

Abstract
To meet the demands for milk calcium, the lactating mother adjusts systemic calcium and bone metabolism by increasing dietary calcium intake, increasing bone resorption, and reducing renal calcium excretion. As part of this adaptation, the lactating mammary gland secretes PTHrP into the maternal circulation to increase bone turnover and mobilize skeletal calcium stores. Previous data have suggested that, during lactation, the breast relies on the calcium-sensing receptor (CaSR) to coordinate PTHrP secretion and milk calcium transport with calcium availability. To test this idea genetically, we bred BLG-Cre mice with CaSR-floxed mice to ablate the CaSR specifically from mammary epithelial cells only at the onset of lactation (CaSR-cKO mice). Loss of the CaSR in the lactating mammary gland did not disrupt alveolar differentiation or milk production. However, it did increase the secretion of PTHrP into milk and decreased the transport of calcium from the circulation into milk. CaSR-cKO mice did not show accelerated bone resorption, but they did have a decrease in bone formation. Loss of the mammary gland CaSR resulted in hypercalcemia, decreased PTH secretion, and increased renal calcium excretion in lactating mothers. Finally, loss of the mammary gland CaSR resulted in decreased calcium accrual by suckling neonates, likely due to the combination of increased milk PTHrP and decreased milk calcium. These results demonstrate that the mammary gland CaSR coordinates maternal bone and calcium metabolism, calcium transport into milk, and neonatal calcium accrual during lactation.
Milk provides all the nutrients required for neonatal growth, including calcium. To meet the demands of milk production, lactating mothers increase gastrointestinal calcium intake, decrease renal calcium excretion, and mobilize skeletal calcium stores (1–4). Bone loss during lactation is impressive in both its magnitude and rapidity. Lactating mice lose up to 25%–30% of their bone mass over 12–15 days of lactation, whereas bone mineral density (BMD) declines by 5%–8% in women nursing full time for 6 months. Lactational bone loss is associated with elevated rates of bone formation and bone resorption (2, 4–6). The degree of bone loss correlates with the amount of milk produced and is, in part, the result of suckling-induced, hypothalamic suppression of circulating estrogen combined with elevated circulating PTHrP secreted from the lactating breast (4–7). However, experimentally lowering estrogen and increasing PTHrP does not fully reproduce the degree of bone loss observed during lactation, so other factors must also contribute to the skeletal response (8).
The calcium sensing receptor (CaSR) is a 7-transmembrane-spanning, G protein-coupled receptor that signals in response to changes in extracellular calcium and other organic and inorganic cations (9, 10). It is expressed in the parathyroid glands, kidneys, and bones and is integral to the regulation of systemic calcium metabolism (10, 11). Genetic experiments have demonstrated that this receptor is responsible for calcium sensing by the parathyroid glands and kidneys and is required for the maintenance of circulating calcium and PTH levels as well as proper renal calcium handling (12–17). In addition, recent experiments have underscored the function of the CaSR in directly regulating osteoblast and osteoclast function (18–20). The CaSR is also expressed in many other tissues, in which it participates in the regulation of ion transport and the control of cell proliferation and differentiation (10, 11). Mirroring its effects on PTH secretion by parathyroid cells, the CaSR has been shown to regulate the production of PTHrP by a variety of cells (10, 11).
One site of CaSR expression is the lactating breast (21–23). Experiments from our laboratory have suggested that activation of the CaSR on mammary epithelial cells (MECs) reduces PTHrP production (22, 24). As noted previously, PTHrP secretion by the breast contributes to increased bone resorption and bone loss during lactation (5, 7, 25). In addition, activation of the CaSR stimulates the transport of calcium into milk by increasing the activity of the plasma membrane calcium ATPase 2b (PMCA2) calcium pump (22–24). The CaSR is expressed on the basolateral membrane of lactating MEC, whereas PMCA2 is expressed on the apical surface of these same cells (22, 23). These data have led us to propose a working model whereby, during lactation, the breast becomes a calcium-sensing organ that contributes to the regulation of systemic calcium and bone metabolism (4). By secreting PTHrP, the breast may help to mobilize skeletal calcium for milk production. We hypothesize that the delivery of calcium to the breast, in turn, stimulates calcium uptake into milk and inhibits further PTHrP secretion, defining a classical negative feedback loop between bone and breast.
The above model is based on pharmacological data using calcium and calcimimetic compounds in mice and data derived from heterozygous global CaSR-knockout mice (22, 24). Given the ubiquitous expression of the CaSR and its importance in regulating calcium homeostasis, we could not rule out confounding effects of abnormal systemic calcium levels or off-target effects on other organs in these previous studies. Therefore, to firmly establish a role for the breast CaSR in modulating systemic calcium and skeletal homeostasis during lactation, we developed a genetic mouse model in which we could disrupt the Casr gene specifically in MECs at the onset of lactation. In this study, we demonstrate that mammary-specific disruption of the CaSR increases PTHrP production but impairs MEC calcium transport. These mice demonstrate alterations in systemic calcium metabolism but not accelerated bone resorption. Finally, increased milk PTHrP causes a reduction in neonatal ash calcium content.
