

Abstract
Endothelial nitric oxide synthase (eNOS) dysfunction induces insulin resistance and glucose intolerance. Tetrahydrobiopterin (BH4) is an essential cofactor of eNOS that regulates eNOS activity. In the diabetic state, BH4 is oxidized to 7,8-dihydrobiopterin, which leads to eNOS dysfunction owing to eNOS uncoupling. The current study investigates the effects of BH4 on glucose metabolism and insulin sensitivity in diabetic mice. Single administration of BH4 lowered fasting blood glucose levels in wild-type mice with streptozotocin (STZ)-induced diabetes and alleviated eNOS dysfunction by increasing eNOS dimerization in the liver of these mice. Liver has a critical role in glucose-lowering effects of BH4 through suppression of hepatic gluconeogenesis. BH4 activated AMP kinase (AMPK), and the suppressing effect of BH4 on gluconeogenesis was AMPK-dependent. In addition, the glucose-lowering effect and activation of AMPK by BH4 did not appear in mice with STZ-induced diabetes lacking eNOS. Consecutive administration of BH4 in ob/ob mice ameliorated glucose intolerance and insulin resistance. Taken together, BH4 suppresses hepatic gluconeogenesis in an eNOS-dependent manner, and BH4 has a glucose-lowering effect as well as an insulin-sensitizing effect in diabetic mice. BH4 has potential in the treatment of type 2 diabetes.
Nitric oxide (NO) is a biological messenger produced by NO synthase (NOS), which includes endothelial (eNOS), inducible (iNOS), and neuronal (nNOS) isoforms. eNOS-derived NO is well-known to have a pivotal role in physiological regulation of endothelial function (1,2). eNOS dysfunction occurs in conditions of diabetes and is known to induce insulin resistance and glucose intolerance (3–5). Insulin resistance caused by eNOS dysfunction is thought to be induced by endothelial dysfunction, leading to decreased skeletal muscle blood flow and glucose uptake (4). On the other hand, glucose transport in isolated skeletal muscle is lower in eNOS-deficient (eNOS−/−) mice, indicating that eNOS expressed in skeletal muscle also regulates its glucose uptake (4). Moreover, eNOS−/− mice are insulin resistant at the level of liver (5). These studies suggest that eNOS plays a central role in the regulation of glucose metabolism and insulin sensitivity and represents several therapeutic targets for type 2 diabetes.
The function of eNOS is regulated by multiple factors such as mRNA expression of eNOS, l-arginine, influx of Ca2+, and tetrahydrobiopterin (BH4) (2,6,7). BH4 is an essential cofactor for eNOS catalysis and functions as an allosteric modulator of arginine binding (7,8). Binding of BH4 to eNOS elicits a conformational change that increases the affinity for binding of arginine-based ligands. BH4 binding also plays a role in dimer formation of the active and stabilized form of eNOS (8). BH4 is converted to 7,8-dihydrobiopterin (BH2) by exposure to oxidative stress such as diabetes (8,9). Increase in BH2 induces dysfunction of eNOS, as BH2 is inactive for NOS cofactor function and competes with BH4 for BH4 binding (8,9). Furthermore, in states of diabetes and high glucose, de novo synthesis of BH4, which is rate limited by GTP cyclohydrolase I (GTPCH I), is impaired (10–13). Thus, the availability of BH4 is reduced and the function of eNOS is altered so that the enzyme produces superoxide anion (O2−) rather than NO, a phenomenon called “eNOS uncoupling” (7,8,14). Supplementation of BH4 can improve endothelial dysfunction by elevating the BH4-to-BH2 ratio, leading to recoupling of eNOS, and has been used in clinical trials with patients with atherosclerotic diseases for the expected vasodilatation effects of BH4 through NO production (15). However, it is unclear whether BH4 improves glucose metabolism and insulin sensitivity in diabetic conditions.
In the current study, we investigated the effects of BH4 on blood glucose levels and insulin sensitivity in diabetic mice. Fasting blood glucose levels are regulated by the level of hepatic gluconeogenesis, elevation of which is the major cause of fasting hyperglycemia in diabetes (16,17). We demonstrate here that BH4 lowers fasting blood glucose levels and suppresses gluconeogenesis in liver in an eNOS-dependent manner. In addition, BH4 has an ameliorating effect on glucose intolerance as well as insulin resistance in diabetic mice. Using primary hepatocytes isolated from mouse liver, we have clarified the mechanism by which BH4 suppresses hepatic gluconeogenesis. These data suggest that BH4 has potential as a novel therapeutic approach to diabetes.
RESEARCH DESIGN AND METHODS
Male C57/BL6 (wild-type) mice and male heterozygous Ins2Akita (diabetic Akita) mice, which exhibit hyperglycemia with reduced β-cell mass caused by a point mutation in the insulin 2 gene that leads to misfolded insulin and severe endoplasmic reticulum stress, were obtained from Shimizu (Kyoto, Japan) (18). Male eNOS−/− mice in the C57/BL6 mice background were obtained from The Jackson Laboratory (Bar Harbor, ME). Male B6.V-Lepob/J (ob/ob) mice were obtained from Charles River Japan (Yokohama, Japan). Mice with streptozotocin (STZ)-induced diabetes were made by injection of STZ (120 mg/kg i.p.) to 7-week-old wild-type or eNOS−/− mice. At 3 weeks after injection of STZ, the animals were confirmed to be diabetic by both high blood glucose levels (≥15 mmol/L) and other diabetic features, including polyuria, polydipsia, and hyperglycemia.
The mice were maintained in a temperature-controlled (25 ± 2°C) environment with a 12-h light/dark cycle with free access to standard laboratory chow and water. All experiments were carried out with mice aged 8–10 weeks. The animals were maintained and used in accordance with the Guidelines for Animal Experiments of Kyoto University. All experiments involving animals were conducted in accordance with the Guidelines for Animal Experiments of Kyoto University and were approved by the Animal Research Committee, Graduate School of Medicine, Kyoto University.
Preparations and cultures of mouse hepatocyte and aortic endothelial cell.
Mouse hepatocytes were isolated by collagenase digestion as previously described (19). Primary hepatocytes were prepared by seeding in sixwell type 1 collagen–coated plates at a density of 1.5 × 106 cells in Dulbecco's modified Eagle's medium (DMEM) (low glucose, 5.6 mmol/L) containing 10% (vol/vol) FBS, 100 nmol/L regular insulin, 50 units/mL penicillin, and 50 μg/mL streptomycin. Hepatocytes were then cultured overnight in a humidified atmosphere (5% CO2) at 37°C. As for mouse endothelial cells (ECs), the aorta was dissected and filled with collagenase type II solution. After incubation for 45 min at 37°C, ECs were removed from the aorta and collected by centrifugation at 1,200 rpm for 5 min. The EC was cultured in a sixwell collagen type I–coated dish for 1 week.
