and vascular reactivity in response to the Rlx intervention. Capillary density (A) quantified with immunohistochemical staining of CD-31. Capillary density is quantified as the number of CD31+ cells. Plasma VEGF concentration (B) after the insulin clamps. Vascular reactivity (C) from excised aortas after the insulin clamps. Endothelial-dependent relaxation (left; carbachol [Cch]), smooth muscle–dependent relaxation (middle; sodium nitroprusside [SNP]), and stress generated from phenylephrine (right; PE). Responses represented as a percent of maximal tension from KCl stimulation. Data are expressed as mean ± SE. n = 5–8. *P ≤ 0.05.
DISCUSSION
These studies demonstrate for the first time that infusion of the hormone Rlx acutely augments muscle perfusion and insulin-stimulated MGU in lean, healthy C57BL/6J mice. There is no such acute Rlx effect in HF-fed mice. However, results show that a 3-week Rlx intervention in HF-fed mice ameliorates the metabolic and cardiovascular dysfunction. It is important to recognize that the potent metabolic effects of Rlx were absent in isolated muscle fibers, regardless of whether Rlx was administered acutely in lean mice or as a chronic intervention in HF-fed mice. The data from isolated muscle support the hypothesis that Rlx diminishes the extramyocellular barriers to MGU during hyperinsulinemia.
Baron et al. (8) demonstrated that the coinfusion of insulin and a vasodilator in healthy subjects resulted in a synergistic effect to enhance limb blood flow and muscle glucose uptake. In congruence with this clinical study, the acute Rlx infusion in the chow-fed mice of protocol 1 increased steady-state Rd and muscle Rg during the insulin clamp. The Rlx-infused mice had greater muscle microsphere deposition at the termination of the insulin clamp and enhanced aortic ring relaxation, suggesting a greater hemodynamic response to insulin. The amplified muscle blood flow increases insulin and glucose access to the muscle interstitium. This would predictably enhance insulin action, as supported by augmented in vivo skeletal muscle insulin signaling. The increased pro-MMP-2 and MMP-9 activities were consistent with the acute vasodilatory mechanisms of Rlx (28).The equivalent basal and insulin-stimulated glucose uptake in isolated muscle suggests that the primary mechanism for the enhanced muscle Rg was independent of direct actions of Rlx on the myocyte.
The 3-week Rlx intervention in protocol 2 was effective in improving insulin action in HF-fed mice. The improvement in glucose homeostasis in both the fasted and insulin-stimulated state is speculated to be a result of the actions unique to Rlx that improve vascular adaptations to the HF diet. The isolated MGU data support this conclusion. Potentially, other extramyocellular factors that require long-term treatment, such as an increase in plasma VEGF, could be necessary for the in vivo glucoregulatory effects of Rlx that are absent ex vivo. Furthermore, Rlx has been shown to antagonize angiotensin II action (39), and the renin-angiotensin system has been implicated in the pathogenesis of skeletal muscle capillary rarefaction and insulin resistance (40). The glucoregulatory and vascular adaptations that are present with the long-term Rlx administration are notably absent in the HF-fed cohort of protocol 1. This may provide insight into the mechanism of Rlx action in HF-fed mice. It would suggest that an adaptive process, rather than acute activation, is required to overcome the impairments in insulin action in HF-fed mice. Rlx intervention previously has been shown to ameliorate endothelial dysfunction in models of hypertension (20,21,41). In the current model of HF diet–induced insulin resistance, Rlx improved endothelial dysfunction. The improvement in endothelial reactivity did not occur in HF-fed mice that underwent an acute Rlx infusion, suggesting long-term administration is necessary to overcome the vascular impairment associated with a HF diet (42). Rlx acts through an endothelial nitric oxide synthase–dependent pathway to cause vascular relaxation. It is speculated that the vascular remodeling and impaired endothelial nitric oxide synthase function present in HF-fed mice nullifies the acute Rlx effect in the HF-fed mice in protocol 1 (43). Consistent with this finding, acute administration of Rlx does not ameliorate hypertension in spontaneously hypertensive rats (39). Additionally, Rlx-treated mice had an approximately two-fold increase in skeletal muscle capillary density. Capillary rarefaction and endothelial dysfunction associated with obesity are critical to the pathogenesis of skeletal muscle insulin resistance (44–47). The actions of Rlx on the vasculature may enhance the hemodynamic response to hyperinsulinemia, thus augmenting microvascular perfusion. An increase in muscle blood volume would increase the surface area for insulin and glucose diffusion, in addition to other hormones and nutrients (9,13,15,48). The likely mechanism for the improved endothelial function and expansion of skeletal muscle capillary density is the elevation in circulating VEGF, which has been shown to be critical to the sustained vasodilatory and angiogenic actions of Rlx (21–23).