Materials and Methods
Animals
Floxed CaSR (CaSRlox/lox) mice were previously reported and were maintained on a C3H background (18, 19). β-Lactoglobulin-Cre (BLG-Cre) mice, a gift from Christine Watson (Cambridge University, Cambridge, United Kingdom), express Cre only in MECs during late pregnancy and lactation (5, 26) and were maintained on a C57B6 background. PTHrPlox/lox and PTHrPlox/− mice (5) were maintained on a 129 background. BLG-Cre;CaSRlox/lox (CaSR-cKO) mice and control littermates (CaSRlox/lox) were generated from crosses between BLG-Cre;CaSRlox/+ and CaSRlox/lox and were therefore of mixed C3H and C57B6 background. Likewise, BLG-Cre; PTHrPlox/−, PTHrPlox/− and PTHrPlox/lox mice were littermates and were of mixed C57B6 and 129 backgrounds. The CaSR−/−; PTH−/− (C−P−), PTH−/− (C+P−), and control wild-type (WT; C+P+) mice were the product of a mixed genetic background including C57B6, 129/svj, and 129/SvEv strains and have been reported previously (13). Eight-week-old female mice were mated with CD1 males obtained from Charles River (Wilmington, Massachusetts). After the mice became pregnant, males were removed and lactating females were studied as detailed in Results. Analyses were done on days 2 and 12 of lactation, unless otherwise stated.
Real-time PCR
RNA was isolated using TRIzol (Invitrogen, Carlsbad, California) and purified on RNeasy minicolumns (QIAGEN, Valencia, California), with on-column deoxyribonuclease digestion, as per the manufacturers' instructions. First-strand cDNA was reverse transcribed using the high-capacity cDNA synthesis kit (Applied Biosystems, Foster City, California). Taq-Man assays (Applied Biosystems) for CaSR (Casr) (Mm00443375_m1 or Mm00443377_m1), PTHrP (Pthlh) (Mm00436057_m1), and β-actin (Actb) (Mm00607939_s1) were used with TaqMan universal master mix (Applied Biosystems). SYBR-Green-based assays were performed for keratin 18 (Krt18), whey acidic protein (Wap), sodium/phosphate transporter type 2b (Slc34a2), sodium/potassium/chloride transporter type 1 (Slc12a2), aquaporin 5 (Aqp5), and γ-casein (Csn1s2a), as previously described (24). Primer sequences are listed in Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org. Relative expression levels were determined using the comparative threshold cycle method (24, 27).
Western blot analysis
Western blots used membrane fractions from lactating mammary glands as described previously (23). Twenty-five- to 30-μg membrane proteins were subjected to SDS-PAGE and immunoblotting using standard methods. Anti-PMCA2 and anti-β-actin antibodies were from Thermo Scientific (Waltham, Massachusetts) and Cell Signaling (Beverly, Massachusetts), respectively. The anti-CaSR antibodies were generated by the laboratory of one of the investigators (W.C.).
Dual-energy x-ray absorptiometry (DEXA)
DEXA was performed on days 2 and 11 of lactation, using a Lunar PIXImus (GE Medical Systems, Lunar Division, Madison, Wisconsin). Mice were anesthetized with 50 mg/kg ketamine (Fort Dodge Animal Health, Fort Dodge, Iowa) and 10 mg/kg xylazine (Lloyd, Shenandoah, Iowa), by ip injection.
Microcomputed tomographic (CT) imaging
Trabecular bone within the proximal tibia and the third lumbar vertebra (L3) were evaluated, ex vivo, using a μCT35 (Scanco Medical AG, Bruttisellen, Switzerland). Specimens were scanned in 70% ethanol at 55 kVp (145 μA), using 1000 cone-beam projections, an integration time of 500 milliseconds, and a 6-μm isometric voxel size. A gray-scale (1–1000) density threshold of 350 was used for trabecular morphometry. Cortical bone morphometry was averaged from 232 serial cross-sectional images (1.4 mm) centered at the longitudinal midpoint of the tibia, applying a gray-scale (1–1000) density threshold of 350. Cortical bone was further analyzed by segmentation into low-mineral-density bone (LMCt), using a lower threshold of 350 and an upper threshold of 630, and high-mineral-density bone (HMCt), by applying a lower threshold of 630 and an upper threshold of 1000.
Histology and histomorphometry
Hematoxylin-and-eosin staining was performed on sections (5 μm) of formalin-fixed, paraffin-embedded tissue. Routine bone histomorphometry was performed on 4-μm, toluidine blue-stained, methylmethacrylate-embedded, nondecalcified sections of the third lumbar vertebra as described (5, 6). Static and dynamic histomorphometric analyses were performed using the Osteomeasure system (OsteoMetrics, Decatur, Georgia).
Biochemical measurements
Blood was collected on day 2 of lactation from the retroorbital venous plexus into heparinized capillary tubes. On day 12 of lactation, blood was collected by cardiac puncture into syringes containing 28 U heparin (Becton Dickinson, Franklin Lakes, New Jersey) and added to tubes containing a protease inhibitor cocktail (Roche, Indianapolis, Indiana) (200 μg/mL aprotinin, 30 μg/mL leupeptin, 7 μg/mL pepstatin, 6 mM EDTA). Plasma was stored at −70°C. Milk was collected 10 minutes after ip administration of 0.1 mIU of oxytocin (Sigma-Aldrich, St Louis, Missouri) on day 12 of lactation (5, 22). Urine was collected on day 12 of lactation and urinary cAMP measured in triplicate using a double-antibody RIA (sensitivity 0.05 pM) (Enzo Life Sciences, Farmingdale, New York) and corrected for creatinine, determined by the picric acid method (BioAssay Systems, Hayward, California). Plasma PTHrP was measured using an immunoradiometric assay (DSL-8100; Beckman Coulter, Webster, Texas), in which we substituted custom rabbit anti-PTHrP (1–36) antibody as capture antibody. The standard curve was constructed using commercial standards diluted in mouse plasma that had been incubated overnight at room temperature to degrade endogenous PTHrP. Milk PTHrP was measured using the unmodified DSL-8100 immunoradiometric assay. Serum cross-linked C-telopeptides of type I collagen (CTX) were measured in duplicate with the RatLaps immunoassay (Nordic Bioscience Diagnostics, Atlanta, Georgia). Osteocalcin was measured using an established RIA (5, 8, 22, 24). Milk was diluted 1:100 in distilled water and calcium measured using the QuantiChrom calcium assay kit (BioAssay Systems, Hayward, California). Pups were weighed prior to cremation at 700°C. Ash was dissolved in 0.1 N hydrochloric acid and calcium was measured by atomic absorption spectroscopy or using the QuantiChrom calcium assay.