Glucose production via gluconeogenesis in hepatocytes.
Freshly isolated hepatocytes from mice fasted for 16 h were treated in 24-well plates (7.5 × 105 cells/well) in buffer A, which consisted of 0.5 mL Krebs-Ringer bicarbonate medium of 119.4 mmol/L NaCl, 3.7 mmol/L KCl, 2.7 mmol/L CaCl2, 1.3 mmol/L KH2PO4, 1.3 mmol/L MgSO4, and 24.8 mmol/L NaHCO3 without glucose; 2% (wt/vol) BSA; 0.24 mmol/L 3-isobutyl-1-methylxanthine; and gluconeogenetic substrates (1 mmol/L pyruvate plus 10 mmol/L lactate). Hepatocytes were treated with BH4 (Schircks Laboratories, Jona, Switzerland), sodium nitroprusside (SNP), NG-nitro-l-arginine methyl ester (Sigma, St. Louis, MO), sepiapterin (Schircks Laboratories), erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) (Wako, Osaka, Japan), and compound C (Sigma). Glucose production was measured by glucose oxidation method as previously described (19).
Immunoblotting analysis of hepatocytes.
Western blotting was performed as previously described (19). Primary hepatocytes cultured overnight were incubated in buffer A treated with BH4, SNP, sepiapterin, and EHNA. Hepatocytes were homogenized in lysis buffer. Cell lysates (50–150 μg protein/lane) were heated at 95°C for 5 min and subjected to electrophoresis on 6–10% (vol/vol) sodium dodecyl sulfate–polyacrylamide gels and transferred onto nitrocellulose membranes. For analysis of eNOS dimerization, the samples were not heated and the temperature was maintained at <15°C during electrophoresis. Primary antibodies used were anti–phosphorylated (phospho-)AMP kinase (AMPK)α (Thr172), anti-AMPKα, anti–phospho-acetyl-CoA carboxylase (ACC) (Ser79), anti-ACC, anti–phospho-eNOS (Ser1177), anti–phospho-Akt (Ser473), anti-Akt (all at 1:1,000 dilution; Cell Signaling Technology, Danvers, MA), anti-eNOS polyclonal antibody (1:500 dilution; BD Transduction Laboratories, San Jose, CA), anti-CD31 monoclonal antibody (1:2,000 dilution; Dianova, Hamburg, Germany), anti–GTPCH I (1:3,000; kind gift from Prof. H. Ichinose, Tokyo Institute of Technology), anti–dihydrofolate reductase (DHFR), anti–α1-antitrypsin (1:500; Santa Cruz, Delaware, CA), and anti–β-actin (1:5,000; Sigma). Secondary antibodies used were horseradish peroxidase–conjugated anti-rabbit, -mouse, -rat, or -goat antibody (GE Healthcare, Buckinghamshire, U.K.). The fluorescent bands were visualized using a detection system (Amersham ECL Plus; GE Healthcare) and quantified by densitometry using Image J software from National Institutes of Health (Bethesda, MD).
Cell transfection and short interfering RNA.
Stealth short interfering RNA (siRNA) of AMPKα1 was purchased from Invitrogen (Carlsbad, CA). The sequences of siRNA for AMPKα1 were 5′-UCUCUUUCCUGAGGACCCAUCUUAU-3′ and 5′-AUAAGAUGGGUCCUCAGGAAAGAGA-3′. The sequences of control siRNAs were 5′-ACCAACAACAGUUUGGGAAUAGGGA-3′and 5′- UCCCUAUUCCCAAACUGUUGUUGGU-3′. Isolated hepatocytes in DMEM (low glucose, 5.6 mmol/L) containing 10% (vol/vol) FBS and 100 nmol/L regular insulin were mixed with Opti-MEM containing siRNA and Lipofectamine RNAi MAX (Invitrogen) and were plated on wells and then incubated at 37°C in a CO2 incubator. The final amounts of hepatocytes, DMEM, Opti-MEM, siRNA, and Lipofectamine RNAi MAX were 5.0 × 105 cells/mL, 75% (vol/vol), 25% (vol/vol), 50 nmol/L, and 0.2%, respectively. Medium was replaced with DMEM 6 h after transfection. Forty-eight hours after transfection, the medium was replaced with buffer A, the cells were incubated for 60 min with or without BH4, and the glucose content of the supernatant was measured.
Nitrite/nitrate analysis.
Primary hepatocytes and liver tissues were homogenized in buffer A, and the amount of nitrite/nitrate in the supernatant was determined by a fluorescence method.
Immunocytochemistry.
The hepatocytes were incubated with rabbit polyclonal anti-nitrotyrosine antibody (1:100 dilution; Millipore, Billerica, MA). Cells were then incubated with goat anti-rabbit IgG fluorescein-conjugated secondary antibody (1:100 dilution, Alexa Fluor 488; Invitrogen). Fluorescence in cells was monitored as previously described (19).
Measurement of adenine nucleotide content.
After primary isolated hepatocytes were incubated in buffer A with or without BH4 and SNP for 30 min, treatment was stopped by rapid addition of 0.1 mL of 2 mol/L HClO4, followed by mixing by vortex and sonication in ice-cold water for 3 min. Adenine nucleotide contents were measured by a luminometric method as previously described (19,20).
Isolation of total RNA and quantitative RT-PCR.
Total RNA was isolated from livers of 10 week-old wild-type mice, wild-type mice with STZ-induced diabetes, and ob/ob mice using Trizol (Invitrogen) as previously described (21). The mouse sequence of forward and reverse primers to detect GTPCH I and DHFR, glucose 6-phosphatase (G6Pase), phosphoenolpyruvate carboxykinase (PEPCK), and glyceraldehyde-3-phosphate dehydrogenase as an inner control are shown in Supplementary Table 1. SYBR Green PCR Master Mix (Applied Biosystems, Foster, CA) was prepared for the quantitative RT-PCR run. The thermal cycling conditions were denaturation at 95°C for 10 min followed by 50 cycles at 95°C for 15 s and 60°C for 1 min. mRNA levels were measured by real-time quantitative RT-PCR using ABI PRISM 7000 Sequence Detection System (Applied Biosystems).
Biopterin analysis.