The efficacy of Rlx to enhance MGU during hyperinsulinemia acutely in lean and chronically in HF-fed mice was independent of a direct interaction with the insulin receptor. Originally, Rlx was characterized to be part of the insulin family of proteins because of their common two-chain structure. More recent data demonstrated that Rlx diverged from the insulin family early in vertebrate evolution, forming a separate protein and receptor family (49). There is no evidence to support the cross-reactivity of Rlx with the insulin receptor because Rlx does not activate protein kinase receptors (50). The metabolic effects demonstrated in the current study were likely attributable to extramyocellular and extrahepatic adaptations to Rlx. Notably, the specific receptor for Rlx, RXFP1, has not been identified in skeletal myocytes or hepatocytes (50).
ECM deposition occurs in skeletal muscle, liver, and cardiac tissue of HF-fed rodents (1,51,52) and insulin-resistant humans (2,51,53,54). The interaction of collagen proteins with the integrin receptors has been linked to the development of hepatic and skeletal muscle insulin resistance (1). In the current studies, Rlx reversed the deposition of hepatic collagen III, although there was no difference in hepatic collagen IV (data not shown). It is speculated that the improved hepatic insulin action was related to the reduction in collagen III and the potential interaction with the hepatic integrin receptors. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis are associated with the development and progression of fibrosis in type 2 diabetes and subsequent impairments in hepatic insulin action (51,55). Previously, collagen III and collagen IV have been shown to be elevated in muscle of HF-fed mice and humans (1,2); however, there was no difference in these collagen species in skeletal muscle in the current model, perhaps relating to differences in diet duration. Although collagen III and collagen IV are major components of the ECM, there are other distinct matrix proteins that could have been altered.
Rlx intervention diminished collagen III and collagen IV accumulation in the heart, which is consistent with the antifibrotic effects of long-term Rlx treatment shown in rodent models of type 1 diabetes (18) and hypertension (19). The decrease in cardiac ECM proteins after the Rlx treatment likely was a consequence of Rlx inhibiting the downstream activation of SMAD2 by transforming growth factor-β (24,25,27). The diminished cardiac collagen III and collagen IV and ameliorated endothelial dysfunction in protocol 2 emphasize that Rlx may be efficacious in the treatment of the broader dysfunction associated with the metabolic syndrome.
Macroangiopathies and microangiopathies correlate with insulin resistance (56,57) leading to impaired tissue perfusion, which is a key component of the etiology of diabetes-related tissue and organ damage (58). Furthermore, endothelial dysfunction and capillary rarefaction contribute to hypertension and insulin resistance, both of which are components of the metabolic syndrome, and increase the risk of cardiovascular mortality (47,58,59). Thus, it is critical to consider the common underlying vascular pathologies to develop novel intervention strategies to treat the metabolic syndrome. However, a critical consideration for the long-term clinical administration of a vascular proliferative compound is the potential exacerbation of tumor development and metastasis. This is particularly important because there is a positive association between insulin resistance and many types of cancers (60). It is possible that chronic exposure to Rlx for the treatment of insulin resistance may exacerbate outcomes in cancer patients. Notably, Rlx is expressed at higher levels in prostate cancer and correlates to metastatic potential and diminished survival (61). The inhibition of RXFP1 has been investigated as a potential therapeutic target (62).