Calcium injections
Mice were fasted for 4–5 hours and then given consecutive, every-other-day ip injections of 500, 750, or 1000 μM calcium gluconate. Calcium injections were administered to virgin mice or to lactating mice (on days 7, 9, and 11 of lactation). Blood was collected, by eye bleed, 30 minutes before and after calcium injections. Calcium levels were determined using the QuantiChrom calcium assay.
Statistical methods
Statistical analyses were performed using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, California, www.graphpad.com). To compare 2 groups, the unpaired, 2-tailed, Student's t test was used with a confidence level of 95%. For quantitative real-time PCR analyses, the 1-sample t test was used to test the difference in fold expression levels from a theoretical value of 1. When comparing 3 or more groups, we used 1-way ANOVA with the Tukey multiple comparison test. For all analyses, values of P < .05 were considered statistically significant.
Results
Disruption of CaSR expression in the mammary glands of BLG-Cre;CaSRlox/lox mice
Generalized disruption of the Casr gene by homologous recombination causes neonatal death, precluding an examination of the effects of the CaSR in the lactating mammary gland (12). Therefore, we used a conditional knockout strategy to target the Casr gene only in MECs during late pregnancy and lactation. BLG-Cre mice use the ovine β-lactoglobulin gene promoter to drive the expression of Cre recombinase specifically within MECs, beginning in the latter half of pregnancy and during lactation (5, 26). The Casr gene was expressed equally well in the mammary glands from virgin, CaSR-cKO and virgin, CaSRlox/lox (control) mice before the BLG-Cre transgene was expressed (Supplemental Figure 1). However, during lactation, when the BLG-Cre transgene was robustly expressed, CaSR mRNA levels were dramatically reduced in mammary glands from CaSR-cKO mice compared with control mice (Figure 1A). CaSR protein was undetectable in membrane preparations from lactating CaSR-cKO glands compared with lactating control glands (Figure 1B). Interestingly, Western analysis performed under nonreducing conditions suggested that mammary gland membranes contain higher molecular weight forms of the CaSR than kidney membranes from the same mice. Similar results have been seen in transfected cells and other tissues and are thought to represent multimeric forms of the CaSR (28–32). These data demonstrate that our gene knockout strategy successfully disrupted CaSR mRNA and protein expression in the lactating mammary gland.
Figure 1.

Loss of CaSR mRNA and protein during lactation in CaSR-cKO mice. A, Quantitative PCR for CaSR mRNA expression in control (CaSRlox/lox) and CaSR-cKO (BLG-Cre;CaSRlox/lox) mammary glands. Note the almost complete loss of CaSR expression in lactating mammary glands in the cKO mice. Bars represent the mean and SEM of 7 mice for each genotype. B, Western blots for CaSR expression in membrane extracts from kidneys and CaSR-cKO and control mammary glands run under nonreducing conditions. The predominant forms of the CaSR in mammary gland membranes appear to be multimers with molecular masses of approximately 160 and 250 kDa. There was no immunoreactive CaSR protein detected in membranes from cKO mammary glands. Kidney expressed CaSR isoforms predominantly of approximately 65 and of 140 kDa.
Loss of CaSR expression did not impair mammary alveolar development or prevent lactation. The histological appearance of mammary glands from lactating CaSR-cKO and control mice was comparable (Supplemental Figure 2, A and B). Furthermore, lactating CaSR-cKO glands expressed normal levels of secretory differentiation markers, demonstrating that MEC differentiation occurred normally (Supplemental Figure 2C). Finally, pups suckling CaSR-cKO dams grew at a rate similar to pups suckling control dams, demonstrating adequate milk production (Supplemental Figure 2D). Together these data show that loss of the CaSR from MECs does not impair overall lactation.
Disruption of the CaSR during lactation increases PTHrP production
Previous studies in WT and global CaSR heterozygous null mice had suggested that activation of the CaSR in lactating MECs suppressed PTHrP mRNA expression and PTHrP secretion (22, 24). Consistent with those observations, PTHrP mRNA levels were increased approximately 6-fold in CaSR-cKO vs control mammary glands (Figure 2A), and there was an approximate 4-fold increase in the amount of PTHrP secreted into the milk of CaSR-cKO mice (Figure 2B). Interestingly, despite the significant increase in milk PTHrP, we did not detect any significant increase in circulating immunoreactive PTHrP in CaSR-cKO mice. As shown in Figure 2C, plasma PTHrP levels in the CaSR-cKO mice were not statistically significantly different from levels measured in the control mice. Given that the levels of circulating PTHrP in both control and CaSR-cKO mice were at the threshold of sensitivity of the current PTHrP immunoassay, we wondered whether the failure to see an increase in PTHrP was a reflection of the assay's limitations at low PTHrP concentrations. Therefore, we also measured urinary cAMP levels, which have previously been used as an indirect measurement of circulating PTH and PTHrP bioactivity (33). As shown in Figure 2D, despite their decreased circulating PTH levels (see Figure 5), urinary cAMP levels were elevated in CaSR-cKO mice compared with controls. These data demonstrate that the CaSR regulates PTHrP gene expression by MECs and PTHrP secretion from MECs into milk. In addition, they suggest that PTHrP bioactivity was increased in the circulation, even though there was no measured increase in PTHrP plasma immunoreactivity.