Tissues or whole blood of wild-type mice and wild-type mice with STZ-induced diabetes was collected. For measurement of uptake of BH4 in liver, BH4 (20 mg/kg) dissolved with 0.9% (wt/vol) sterile saline was administrated intraperitoneally to wild-type mice. After cervical dislocation, the mice were abdominally dissected and liver tissues were collected at 0, 30, 60, 120, and 180 min after injection. The organs were weighed, frozen immediately in liquid N2, and then stored at −80°C. Total biopterin, BH4, and BH2 were measured as previously described (22).
Effect of BH4 on blood glucose levels of wild-type mice with STZ-induced diabetes, eNOS−/− mice with STZ-induced diabetes, and diabetic Akita mice.
Blood glucose levels were measured in wild-type mice with STZ-induced diabetes, eNOS−/− mice with STZ-induced diabetes, and diabetic Akita mice fasted for 16 h, and BH4 (20 mg/kg) or metformin (250 mg/kg; Sigma) in 0.9% (wt/vol) sterile saline or 0.9% sterile saline alone was injected intraperitoneally. Blood glucose levels were measured again 2 h after injection.
Effect of BH4 on blood glucose levels of ob/ob mice.
Blood glucose levels and body weight of ob/ob mice were measured. The mice were divided into two groups shown in Supplementary Table 2, and 0.9% (wt/vol) sterile saline with or without BH4 (10 mg/kg) was injected intraperitoneally twice a day for 10 days. Fed blood glucose levels were measured. After fasting overnight for 16 h, fasting blood glucose levels were measured.
Intraperitoneal glucose tolerance test.
Wild-type mice were fasted overnight for 16 h, and glucose (2 g/kg) was injected intraperitoneally with BH4 (20 mg/kg) in 0.9% (wt/vol) sterile saline or 0.9% sterile saline alone. After 10 days’ treatment of saline with or without BH4 (20 mg/kg), ob/ob mice were fasted overnight for 16 h, and glucose (1 g/kg) was injected intraperitoneally. Blood glucose levels and plasma insulin concentrations were measured at 0, 30, 60, 90, and 120 min after injection. Plasma insulin concentrations were determined by using an ELISA kit (Shibayagi, Gunma, Japan). Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated with the following formula: [fasting insulin (mU/L) × fasting plasma glucose (mmol/L)]/22.5.
Pyruvate tolerance test.
Pyruvate, BH4, and sepiapterin were dissolved with 0.9% (wt/vol) sterile saline. Wild-type, eNOS−/−, and ob/ob mice were fasted overnight for 16 h, and pyruvate (1 g/kg) was injected intraperitoneally with or without BH4 (20 mg/kg) and sepiapterin (20 mg/kg). Blood glucose levels were measured at 0, 30, 60, 90, and 120 min after injection.
Insulin tolerance test.
After 10 days’ treatment of saline with or without BH4 (20 mg/kg), ob/ob mice were fasted for 6 h, and regular insulin (1 units/kg i.p.) was injected with 0.9% sterile saline. Blood glucose levels were measured at 0, 30, 60, 90, and 120 min after injection.
Statistics.
Comparison between two groups was performed using unpaired Student t test (not noted) and paired Student t test. For more than two groups, one-way or two-way ANOVA followed by post hoc Bonferroni testing was performed. A value of P < 0.05 was considered statistically significant.
RESULTS
Biopterin dynamics and effects of BH4 on blood glucose levels in diabetic mice.
In STZ diabetic wild-type mice, the content of BH2 was increased and the BH4-to-BH2 ratio was decreased in blood and respective tissues (Fig. 1A–D). For investigation of whether BH4 lowers blood glucose levels, BH4 (20 mg/kg) in saline was injected intraperitoneally to STZ diabetic wild-type mice. Blood glucose levels were not changed 2 h after administration of BH4 in fed STZ diabetic wild-type mice, while blood glucose levels were lowered by ~2.4 mmol/L in overnight-fasted STZ diabetic wild-type mice—a change similar to that with metformin (Fig. 1E and F and Supplementary Fig. 1A). The same effects also were found in diabetic Akita mice (Supplementary Fig. 1B).
FIG. 1.

Biopterin dynamics and effects of BH4 on blood glucose levels in diabetic mice. A–D: BH2 levels and BH4-to-BH2 ratio of liver, blood, kidney, and spleen. Values are means ± SE. n = 7. *P < 0.05, **P < 0.01, ***P < 0.001 vs. without STZ. E and F: Fed blood glucose levels were not changed 2 h after injection of BH4 (20 mg/kg i.p.) to STZ diabetic wild-type mice; fasting blood glucose levels were significantly decreased. Values are means ± SE. n = 8. *P < 0.05 vs. the value of preinjection of saline with BH4 intraperitoneally; paired t test. No significant difference of fed and fasting blood glucose levels 2 h after intraperitoneal injection of saline to mice with STZ-induced diabetes.
Liver tissue has an important role in glucose-lowering effects of BH4.
Although the intraperitoneal glucose tolerance test (IPGTT) data in wild-type mice revealed no effects of BH4 on blood glucose levels and plasma insulin levels, the pyruvate tolerance test (PTT) data showed that BH4 decreased hepatic glucose production (Fig. 2A–C), suggesting that the suppressing effect on hepatic gluconeogenesis has a critical role in the glucose-lowering effect of BH4. The mRNA and protein expression levels of GTPCH I, a rate-limiting enzyme of the BH4 de novo synthesis pathway, were decreased in liver tissues of STZ diabetic wild-type mice (Fig. 2D and E). On the other hand, uptake of BH4 into liver by its supplementation is regulated by DHFR, a rate-limiting enzyme of the BH4 salvage synthesis pathway (23), and the expression of DHFR in liver tissues of STZ diabetic wild-type mice was not changed (Fig. 2F and G). The uptake of BH4 in liver of wild-type mice was confirmed with a peak at 30 min by administration of BH4 (20 mg/kg) as previously described (22,23) (Supplementary Fig. 2A). After 2-h administration of BH4, the mRNA expression levels of PEPCK were significantly decreased, while those of G6Pase were not changed, and the eNOS dimerization and NO content were increased in the liver of STZ diabetic wild-type mice (Fig. 2H–K). The mRNA expression levels of PEPCK and G6Pase in the liver of wild-type mice were not changed (Supplementary Fig. 2B and C).
FIG. 2.