These studies demonstrate the effectiveness of Rlx in targeting the extramyocellular barriers to MGU for the treatment of insulin resistance. Intervention with the hormone Rlx targets multiple physiological systems to ameliorate cardiac and hepatic collagen accumulation, increase skeletal muscle capillary density, and improve diet-induced endothelial dysfunction. These effects contribute to the enhancement of in vivo insulin action in HF-fed mice and require an extended treatment period to mitigate these extramyocellular barriers to insulin-stimulated MGU. The results not only highlight the efficacy of Rlx in the correction of muscle insulin resistance but also demonstrate the potential therapeutic value of Rlx in reversing fibrosis and vascular dysfunction associated with a HF diet.
ACKNOWLEDGMENTS
This work was funded by National Institutes of Health grants DK054902 andDK059637 (Mouse Metabolic Phenotyping Center). The Diabetes Research and Training Center (DK20593) also provided support for this work.
The authors thank Dr. Dennis Stewart of Corthera, Inc., a subsidiary of Novartis Pharmaceuticals Corp., for supplying recombinant H-2 relaxin and for helpful insight. No other potential conflicts of interest relevant to this article were reported.
J.S.B. designed experiments, researched data, and wrote the manuscript. L.L. contributed to the research of data and reviewed the manuscript. K.M.H. designed experiments and reviewed the manuscript. L.K. contributed to the research of data and reviewed the manuscript. M.O. researched data. F.D.J. and D.P.B. contributed to the research of data. C.M.B. designed experiments and reviewed the manuscript. D.H.W. designed experiments, contributed to the discussion, and reviewed the manuscript. D.H.W. is the guarantor of this work, had full access to data, and takes full responsibility for the integrity of data and accuracy of data analysis.
Parts of this study were presented in abstract form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011; the 72nd Scientific Sessions of the American Diabetes Association, Philadelphia, Pennsylvania, 8–12 June 2012; and the 73rd Scientific Sessions of the American Diabetes Association, Chicago, Illinois, 21–25 June 2013.
The authors thank ZhiZhang Wang of the Vanderbilt Mouse Metabolic Phenotyping Center Cardiovascular Pathophysiology Core for performing echocardiography in these studies and Melissa B. Downing of the Vanderbilt University Mouse Pathology Core Laboratory of the Mouse Metabolic Phenotyping Center.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0033/-/DC1.
REFERENCES
- 1.Kang L, Ayala JE, Lee-Young RS, et al. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 2011;60:416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berria R, Wang L, Richardson DK, et al. Increased collagen content in insulin-resistant skeletal muscle. Am J Physiol Endocrinol Metab 2006;290:E560–E565 [DOI] [PubMed] [Google Scholar]
- 3.Gavin TP, Stallings HW, 3rd, Zwetsloot KA, et al. Lower capillary density but no difference in VEGF expression in obese vs. lean young skeletal muscle in humans. J Appl Physiol 2005;98:315–321 [DOI] [PubMed] [Google Scholar]
- 4.Chou E, Suzuma I, Way KJ, et al. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation 2002;105:373–379 [DOI] [PubMed] [Google Scholar]
- 5.Baron AD, Clark MG. Role of blood flow in the regulation of muscle glucose uptake. Annu Rev Nutr 1997;17:487–499 [DOI] [PubMed] [Google Scholar]
- 6.Baron AD. Hemodynamic actions of insulin. Am J Physiol 1994;267:E187–E202 [DOI] [PubMed] [Google Scholar]
- 7.Baron AD, Tarshoby M, Hook G, et al. Interaction between insulin sensitivity and muscle perfusion on glucose uptake in human skeletal muscle: evidence for capillary recruitment. Diabetes 2000;49:768–774 [DOI] [PubMed] [Google Scholar]
- 8.Baron AD, Steinberg H, Brechtel G, Johnson A. Skeletal muscle blood flow independently modulates insulin-mediated glucose uptake. Am J Physiol 1994;266:E248–E253 [DOI] [PubMed] [Google Scholar]
- 9.Vincent MA, Clerk LH, Lindner JR, et al. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 2004;53:1418–1423 [DOI] [PubMed] [Google Scholar]