Figure 2.

Loss of the CaSR increases PTHrP production by the mammary gland. A, PTHrP mRNA levels in lactating mammary glands as measured by quantitative PCR. Bars represent the mean ± SEM for 6 mice of each genotype. B, Immunoreactive PTHrP levels measured in milk harvested from control and CaSR-cKO dams. Bars represent the mean ± SEM for 5 mice of each genotype. C, Circulating plasma immunoreactive PTHrP levels in control and CaSR-cKO mice on day 12 of lactation. Bars represent the mean ± SEM for 6 mice of each genotype. D, Urinary cAMP concentrations corrected for creatinine excretion in control and CaSR-cKO mice on day 12 of lactation. Bars represent the mean ± SEM for 13 mice of each genotype.
Figure 5.

Loss of the mammary CaSR alters maternal calcium homeostasis during lactation. A, Serum calcium in control and CaSR-cKO mice on days 2 and 11 of lactation. The CaSR-cKO mice are transiently hypercalcemic in early lactation. Bars represent the mean ± SEM for 7 mice of each genotype. B, Circulating PTH levels in control and CaSR-cKO mice on day 12 of lactation. PTH levels are suppressed in lactating cKO mice. Bars represent the mean ± SEM for 7 mice of each genotype. C, Urinary calcium levels corrected for creatinine in control and CaSR-cKO mice on day 12 of lactation. Urinary calcium excretion is increased in lactating cKO mice. Bars represent the mean ± SEM for 13 mice of each genotype. D, 1,25 Dihydroxyvitamin D levels in control and CaSR-cKO mice on day 12 of lactation. Vitamin D levels were unchanged in lactating CaSR-cKO mice. Bars represent the mean ± SEM for 6 mice of each genotype. E, Serum calcium in rescued CaSR−/− mice on day 12 of lactation. C+P+ represents WT controls; C+P− represents PTH−/− mice; and C−P− represents CaSR−/−;PTH−/− mice. Note the persistent hypercalcemia in the C−P− cohort. Bars represent the mean ± SEM for 5 mice of each genotype. F, Urinary calcium corrected for creatinine in rescued CaSR−/− mice on day 12 of lactation. Note that relative to C+P+ mice, urinary calcium levels are increased in C+P− mice but not in C−P− mice. Bars represent the mean ± SEM for 5 mice of each genotype.
Loss of the mammary CaSR alters bone mineralization but not bone loss during lactation
Our hypothesis predicted that increased mammary PTHrP production would accelerate bone resorption and magnify bone loss. Therefore, we measured longitudinal changes in BMD by DEXA and bone mass and microarchitecture by micro-CT. As seen in Supplemental Figure 3, BMD in control mice declined significantly over 9 days of lactation as expected. However, despite the increased production of PTHrP by the mammary glands, the rate of decline in BMD by DEXA in CaSR-cKO mothers was the same as in control mice at all sites. Similar results were obtained comparing bone mass by micro-CT on day 12 of lactation. As shown in Figure 3, A–D, trabecular bone volume/total volume (BV/TV) and trabecular number were comparable between CaSR-cKO and control mice, although trabecular thickness was decreased in the CaSR-cKO dams (Supplemental Table 2). In addition, BMD as measured by micro-CT was lower in CaSR-cKO mice. Figure 3, E–P, and Supplemental Table 3 demonstrate micro-CT measurements for cortical bone. Overall cortical BV/TV was identical in CaSR-cKO and control mice although, as with trabecular bone, BMD was significantly lower in CaSR-cKO mice.
Figure 3.

Micro-CT results for CaSR-cKO and control mice. A–D, Trabecular micro-CT results. A, Results for bone mass as measured by BV/TV. There is no difference between the CaSR-cKO and control mice. B, Results for BMD in CaSR-cKO and control mice. Note that BMD is significantly reduced in the CaSR-cKO bones. C and D, Three-dimensional reconstructions of trabecular bone from the proximal tibia of control and CaSR-cKO lactating mice. E–P, Cortical micro-CT results. E, Results for overall cortical bone mass (micro-CT threshold 350–1000) as measured by BV/TV. There is no difference between the CaSR-cKO and control mice. F, Results for cortical BMD in CaSR-cKO and control mice. Note that BMD is significantly reduced in the CaSR-cKO bones. G and H, Three-dimensional reconstructions of representative cortical bone from the tibia of control and CaSR-cKO lactating mice. I, Results for low-mineral-density (micro-CT threshold 350–630) cortical bone mass (Bv/TV). There is an increase in low-mineral-density bone in the CaSR-cKO mice. J, Results for BMD of low-density bone in CaSR-cKO and control mice. BMD is significantly reduced in the CaSR-cKO bones. K and L, Three-dimensional reconstructions of representative low-mineral-density bone in the tibial cortex of control and CaSR-cKO lactating mice. In controls the low-mineral-density bone is located mostly at the endosteal and periosteal surfaces, but in the CaSR-cKO mice, it is more evenly distributed throughout the entire cortex. M, Results for high-mineral-density (micro-CT threshold 630–1000) cortical bone mass (Bv/TV). There is a decrease in high-mineral-density bone in the CaSR-cKO mice. N, Results for BMD of high-mineral-density bone in CaSR-cKO and control mice. BMD is significantly reduced in the CaSR-cKO bones. O and P, Three-dimensional reconstructions of representative high-density bone in the tibial cortex of control and CaSR-cKO lactating mice. There is a clear and uniform reduction in high-mineral-density bone throughout the cortex. In all instances, values represent the means ± SEM for 12 controls and 9 CaSR-cKO mice. *, P < .05; **, P < .01; ***, P < .001.