Role of liver tissue in glucose-lowering effects of BH4. A and B: IPGTT to wild-type mice. Blood glucose levels and plasma insulin levels after administration of glucose (2 g/kg i.p.) with or without BH4 (20 mg/kg). Values are means ± SE (n = 6). C: PTT to wild-type mice. Elevation of blood glucose levels after intraperitoneal administration of pyruvate with BH4 (20 mg/kg) to wild-type mice was suppressed compared with those without BH4. Values are means ± SE (n = 6). *P < 0.05 vs. saline. D: In mice with STZ-induced diabetes, mRNA levels of GTPCH I expression were significantly decreased compared with those in nondiabetic wild-type mice liver. Values are means ± SE (n = 5). **P < 0.01 vs. nondiabetic wild-type mice liver. E: In wild-type mice with STZ-induced diabetes, protein expression levels of GTPCH I were significantly decreased compared with those in nondiabetic wild-type mice liver. Values are means ± SE (n = 5). **P < 0.01 vs. nondiabetic wild-type mice liver. F: No significant difference of mRNA expression levels of DHFR in liver was detected between nondiabetic mice and mice with STZ-induced diabetes. Values are means ± SE (n = 10). G: No significant difference of protein expression levels of DHFR in liver was detected between nondiabetic mice and mice with STZ-induced diabetes. Values are means ± SE (n = 5). H and I: In liver tissues of wild-type mice with STZ-induced diabetes treated with BH4, mRNA levels of PEPCK were significantly decreased compared with those treated without BH4. The mRNA levels of G6Pase were not changed. Values are means ± SE (n = 6), *P < 0.05 vs. saline. J: Liver tissues of eNOS dimer and monomer expression 2 h after intraperitoneal injection of saline with or without BH4 (20 mg/kg) to wild-type mice with STZ-induced diabetes. Densitometric analysis of the ratio of eNOS dimer to monomer. Values are means ± SE (n = 5). *P < 0.05 vs. saline. K: In liver tissues of wild-type mice with STZ-induced diabetes treated with BH4, NO content was significantly increased compared with those treated without BH4. Values are means ± SE (n = 5). *P < 0.05 vs. saline.
BH4 suppresses gluconeogenesis and increases AMPKα phosphorylation in wild-type mouse hepatocytes.
As eNOS expression was confirmed in isolated hepatocytes from wild-type mice (Supplementary Fig. 3), we examined the direct effect of BH4 in suppression of hepatic gluconeogenesis using hepatocytes isolated from wild-type mice fasted for 16 h. In a time course study of exposure to BH4, the suppressing effect on gluconeogenesis appeared after 60 min (P < 0.01 vs. corresponding control) (Fig. 3A). We then investigated the increment of AMPKα phosphorylation by time course exposure of BH4 to hepatocytes. AMPK was activated after 30 min by BH4 (Fig. 3B). After 60 min exposure to BH4, gluconeogenesis was dose-dependently suppressed at doses of 50 and 100 μmol/L BH4 (control, 101.7 ± 3.7 nmol/mg protein; 50 μmol/L BH4, 72.4 ± 7.1 nmol/mg protein, P < 0.01 vs. control; 100 μmol/L BH4, 60.6 ± 4.1 nmol/mg protein, P < 0.001 vs. control) (Fig. 3C). AMPK was activated at doses of 50 and 100 μmol/L BH4 by 30 min exposure (Fig. 3D). In accordance with the activation of AMPK, an increase in phosphorylation of ACC by BH4 was confirmed (Fig. 3B and D). For determination of whether BH4 suppresses gluconeogenesis in an AMPK-dependent manner, the effect of silencing AMPK was examined (Fig. 3E). By transfection of AMPKα1 siRNA, the suppressing effect of BH4 on gluconeogenesis disappeared (Fig. 3F). The suppressing effect of BH4 on gluconeogenesis also disappeared in the presence of compound C, an AMPK inhibitor (Fig. 3G).
FIG. 3.

BH4 suppressed gluconeogenesis and increased AMPKα phosphorylation in hepatocytes isolated from wild-type mice. A: Time course of gluconeogenesis with exposure to BH4. Suppressing effect on gluconeogenesis by 50 μmol/L BH4 compared with control was detected after 60 min in hepatocytes isolated from wild-type mice. Values are means ± SE (n = 6). **P < 0.01 vs. control. B: Time course of phosphorylation of AMPKα and ACC upon exposure to BH4 (50 μmol/L). Both AMPKα and ACC phosphorylation were stimulated after 30 min exposure to BH4 in hepatocytes isolated from wild-type mice. Data are expressed as fold stimulation over control. Values are means ± SE (n = 3). *P < 0.05, **P < 0.01 vs. control. C: Suppressing effect on gluconeogenesis after 1 h exposure of BH4 was detected ranging over 50 μmol/L in hepatocytes isolated from wild-type mice. Values are means ± SE (n = 6). **P < 0.01, ***P < 0.001 vs. control. D: Effect of BH4 on phosphorylation of AMPK and ACC. After 30 min exposure to BH4, both AMPKα and ACC phosphorylation were increased by BH4 dose dependently ranging over 50 μmol/L in hepatocytes isolated from wild-type mice. Data are expressed as fold stimulation over control. Values are means ± SE (n = 3). *P < 0.05, **P < 0.01 vs. control. E: With transfection with AMPKα1 siRNA, protein expression of AMPKα was decreased compared with that of transfection with control siRNA. Values are means ± SE (n = 3). ***P < 0.001 vs. control siRNA. F: Transfected with AMPKα1 siRNA, suppressing effect of BH4 (50 μmol/L) on hepatic glucose production was inhibited. Values are means ± SE (n = 6). ***P < 0.001 vs. values transfected with control siRNA without BH4. G: Compound C (20 μmol/L), an AMPK inhibitor, abolished the suppressing effect of BH4 (50 μmol/L) on gluconeogenesis. Values are means ± SE (n = 6). *P < 0.05 vs. values without BH4 and without compound C.
BH4 suppresses gluconeogenesis and increases AMPKα phosphorylation eNOS dependently in hepatocytes.
Exposure to BH4 in hepatocytes increased NO production and eNOS phosphorylation (Fig. 4A and B). To examine whether BH4 suppresses hepatic gluconeogenesis and activates AMPK in the absence of eNOS, we performed experiments using mouse hepatocytes lacking eNOS. In hepatocytes isolated from eNOS−/− mice, BH4 did not suppress gluconeogenesis (control, 103.9 ± 10.8 nmol/mg protein; 50 μmol/L BH4, 98.5 ± 11.3 nmol/mg protein; 100 μmol/L BH4, 89.1 ± 10.9 nmol/mg protein, P = NS vs. control) (Fig. 4C). BH4 did not alter AMPKα and ACC phosphorylation in hepatocytes lacking eNOS (Fig. 4D). The suppressing effect of BH4 on gluconeogenesis and activation of AMPK also disappeared in the presence of NG-nitro-l-arginine methyl ester, an NOS inhibitor (Supplementary Fig. 4A and B). SNP, an NO donor, has suppressing effects on gluconeogenesis and increases the effects on AMPK activation both in wild-type and eNOS−/− hepatocytes (Supplementary Fig. 5A–D). Immunocytochemical staining of primary cultured hepatocytes from wild-type mice with anti-nitrotyrosine antibody, which detects ONOO−, showed that ONOO− production was not increased by exposure with BH4 or SNP (Supplementary Fig. 5E).