- 10.Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest 1990;85:1844–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baron AD, Laakso M, Brechtel G, Edelman SV. Mechanism of insulin resistance in insulin-dependent diabetes mellitus: a major role for reduced skeletal muscle blood flow. J Clin Endocrinol Metab 1991;73:637–643 [DOI] [PubMed] [Google Scholar]
- 12.Fueger PT, Shearer J, Bracy DP, et al. Control of muscle glucose uptake: test of the rate-limiting step paradigm in conscious, unrestrained mice. J Physiol 2005;562:925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wasserman DH. Four grams of glucose. Am J Physiol Endocrinol Metab 2009;296:E11–E21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim F, Pham M, Maloney E, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 2011;13:294–307 [DOI] [PubMed] [Google Scholar]
- 16.Conrad KP. Emerging role of relaxin in the maternal adaptations to normal pregnancy: implications for preeclampsia. Semin Nephrol 2011;31:15–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuane JT, Debrah JE, Sautina L, et al. Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 2011;152:2786–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel CS, Hewitson TD, Zhang Y, Kelly DJ. Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology 2008;149:3286–3293 [DOI] [PubMed] [Google Scholar]
- 19.Lekgabe ED, Kiriazis H, Zhao C, et al. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 2005;46:412–418 [DOI] [PubMed] [Google Scholar]
- 20.Conrad KP. Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension 2010;56:2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP. Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 2011;57:1151–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segal MS, Sautina L, Li S, et al. Relaxin increases human endothelial progenitor cell NO and migration and vasculogenesis in mice. Blood 2012;119:629–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unemori EN, Lewis M, Constant J, et al. Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. Wound Repair Regen 2000;8:361–370 [DOI] [PubMed] [Google Scholar]
- 24.Unemori EN, Pickford LB, Salles AL, et al. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest 1996;98:2739–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida T, Kumagai H, Suzuki A, et al. Relaxin ameliorates salt-sensitive hypertension and renal fibrosis. Nephrol Dial Transplant 2012;27: [DOI] [PubMed] [Google Scholar]
- 26.Hewitson TD, Ho WY, Samuel CS. Antifibrotic properties of relaxin: in vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology 2010;151:4938–4948 [DOI] [PubMed] [Google Scholar]
- 27.Heeg MH, Koziolek MJ, Vasko R, et al. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int 2005;68:96–109 [DOI] [PubMed] [Google Scholar]
- 28.Jeyabalan A, Novak J, Doty KD, et al. Vascular matrix metalloproteinase-9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short-term administration of relaxin. Endocrinology 2007;148:189–197 [DOI] [PubMed] [Google Scholar]
- 29.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 30.Berglund ED, Li CY, Poffenberger G, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 2008;57:1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007;56:1025–1033 [DOI] [PubMed] [Google Scholar]
- 32.Wasserman DH, Ayala JE, McGuinness OP. Lost in translation. Diabetes 2009;58:1947–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol 1956;187:15–24 [DOI] [PubMed] [Google Scholar]
- 34.Lee-Young RS, Griffee SR, Lynes SE, et al. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem 2009;284:23925–23934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jørgensen SB, Viollet B, Andreelli F, et al. Knockout of the α2 but not α1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 2004;279:1070–1079 [DOI] [PubMed] [Google Scholar]
- 36.Eagle S, Brophy CM, Komalavilas P, et al. Surgical skin markers impair human saphenous vein graft smooth muscle and endothelial function. Am Surg 2011;77:922–928 [PMC free article] [PubMed] [Google Scholar]
- 37.Hocking KM, Brophy C, Rizvi SZ, et al. Detrimental effects of mechanical stretch on smooth muscle function in saphenous veins. J Vasc Surg 2011;53:454–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muhs BE, Gagne P, Plitas G, Shaw JP, Shamamian P. Experimental hindlimb ischemia leads to neutrophil-mediated increases in gastrocnemius MMP-2 and -9 activity: a potential mechanism for ischemia induced MMP activation. J Surg Res 2004;117:249–254 [DOI] [PubMed] [Google Scholar]