To further examine the apparent decrease in bone mineral in the CaSR-cKO mice, we divided the cortical bone compartment into HMCt bone, defined by a micro-CT threshold between 630 and 1000, and LMCt bone, defined by a micro-CT threshold between 350 and 630. As shown in Figure 3, relative to controls, the CaSR-cKO mice had an increased amount of LMCt and a decreased amount of HMCt. However, BMD was decreased in the CaSR-cKO bones, regardless of threshold. Therefore, the hydroxyapatite content of cortical bone appears to be uniformly reduced in the lactating CaSR-cKO mice as compared with lactating controls, although the absolute changes are relatively small.
We further measured static and dynamic bone parameters by histomorphometry (Table 1). Trabecular BV/TV was lower in the CaSR-cKO mice as compared with controls. This was associated with an increase in trabecular spacing and a decrease in trabecular number. Moreover, there was an increase in the amount of osteoid in CaSR-cKO mice as reflected by a significantly higher osteoid volume/bone volume and osteoid surface/bone surface (OS/BS), indicating a reduction in mineralization. Static measurements demonstrated an increase in the numbers of osteoblasts (osteoblast surface/bone surface, ObS/BS, and osteoblast number/bone perimeter) in CaSR-cKO mice but no differences in the numbers of osteoclasts. Finally, dynamic histomorphometry revealed significantly lower mineral apposition rates and bone formation rates in CaSR-cKO mice as compared with controls, consistent with the increased osteoid, both suggesting decreased osteoblast activity. Together these data suggest a decrease in the mineralizing activity of osteoblasts in the lactating cKO mice.
Table 1.
Histomorphometry of Proximal Tibia in Lactating Control and CaSR-cKO Mice
| Control | cKO | P Value | |
|---|---|---|---|
| BV/TV | 12.03 ± 0.87 | 7.34 ± 0.56a | .00083 |
| TbN | 3.46 ± 0.23 | 2.31 ± 0.18a | .0018 |
| TbTh | 34.79 ± 1.07 | 31.84 ± 0.98 | .072 |
| TbSp | 269.3 ± 21.67 | 419.2 ± 35.1a | .0012 |
| OV/BV | 7.1 ± 0.54 | 12.36 ± 0.8a | .000023 |
| OS/BS | 35.58 ± 2.3 | 53.72 ± 3.45a | .00024 |
| ObS/BS | 20.32 ± 1.61 | 26.76 ± 2.02a | .0220075 |
| OcS/BS | 10.62 ± 1.04 | 9.82 ± 1.22 | .62309 |
| NOb/BPm | 19.93 ± 1.84 | 26.36 ± 2.05a | .035 |
| NOc/BPm | 2.89 ± 0.24 | 2.73 ± 0.34 | .67 |
| MAR | 2.95 ± 0.36 | 2.07 ± 0.2a | .038 |
| BFR/BV | 2917 ± 206 | 1855 ± 167a | .0021 |
Abbreviations: BFR/BV, bone formation rate; MAR, mineral apposition rate; NOb/BPm, osteoblast number/bone perimeter; ObS/BS, osteoblast surface/bone surface; OV/BV, osteoid volume/bone volume; TbN, number of trabeculae; TbTh, trabecular thickness; TbSp, trabecular spacing; OcS/BS, osteoclast surface/bone surface; NOc/BPm, number of osteoclasts/bone perimeter.
Statistical significance.
Circulating collagen type 1 CTX levels, a biochemical marker of bone resorption, were similar in CaSR-cKO and control mice (Figure 4). In contrast, consistent with the bone formation estimates, circulating levels of type 1 procollagen N terminal (P1NP) and osteocalcin were decreased when the mammary gland CaSR was disrupted.
Figure 4.

Loss of the mammary gland CaSR reduces biochemical indices of bone formation but does not increase biochemical markers of bone resorption during lactation. A, Results for serum CTX in control and CaSR-cKO mice on day 12 of lactation. There was no significant increase in rates of bone resorption in CaSR-cKO mice. Bars represent the mean ± SEM for 6 mice of each genotype. B, Serum osteocalcin levels in control and CaSR-cKO mice on day 12 of lactation. There was a significant decrease in circulating osteocalcin levels in cKO as compared with control mice. Bars represent the mean ± SEM for 6 mice of each genotype. C, Serum P1NP levels in control and CaSR-cKO mice on day 12 of lactation. There was a significant decrease in circulating P1NP levels in cKO as compared with control mice. Bars represent the mean ± SEM for 4 control mice and 6 CaSR-cKO mice.