FIG. 4.

Lack of the effect of BH4 on suppression of gluconeogenesis in eNOS−/− mouse hepatocytes. A: BH4 (50 μmol/L) significantly increased NO production in hepatocytes from wild-type mice. SNP (20 μmol/L) was used as positive control. Values are means ± SE (n = 5). *P < 0.05 vs. control. B: BH4 (ranging from 10 to 50 μmol/L) increased eNOS phosphorylation at Ser1177 in hepatocytes from wild-type mice. Values are means ± SE (n = 5). *P < 0.05 vs. control. C: BH4 (ranging from 10 to 100 μmol/L) did not suppress gluconeogenesis after 1 h exposure in hepatocytes from eNOS−/− mice. Values are means ± SE (n = 6). D: After 30 min exposure to BH4 ranging from 10 to 100 μmol/L, AMPKα and ACC phosphorylation were not increased by BH4 in hepatocytes from eNOS−/− mice. Data are expressed as fold stimulation over control. Values are means ± SE (n = 3).
Effect of BH4 on adenine nucleotide content in hepatocytes.
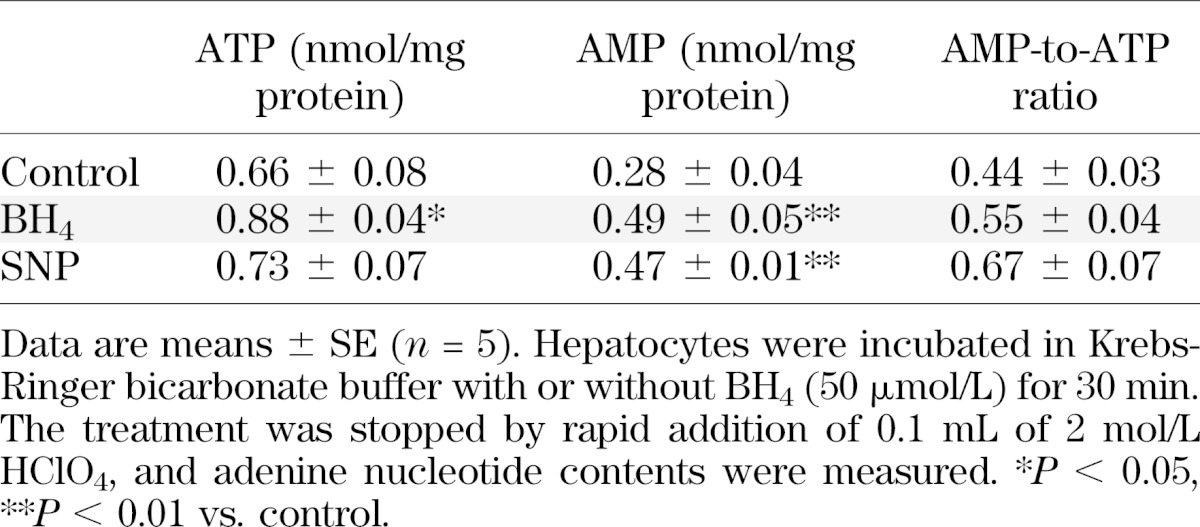
For investigation of the mechanism of AMPK activation by BH4 in hepatocytes, the adenine nucleotide content with exposure of BH4 to hepatocytes was measured. BH4 and SNP significantly increased AMP content in wild-type mouse hepatocytes (Table 1). Unexpectedly, BH4 also significantly increased ATP content. To clarify the mechanism by which BH4 increases AMP content and activates AMPK in hepatocytes, we examined the effect of AMP deaminase (AMPD) on activation of AMPK and suppression of gluconeogenesis by BH4. Although EHNA, a known AMPD inhibitor, activated AMPK and suppressed hepatic gluconeogenesis, BH4 did not have an additive effect on EHNA (Supplementary Fig. 6A and B). These results indicate that inhibition of AMPD, at least in part, contributes to AMP accumulation by BH4 in hepatocytes.
TABLE 1.
Effects of BH4 on ATP, AMP, and AMP-to-ATP ratio in wild-type mouse hepatocytes
Sepiapterin, a BH4 precursor, suppresses gluconeogenesis and increases AMPK activation.
Similarly to BH4, sepiapterin is absorbed in hepatocytes and immediately converted to BH4 via a salvage pathway of BH4 biosynthesis (23). Sepiapterin was found to suppress gluconeogenesis and activate AMPK (Fig. 5A and B). However, these effects were abolished in hepatocytes lacking eNOS (Fig. 5A and B).
FIG. 5.

Effect of sepiapterin, a BH4 precursor, on gluconeogenesis and AMPK activation. A: After 1 h exposure, sepiapterin (50 μmol/L) significantly suppressed gluconeogenesis in hepatocytes isolated from wild-type mice. This effect was not observed in hepatocytes isolated from eNOS−/− mice. Values are means ± SE (n = 6). *P < 0.05 vs. control. B: After 30 min exposure to sepiapterin (50 μmol/L), AMPKα phosphorylation was increased in hepatocytes isolated from wild-type mice. AMPKα phosphorylation was not increased by sepiapterin in hepatocytes isolated from eNOS−/− mice. Data are expressed as fold stimulation over control. Values are means ± SE (n = 3). **P < 0.01 vs. control.
Role of eNOS in in vivo action of BH4 on glucose metabolism.
The lowering effect of BH4 on fasting blood glucose levels disappeared in STZ-induced diabetic eNOS−/− mice (Fig. 6A). The PTT data showed that BH4 did not decrease hepatic glucose production in eNOS−/− mice (Fig. 6B). Similar results were also obtained in sepiapterin administration (Supplementary Fig. 7A and B). We then compared the effects of BH4 on phosphorylation of AMPKα in liver tissues of these diabetic mice. BH4 activated AMPK in both STZ diabetic wild-type mice liver and diabetic Akita mice liver but not in STZ diabetic eNOS−/− mice liver (Fig. 6C and D and Supplementary Fig. 8A). AMPKα phosphorylation was not changed by fasting for 16 h in liver tissues of wild-type mice (Supplementary Fig. 8B).
FIG. 6.