- 39.Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG. Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 2005;46:745–750 [DOI] [PubMed] [Google Scholar]
- 40.Guo Q, Mori T, Jiang Y, et al. Losartan modulates muscular capillary density and reverses thiazide diuretic-exacerbated insulin resistance in fructose-fed rats. Hyperten Res 2013;35:48–54. doi: 10.1038/hr.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasser JM, Molnar M, Baylis C. Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(ω)-nitro-L-arginine methyl ester hypertensive rats. Hypertension 2011;58:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky HM. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ Res 2005;96:1178–1184 [DOI] [PubMed] [Google Scholar]
- 43.Ma L, Ma S, He H, et al. Perivascular fat-mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high-fat diet-induced obese rats. Hypertens Res 2010;33:446–453 [DOI] [PubMed] [Google Scholar]
- 44.Lillioja S, Young AA, Culter CL, et al. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest 1987;80:415–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mårin P, Andersson B, Krotkiewski M, Björntorp P. Muscle fiber composition and capillary density in women and men with NIDDM. Diabetes Care 1994;17:382–386 [DOI] [PubMed] [Google Scholar]
- 46.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest 1996;97:2601–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonner JS, Lantier L, Hasenour CM, James FD, Bracy DP, Wasserman DH. Muscle-specific vascular endothelial growth factor deletion induces muscle capillary rarefaction creating muscle insulin resistance. Diabetes 2013;62:572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vincent MA, Barrett EJ, Lindner JR, Clark MG, Rattigan S. Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am J Physiol Endocrinol Metab 2003;285:E123–E129 [DOI] [PubMed] [Google Scholar]
- 49.Halls ML, van der Westhuizen ET, Bathgate RA, Summers RJ. Relaxin family peptide receptors–former orphans reunite with their parent ligands to activate multiple signaling pathways. Br J Pharmacol 2007;150:677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev 2013;93:405–480 [DOI] [PubMed] [Google Scholar]
- 51.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001;34:738–744 [DOI] [PubMed] [Google Scholar]
- 52.Zaman AK, Fujii S, Goto D, et al. Salutary effects of attenuation of angiotensin II on coronary perivascular fibrosis associated with insulin resistance and obesity. J Mol Cell Cardiol 2004;37:525–535 [DOI] [PubMed] [Google Scholar]
- 53.Shimizu M, Umeda K, Sugihara N, et al. Collagen remodelling in myocardia of patients with diabetes. J Clin Pathol 1993;46:32–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang DJ, Pritchard MT, Nagy LE. Obesity, diabetes mellitus, and liver fibrosis. Am J Physiol Gastrointest Liver Physiol 2011;300:G697–G702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacDonald GA, Bridle KR, Ward PJ, et al. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. J Gastroenterol Hepatol 2001;16:599–606 [DOI] [PubMed] [Google Scholar]
- 56.Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol 2005;16:2134–2140 [DOI] [PubMed] [Google Scholar]
- 57.Hedblad B, Nilsson P, Engström G, Berglund G, Janzon L. Insulin resistance in non-diabetic subjects is associated with increased incidence of myocardial infarction and death. Diabet Med 2002;19:470–475 [DOI] [PubMed] [Google Scholar]
- 58.De Boer MP, Meijer RI, Wijnstok NJ, et al. Microvascular dysfunction: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Microcirculation 2012;19:5–18 [DOI] [PubMed] [Google Scholar]
- 59.Utsunomiya KA. Treatment strategy for type 2 diabetes from the perspective of systemic vascular protection and insulin resistance. Vasc Health Risk Manag 2012;8:429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inoue M, Tsugane S. Insulin resistance and cancer: epidemiological evidence. Endocr Relat Cancer 2012;19:F1–F8 [DOI] [PubMed] [Google Scholar]
- 61.Feng S, Agoulnik IU, Bogatcheva NV, et al. Relaxin promotes prostate cancer progression. Clin Cancer Res 2007;13:1695–1702 [DOI] [PubMed] [Google Scholar]
- 62.Feng S, Agoulnik AI. Expression of LDL-A module of relaxin receptor in prostate cancer cells inhibits tumorigenesis. Int J Oncol 2011;39:1559–1565 [DOI] [PubMed] [Google Scholar]