Disruption of the mammary CaSR alters systemic calcium metabolism during lactation
Loss of the CaSR on MECs caused transient hypercalcemia (Figure 5). Circulating calcium levels were elevated in CaSR-cKO mice on day 2 of lactation, but on day 11 of lactation, calcium levels had returned to the normal range (Figure 5A). In contrast, on day 11 of lactation, circulating levels of PTH were significantly reduced in CaSR-cKO mice (Figure 5B). This was associated with a significant increase in renal calcium excretion in CaSR-cKO mothers as assessed by the urinary calcium to creatinine ratio (Figure 5C). Interestingly, despite the lower PTH levels, circulating 1,25 dihydroxyvitamin D3 levels were not different between the CaSR-cKO and control mice (Figure 5D), perhaps reflecting higher PTHrP plasma bioactivity (Figure 2). These data suggest that normalization of systemic calcium levels as lactation progressed resulted from a compensatory decrease in PTH secretion and an increase in urinary calcium excretion triggered, in part, by stimulation of the parathyroid and renal CaSR. To test this idea, we measured serum and urine calcium levels in rescued global CaSR-knockout mice, generated by breeding CaSR−/− mice to PTH−/− mice. As shown in Figure 5E, serum calcium levels were equivalent in lactating WT (C+P+) vs PTH−/− (C+P−) mice. However, global disruption of both the PTH and CaSR genes in CaSR−/−; PTH−/− (C−P−) mice resulted in persistent hypercalcemia on day 12 of lactation. Lactating PTH−/− controls had a significant increase in urinary calcium excretion as compared with WT mice. However, the additional loss of the renal CaSR in CaSR−/−; PTH−/− mice reduced urinary calcium excretion to baseline, reversing the elevated urinary calcium excretion caused by mammary-specific disruption of the CaSR (Figure 5F). These data suggest that the loss of the CaSR in the lactating mammary gland causes transient hypercalcemia, which triggers a CaSR-mediated compensatory renal calcium wasting. The concurrent loss of the renal and mammary CaSR inhibits urinary calcium excretion and results in persistent hypercalcemia.
Loss of the mammary gland CaSR lowers milk calcium content
Previous data suggested that the CaSR stimulates calcium transport into milk (22–24). Therefore, we measured the calcium content of milk from CaSR-cKO vs control mice. Disrupting the MECs, the CaSR in MECs reduced milk calcium concentrations by approximately 50% (Figure 6A). The calcium pump, PMCA2, is responsible for up to 70% of the calcium transported into milk (23, 34). Therefore, we examined whether the loss of the CaSR affected PMCA2 expression within the lactating mammary gland. As shown in Supplemental Figure 4, disrupting the CaSR did not reduce the levels of PMCA2 mRNA or protein.
Figure 6.
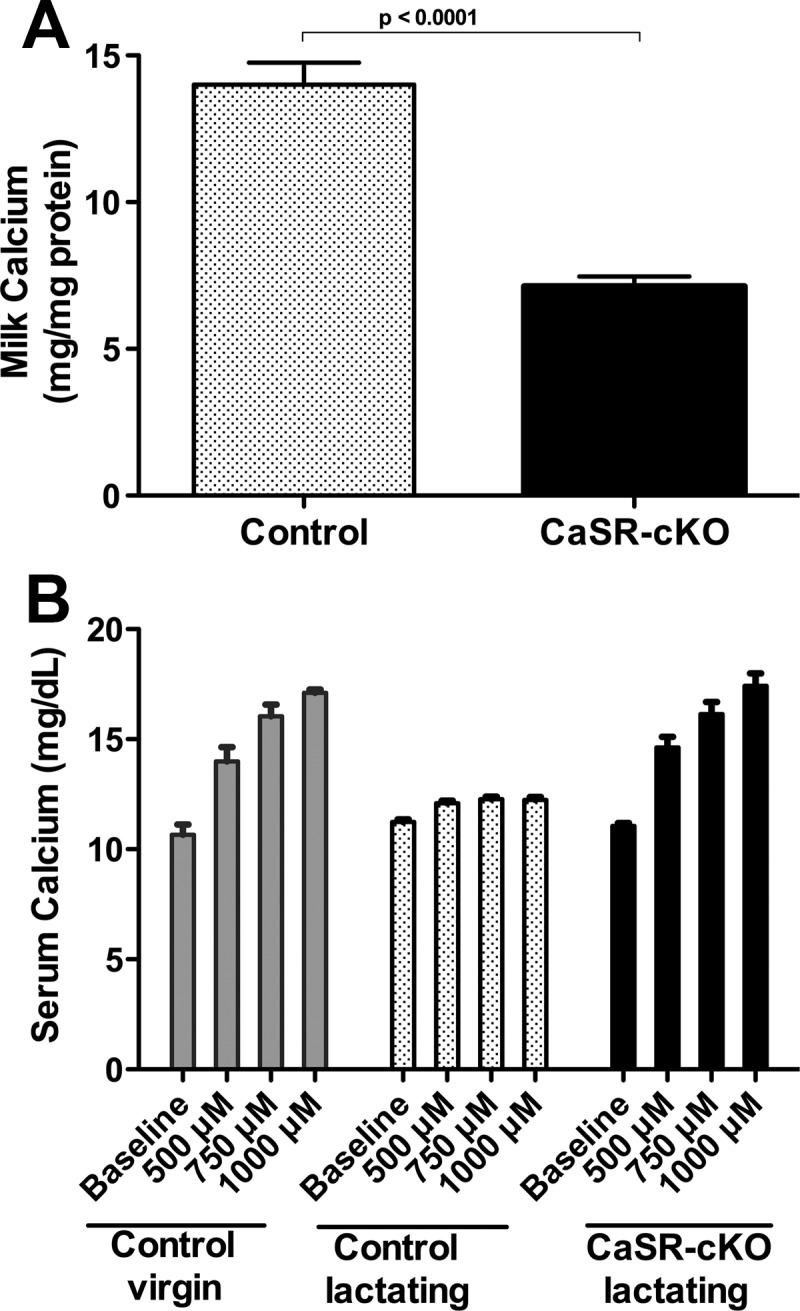
Loss of the mammary CaSR inhibits calcium transport into milk and clearance of calcium from the circulation in lactating mice. A, Milk calcium concentrations in control and CaSR-cKO mice on day 12 of lactation. Milk calcium is reduced in CaSR-cKO mice. Bars represent the mean ± SEM for 5 mice of each genotype. B, Shown are the serum calcium levels measured 30 minutes after IP calcium doses in control virgin mice, control lactating mice, and CaSR-cKO mice. In virgin mice, the administration of increasing doses of ip calcium results in progressive hypercalcemia. However, in lactating controls, calcium is quickly cleared from the circulation. Loss of the mammary CaSR impairs calcium clearance and reverts the lactating pattern back to that of a virgin mouse, demonstrating that the enhanced calcium clearance during lactation depends on the CaSR in the mammary gland. Bars represent the mean ± SEM for 3 mice of each genotype at each dose of ip calcium.