Effects of BH4 in eNOS−/− mice with STZ-induced diabetes. A: No significant difference of fasting blood glucose levels 2 h after intraperitoneal injection of saline with or without BH4 (20 mg/kg) to eNOS−/− mice with STZ-induced diabetes. Values are means ± SE (n = 7). B: PTT to eNOS−/− mice. No effects of BH4 (20 mg/kg) on suppressing hepatic gluconeogenesis were detected in PTT in eNOS−/− mice. Values are means ± SE (n = 6). C: AMPKα phosphorylation in liver of eNOS−/− mice with STZ-induced diabetes was not changed by BH4 administration. Data are expressed as fold stimulation over saline. Values are means ± SE (n = 3). D: AMPKα phosphorylation in liver of wild-type mice with STZ-induced diabetes was significantly increased by BH4 (20 mg/kg) administration. Data are expressed as fold stimulation over saline. Values are means ± SE (n = 3). **P < 0.01 vs. saline.
Effects of BH4 on glucose metabolism and insulin sensitivity in ob/ob mice.
Our PTT data show that the suppressing effect on gluconeogenesis is also confirmed by single administration of BH4 in ob/ob mice (Fig. 7A), while the mRNA expression levels of PEPCK and G6Pase in the liver (Supplementary Fig. 9A and B), fasting and fed blood glucose levels, and IPGTT data were not changed (data not shown). By consecutive administration of BH4 (20 mg/kg) in saline for 10 days to ob/ob mice, fasting blood glucose levels were significantly lowered by 3.9 mmol/L and fed blood glucose levels tended to be decreased compared with those in ob/ob mice treated with saline alone (Fig. 7B and C). Our IPGTT, HOMA-IR, and insulin tolerance test data suggest that consecutive administration of BH4 ameliorates glucose intolerance as well as insulin resistance (Fig. 7D–G). Phosphorylation of AMPKα, ACC, and Akt was increased in liver tissues of BH4-treated ob/ob mice compared with those in saline-treated mice (Fig. 7H and I).
FIG. 7.

Effects of BH4 in ob/ob mice. A: PTT to ob/ob mice with or without single administration of BH4 (20 mg/kg). Values are means ± SE (n = 6). *P < 0.05 vs. the value of saline. B: Fasting blood glucose levels of ob/ob mice treated with BH4 (20 mg/kg/day) for 10 days were significantly decreased compared with those treated without BH4. Values are means ± SE (n = 6). *P < 0.05 vs. the value of saline. C: Fed blood glucose levels in ob/ob mice treated with or without BH4 for 10 days. P = 0.07 vs. the value of saline. Values are means ± SE (n = 6). D and E: IPGTT to ob/ob mice. Blood glucose levels and plasma insulin levels after administration of glucose (1 g/kg i.p.) with or without BH4 for 10 days. Values are means ± SE (n = 6). *P < 0.05, **P < 0.01 vs. without BH4. F: HOMA-IR calculated from fasting blood glucose and insulin levels from IPGTT data in ob/ob mice treated with or without BH4 for 10 days. Values are means ± SE (n = 6). **P < 0.01 vs. the value of saline. G: Insulin tolerance test (ITT) to ob/ob mice treated with or without BH4 for 10 days. Values are means ± SE (n = 6). *P < 0.05 vs. the value of saline. H and I: AMPKα, ACC, and Akt phosphorylation in liver tissues of ob/ob mice was increased by 10 days’ administration of BH4. Data are expressed as fold stimulation over saline. Values are means ± SE (n = 3). *P < 0.05 vs. saline.
DISCUSSION
The current study shows that BH4, known as a cofactor of eNOS, has a glucose-lowering effect in diabetic mice. The BH4-to-BH2 ratio was found to be decreased in various tissues of mice in the diabetic state, indicating deterioration of eNOS bioactivity by eNOS uncoupling. Previous studies have shown that impairment of eNOS function is involved in glucose dysmetabolism and insulin resistance (4,5), which lends support to the notion that alleviation of eNOS dysfunction such as by supplementation of BH4 ameliorates glucose dysmetabolism and insulin resistance. In addition, we found that supplementation of BH4 increased dimerization of eNOS and NO production in the liver of diabetic mice, which strongly suggests alleviation of eNOS dysfunction by recoupling of eNOS. Simultaneously with the restoration of eNOS activity, BH4 elicited a glucose-lowering effect in these mice. No such glucose-lowering effect by BH4 appeared in diabetic mice lacking eNOS. These findings clearly implicate recoupling of eNOS in the glucose-lowering effect of BH4.
We have shown that the liver plays a critical role in the glucose-lowering effect of BH4 through suppression of hepatic gluconeogenesis. It is well-known that BH4 is synthesized mainly in liver (24) and that this is impaired by oxidative stress such as liver cirrhosis and diabetes (25,26). Single administration of BH4 is known to accumulate at higher levels in liver than other tissues including skeletal muscle (24), which also lends support to the view that BH4 readily elevates BH4-to-BH2 ratio and regulates glucose metabolism in the liver.
We then investigated the molecular mechanism of suppression of hepatic gluconeogenesis by BH4 using isolated mouse hepatocytes. BH4 acts directly on hepatocytes and suppresses hepatic gluconeogenesis eNOS dependently. Several studies reported that eNOS is found in hepatic sinusoidal and venous endothelial cells and not in hepatocytes (27,28), whereas other studies claim detection of eNOS in hepatocytes (29,30). We confirmed that eNOS is expressed in hepatocytes, which suggests that intrahepatocellular eNOS is essential for the effect of BH4 in suppression of hepatic gluconeogenesis. In addition, BH4 activated AMPK, and the suppressing effect of BH4 on gluconeogenesis disappeared by siRNA silencing of AMPKα1 subunits in hepatocytes, indicating that AMPK is involved in the suppressing effect of BH4 on hepatic gluconeogenesis. AMPK activation by BH4 was not observed in eNOS−/− mouse hepatocytes or in the presence of NOS inhibitor, suggesting that eNOS acts upstream of AMPK activation in suppression of hepatic gluconeogenesis by BH4. AMPK is a Ser/Thr kinase that acts as an energy sensor and is activated by an increase in the AMP-to-ATP ratio and/or AMP in response to a variety of metabolic stresses, such as hypoxia, ischemia, and exercise (31,32). In our data, BH4 significantly increased AMP content and tended to increase the AMP-to-ATP ratio. It is known that inhibition of AMPD increases AMP in isolated hepatocytes (33). Recently, Ouyang et al. (34) reported that inhibition of AMPD might be involved in increased production of AMP and activation of AMPK by metformin. In the current study, the AMPD inhibitor EHNA was found to activate AMPK, but BH4 did not elicit an additional effect on AMPK activation in the presence of EHNA, suggesting that AMPD might be inhibited by BH4 in hepatocytes. Interestingly, BH4 significantly increased ATP content along with the increase in AMP. This effect was not found in exposure to other potent AMPK activators, as previously reported (35). The reason why BH4 increases ATP content is unclear, but BH4 is known to work as an antioxidant (36). It has been reported that BH4 preserves ATP content and has a cytoprotective effect from hypoxia on neuronal cells (37). BH4 might thus prevent cytotoxic damage from reactive oxygen species/reactive nitrogen species (RNS) as a scavenger, keeping ATP content higher than in the absence of BH4. We therefore cannot exclude the possibility that BH4 acts as a reactive oxygen species/RNS scavenger in ameliorating glucose dysmetabolism, but such an effect would be limited in terms of suppressing hepatic gluconeogenesis because the effect of BH4 was not observed in mice lacking eNOS. Previous studies found that NO has an activating effect on AMPK (38,39). Also, in our results SNP, an NO donor, activated AMPK in hepatocytes just as BH4 does. Regarding the mechanism of AMPK activation by BH4 via eNOS, it is possible that NO itself generated by eNOS activates AMPK; another possibility is that the RNS peroxynitrite (ONOO−), an adduct of NO with superoxide, works intermediately as the activator of AMPK by BH4 (19,40). The involvement of RNS on AMPK activation by BH4 was not suggested by our present data.
Our data using ob/ob mice, a mouse model of insulin resistance, suggest that the primary physiological action of BH4 is a suppressing effect of hepatic gluconeogenesis. In addition to this effect, consecutive administration of BH4 ameliorated glucose intolerance as well as insulin resistance. A possible mechanism of these additive effects of BH4 is induction by the subsequent downstream targets of AMPK activated by BH4 such as metformin, which are known to have insulin-sensitizing effects, e.g., by modulating carbohydrate and lipid metabolism via the downstream signals of AMPK (41). It is generally known that increase in Akt phosphorylation represents an amelioration of hepatic insulin resistance. This may be applicable to the effect of BH4, while it raises the possibility that Akt-dependent signaling is involved in the suppressing effect of BH4 on hepatic gluconeogenesis in ob/ob mice. Another possible mechanism of BH4 ameliorating insulin resistance would be via a direct effect of BH4 on endothelial cells. Similar to several NO donors and NO-moderating compounds (42), BH4 might also exert an insulin-sensitizing effect by augmenting the delivery of insulin and glucose to skeletal muscle via capillary recruitment. Since the role of eNOS in vivo was assessed using global eNOS−/− mice, it is difficult to exclude the possibility of indirect effects of eNOS on the liver. Therefore, limitations of the current study must be considered. Further investigations, e.g., by using liver-specific eNOS−/− mice, are required to elucidate the pleiotropic effects of BH4 in lowering blood glucose levels.
The glucose-lowering effect of BH4 by single administration intraperitoneally on fasting blood glucose levels in STZ diabetic mice was similar to that of metformin (250 mg/kg). The dose of metformin that we used was adjusted to previous studies in mice (43) and is more than fivefold higher than that in clinical use for type 2 diabetic patients (44). We demonstrate here the lowering effects of BH4 on blood glucose levels using a dosage similar to that of BH4 used in patients with phenylketonuria as a cofactor of phenylalanine hydroxylase (45).
Numerous clinical trials have been performed on the effect of BH4 as a cofactor of eNOS on endothelial dysfunction in a variety of vascular diseases including coronary artery disease (15). While many of the results are disappointing (46), BH4 remains a viable candidate for clinical use if the design of the various trials is reconsidered. Several of the studies reported that BH4 levels are plainly decreased and that uncoupled eNOS is found in the diabetic state and not in nondiabetic states (47). Moreover, nondiabetic patients were included in most of the clinical trials (46); those trials should be performed in patients with diabetes. The current study, furthermore, clarifies a novel concept of the relationship between BH4 and glucose metabolism and insulin resistance that suggests a new approach to the prevention of macrovascular complications of diabetes induced by endothelial dysfunction as well as amelioration of the disease itself.
In conclusion, BH4 has a glucose-lowering effect by suppressing hepatic gluconeogenesis in an eNOS-dependent manner and ameliorates glucose intolerance as well as insulin resistance in diabetic mice, suggesting that BH4 has potential in the treatment of type 2 diabetes.
ACKNOWLEDGMENTS
This study was supported by Scientific Research grants; a grant for Leading Project for Biosimulation from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; a grant from Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Cooperation; a grant from the Ministry of Health, Labor, and Welfare, Japan; and a grant from Kyoto University Global Center of Excellence (COE) Program “Center for Frontier Medicine.”
No potential conflicts of interest relevant to this article were reported.
A.A. and Y.F. researched data, contributed to discussion, and wrote, reviewed, and edited the manuscript. A.Ob. and A.Oh. researched data and contributed to discussion. T.F., Y.S., M.O., Y.N., S.F., and M.H. contributed to discussion. H.H. researched data and contributed to discussion. N.I. contributed to discussion and wrote, reviewed, and edited the manuscript. N.I. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011.
The authors thank Ryo Tanaka and Miho Nishimura for their efforts in collaboration in the Student Research Program, Department of Biosciences, Teikyo University of Science and Technology.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1242/-/DC1.