The above data suggested that ablating the MEC CaSR blunted the ability of the mammary gland to transport calcium from the circulation into milk. To test this notion, we administered increasing doses of calcium by ip injections to virgin control mice and to lactating CaSR-cKO and control mice. As shown in Figure 6B, increasing doses of ip calcium cause a dose-responsive increase in circulating calcium concentrations in virgin controls. The same doses of calcium administered to lactating controls had little effect on circulating calcium, likely due to the ability of the CaSR to stimulate calcium transport into milk. However, removal of the MEC CaSR in lactating CaSR-cKO mice resulted in a reversion to the pattern seen in virgin controls, in which increasing doses of calcium lead to progressive hypercalcemia. Together these data demonstrate that activation of the CaSR on MECs during lactation stimulates calcium transport by these cells, preventing any increase in circulating calcium during lactation, when bone resorption is increased and renal calcium excretion is decreased.
The mammary CaSR regulates neonatal calcium accrual
Given the decreased calcium content and the increased PTHrP concentrations of milk produced by the CaSR-cKO mice, we next examined the calcium content of neonatal mice suckling these mothers. Twelve-day-old mice taken from CaSR-cKO and control dams were cremated and the calcium content of the ash was measured and corrected for body weight. The calcium content of the pups suckling CaSR-cKO mothers was significantly reduced compared with the calcium content of pups suckling control mothers (Figure 7A). To determine whether this resulted from the ingestion of milk with less calcium or of milk with higher PTHrP levels, similar measurements were made in neonates suckling from mothers in which the PTHrP gene was disrupted specifically in lactating MECs. As previously reported, lactating BLG-Cre;PTHrPlox/− (PTHrP-cKO) mice produce milk with almost no PTHrP (Supplemental Figure 5) but with normal calcium levels (5). Therefore, we measured the ash calcium content of 12-day-old pups from PTHrPlox/− and BLG-Cre; PTHrPlox/− mothers, which produce milk with reduced or no PTHrP, respectively. We found a dose-responsive, inverse relationship between the PTHrP content of the milk and the ash calcium content of the suckling pups. As shown in Figure 7B, pups taken from BLG-Cre;PTHrPlox/− mothers had a significantly higher ash calcium content than pups taken from control mothers. Pups suckling PTHrPlox/− mothers had intermediate ash calcium contents. These results suggest that milk PTHrP affects calcium accrual in suckling mice independently of milk calcium content.
Figure 7.

Loss of the mammary CaSR impairs neonatal calcium accrual. A, Ash calcium content expressed as the percentage of wet pup weight for pups suckling on control or CaSR-cKO dams. Note that total body calcium content is reduced for pups consuming milk from the CaSR-cKO mothers. Bars represent the mean ± SEM for the pups from 8 mothers of each genotype. B, Ash calcium content of pups suckling on PTHrPlox/lox, PTHrP lox/−, and BLG-Cre;PTHrPlox/− dams. Note that there is a dose-responsive increase in total body calcium content of the pups as the PTHrP levels in milk decline. Bars represent the mean ± SEM for the pups from 6 mothers of each genotype.
Discussion
In this study, we provide genetic confirmation that the CaSR regulates PTHrP production and milk calcium transport by MECs during lactation. Disruption of the CaSR specifically on breast cells at the onset of lactation resulted in increased production and secretion of PTHrP by the mammary gland and increased milk PTHrP content. Loss of the breast calcium-sensing receptor also blunted calcium transport from the circulation into milk and reduced milk calcium concentrations. Due to these changes, lactating mice developed transient hypercalcemia that returned to normal by midlactation as a result of compensatory changes in PTH secretion and renal calcium excretion. These data confirm that, during lactation, the breast regulates systemic calcium homeostasis and that the MEC CaSR controls breast PTHrP production and calcium uptake in response to changes in the supply of calcium available for milk production.