REFERENCES
- 1.Triggle CR, Ding H. A review of endothelial dysfunction in diabetes: a focus on the contribution of a dysfunctional eNOS. J Am Soc Hypertens 2010;4:102–115 [DOI] [PubMed] [Google Scholar]
- 2.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab 2009;20:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 2006;113:1888–1904 [DOI] [PubMed] [Google Scholar]
- 4.Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342–345 [DOI] [PubMed] [Google Scholar]
- 5.Shankar RR, Wu Y, Shen HQ, et al. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 2000;49:684–687 [DOI] [PubMed] [Google Scholar]
- 6.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem 2001;276:14533–14536 [DOI] [PubMed] [Google Scholar]
- 7.Vásquez-Vivar J, Kalyanaraman B, Martásek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA 1998;95:9220–9225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crabtree MJ, Channon KM. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 2011;25:81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003;111:1201–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meininger CJ, Marinos RS, Hatakeyama K, et al. Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J 2000;349:353–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Wu Y, Song P, et al. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 2007;116:944–953 [DOI] [PubMed] [Google Scholar]
- 12.Meininger CJ, Cai S, Parker JL, et al. GTP cyclohydrolase I gene transfer reverses tetrahydrobiopterin deficiency and increases nitric oxide synthesis in endothelial cells and isolated vessels from diabetic rats. FASEB J 2004;18:1900–1902 [DOI] [PubMed] [Google Scholar]
- 13.Ding QF, Hayashi T, Packiasamy AR, et al. The effect of high glucose on NO and O2- through endothelial GTPCH1 and NADPH oxidase. Life Sci 2004;75:3185–3194 [DOI] [PubMed] [Google Scholar]
- 14.Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab 2012;302:E481–E495 [DOI] [PubMed] [Google Scholar]
- 15.Katusic ZS, d’Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci 2009;30:48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wajngot A, Chandramouli V, Schumann WC, et al. Quantitative contributions of gluconeogenesis to glucose production during fasting in type 2 diabetes mellitus. Metabolism 2001;50:47–52 [DOI] [PubMed] [Google Scholar]
- 17.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 2011;14:9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita Y, Hosokawa M, Fujimoto S, et al. Metformin suppresses hepatic gluconeogenesis and lowers fasting blood glucose levels through reactive nitrogen species in mice. Diabetologia 2010;53:1472–1481 [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto S, Mukai E, Hamamoto Y, et al. Prior exposure to high glucose augments depolarization-induced insulin release by mitigating the decline of ATP level in rat islets. Endocrinology 2002;143:213–221 [DOI] [PubMed] [Google Scholar]
- 21.Ogura M, Nakamura Y, Tanaka D, et al. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem Biophys Res Commun 2010;393:73–78 [DOI] [PubMed] [Google Scholar]
- 22.Sawabe K, Wakasugi KO, Hasegawa H. Tetrahydrobiopterin uptake in supplemental administration: elevation of tissue tetrahydrobiopterin in mice following uptake of the exogenously oxidized product 7,8-dihydrobiopterin and subsequent reduction by an anti-folate-sensitive process. J Pharmacol Sci 2004;96:124–133 [DOI] [PubMed] [Google Scholar]
- 23.Sawabe K, Suetake Y, Nakanishi N, et al. Cellular accumulation of tetrahydrobiopterin following its administration is mediated by two different processes; direct uptake and indirect uptake mediated by a methotrexate-sensitive process. Mol Genet Metab 2005;86(Suppl. 1):S133–S138 [DOI] [PubMed] [Google Scholar]
- 24.Hoshiga M, Hatakeyama K, Watanabe M, et al. Autoradiographic distribution of [14C]tetrahydrobiopterin and its developmental change in mice. J Pharmacol Exp Ther 1993;267:971–978 [PubMed] [Google Scholar]
- 25.Matei V, Rodríguez-Vilarrupla A, Deulofeu R, et al. Three-day tetrahydrobiopterin therapy increases in vivo hepatic NOS activity and reduces portal pressure in CCl4 cirrhotic rats. J Hepatol 2008;49:192–197 [DOI] [PubMed] [Google Scholar]
- 26.Elrod JW, Duranski MR, Langston W, et al. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: a role for eNOS uncoupling. Circ Res 2006;99:78–85 [DOI] [PubMed] [Google Scholar]
- 27.Shah V, Cao S, Hendrickson H, et al. Regulation of hepatic eNOS by caveolin and calmodulin after bile duct ligation in rats. Am J Physiol Gastrointest Liver Physiol 2001;280:G1209–G1216 [DOI] [PubMed] [Google Scholar]
- 28.Wei CL, Khoo HE, Lee KH, et al. Differential expression and localization of nitric oxide synthases in cirrhotic livers of bile duct-ligated rats. Nitric Oxide 2002;7:91–102 [DOI] [PubMed] [Google Scholar]
- 29.McNaughton L, Puttagunta L, Martinez-Cuesta MA, et al. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc Natl Acad Sci USA 2002;99:17161–17166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mei Y, Thevananther S. Endothelial nitric oxide synthase is a key mediator of hepatocyte proliferation in response to partial hepatectomy in mice. Hepatology 2011;54:1777–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 2009;9:407–416 [DOI] [PubMed] [Google Scholar]
- 32.Hardie DG. The AMP-activated protein kinase pathway—new players upstream and downstream. J Cell Sci 2004;117:5479–5487 [DOI] [PubMed] [Google Scholar]
- 33.Carabaza A, Ricart MD, Mor A, et al. Role of AMP on the activation of glycogen synthase and phosphorylase by adenosine, fructose, and glutamine in rat hepatocytes. J Biol Chem 1990;265:2724–2732 [PubMed] [Google Scholar]
- 34.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 2011;286:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta 2010;1804:581–589 [DOI] [PubMed]
- 36.Kojima S, Ona S, Iizuka I, et al. Antioxidative activity of 5,6,7,8-tetrahydrobiopterin and its inhibitory effect on paraquat-induced cell toxicity in cultured rat hepatocytes. Free Radic Res 1995;23:419–430 [DOI] [PubMed] [Google Scholar]
- 37.Delgado-Esteban M, Almeida A, Medina JM. Tetrahydrobiopterin deficiency increases neuronal vulnerability to hypoxia. J Neurochem 2002;82:1148–1159 [DOI] [PubMed] [Google Scholar]
- 38.Kang KT, Sullivan JC, Spradley FT, et al. Antihypertensive therapy increases tetrahydrobiopterin levels and NO/cGMP signaling in small arteries of angiotensin II-infused hypertensive rats. Am J Physiol Heart Circ Physiol 2011;300:H718–H724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higaki Y, Hirshman MF, Fujii N, et al. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 2001;50:241–247 [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Xie Z, Dong Y, et al. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem 2008;283:27452–27461 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Viollet B, Guigas B, Leclerc J, et al. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cook S, Scherrer U. Insulin resistance, a new target for nitric oxide-delivery drugs. Fundam Clin Pharmacol 2002;16:441–453 [DOI] [PubMed] [Google Scholar]
- 43.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005;310:1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inzucchi SE, Maggs DG, Spollett GR, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 1998;338:867–872 [DOI] [PubMed] [Google Scholar]
- 45.Blau N. Defining tetrahydrobiopterin (BH4)-responsiveness in PKU. J Inherit Metab Dis 2008;31:2–3 [DOI] [PubMed] [Google Scholar]
- 46.Moens AL, Kietadisorn R, Lin JY, et al. Targeting endothelial and myocardial dysfunction with tetrahydrobiopterin. J Mol Cell Cardiol 2011;51:559–563 [DOI] [PubMed] [Google Scholar]
- 47.Heitzer T, Krohn K, Albers S, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 2000;43:1435–1438 [DOI] [PubMed] [Google Scholar]