Our data confirm that the CaSR regulates PTHrP production by breast cells. In normal breast cells in vivo, PTHrP is expressed at high levels only during embryonic development and again during lactation (35–37). Previous studies had shown that treatment with calcium and/or calcimimetics suppressed PTHrP mRNA levels and PTHrP secretion in cultured mouse MECs. In addition, heterozygous global deletion of the CaSR gene resulted in increased PTHrP production by cultured cells (24). We now demonstrate that loss of the CaSR specifically in MECs led to increased PTHrP mRNA levels in the intact mammary gland and PTHrP levels in milk. Circulating PTHrP levels during lactation hover near the limit of detection of current immunoassays, and plasma levels of immunoreactive PTHrP in lactating CaSR-cKO mice were not significantly different from the levels in lactating control mice. Nevertheless, urinary cAMP levels in these mice were significantly increased despite the suppression of circulating PTH levels in CaSR-cKO mice. Urinary cAMP levels reflect the activity of the renal PTH receptor 1 (PTHR1) and have long been used as an indirect, although not strictly quantitative, measure of PTH and PTHrP bioactivity (33). Given that these are the two recognized ligands for this receptor, the elevated urinary cAMP levels in the face of suppressed PTH levels are consistent with elevated PTHrP bioactivity in the CaSR-cKO mice, even though we could not detect an increase in circulating immunoreactive PTHrP. However, it is also possible that increased urinary cAMP levels in the CaSR-cKO mice are the result of some other factor.
Loss of the mammary gland CaSR was not associated with significantly accelerated bone resorption or excessive bone loss as detected by DEXA. There were small reductions in trabecular thickness on micro-CT and bone mass and trabecular numbers were reduced in the lactating CaSR-cKO mice on histomorphometry, but these changes were not reflected in greater rates of bone loss by DEXA. There were also subtle differences in mineral density by micro-CT and an apparent shift toward less densely mineralized cortical bone in the CaSR-cKO mice. These results stand in contrast to the effects of dietary calcium restriction during lactation, which leads to hypocalcemia, increased levels of PTHrP and PTH, and a clear acceleration of maternal bone loss as measured by DEXA (22, 38). The explanation for these differences is likely to be that the CaSR-cKO mice have elevated circulating calcium levels, which leads to a suppression of PTH secretion. As a result, the effective biological drive for the PTHR1 is relatively reduced as compared with calcium restriction, although the elevated urinary cAMP levels in the CaSR cKO mice suggest that renal PTHR1 activation is still absolutely increased as compared with the lactating controls. Another possibility is that hypercalcemia itself may directly affect the activity of osteoclasts, blunting any increased bone resorption. Several studies have shown that CaSR signaling can inhibit osteoclast formation and activity (39–45). Finally, hypercalcemia could have increased calcitonin secretion, which has been shown to restrain excessive bone resorption during lactation (46). Whatever the exact explanation, the interconnectedness of calcium transport and PTHrP secretion as well as the robust nature of systemic compensation all likely limited the ability of bone resorption to increase further in the setting of systemic hypercalcemia.
Although bone resorption was not changed in CaSR-cKO mice, the rates of bone formation were reduced. We found increased osteoid as well as decreased bone formation rates by dynamic histomorphometry and a decreased level of circulating bone formation markers in lactating CaSR-cKO mice as compared with controls. However, osteoblast numbers were actually slightly higher in CaSR-cKO mice, suggesting that disruption of the CaSR gene in the mammary gland somehow impaired osteoblast function. These observations may also help explain the slightly lower BMD on micro-CT. These findings were unexpected and we do not have a clear explanation. However, there are several possibilities. First, this may be a reflection of elevated PTHrP bioactivity in these mice because Horwitz et al (47) have shown that prolonged infusions of PTHrP in human subjects reduces rather than increases bone formation rates as measured by biochemical indices such as circulating P1NP levels. Treatment of cells in culture with PTH or continuous infusion of PTH in animals have also shown that although PTHR1 signaling increases the proliferation of osteoblast progenitors, it can inhibit the differentiation of these cells into mature osteoblasts and lead to the accumulation of unmineralized osteoid (48–51). Second, it is possible that the transient hypercalcemia noted in our study inhibited mineralization in these mice. However, this is unlikely, given that the preponderance of evidence suggests that extracellular calcium stimulates the CaSR to promote osteoblast differentiation, bone formation, and bone matrix mineralization (18–20, 43). Finally, it is always possible that the loss of the mammary gland CaSR resulted in the secretion of some additional unknown factor from the mammary gland that affects osteoblast function.
Our experiments demonstrate that the mammary gland CaSR is necessary for the transport of calcium from the circulation into milk. When the Casr gene is disrupted, milk calcium levels are significantly reduced, and the mice become hypercalcemic. Furthermore, the CaSR mediates the impressive increase in the ability of breast epithelial cells to clear calcium from the circulation during lactation. Most milk calcium is transported across the apical membrane of MECs through the actions of PMCA2 (23, 34). Consistent with previous data demonstrating that CaSR signaling stimulated the transport activity of PMCA2, but not its expression, genetic ablation of the CaSR did not affect the levels of PMCA2 mRNA or protein expression (23). These results suggest a model by which calcium is mobilized and supplied to the lactating breast, which then adjusts its transport of calcium to match the supply. If this is the case, milk calcium levels should vary with maternal calcium intake, a prediction consistent with the data of Cao et al (52) and Shu et al (20), who reported that milk calcium content could be raised in mice by increasing the calcium content of the diet.
A final finding of our study was that the mammary gland CaSR appears to coordinate neonatal calcium accrual with maternal calcium and bone metabolism by changing milk calcium and/or PTHrP content. Loss of the CaSR on breast epithelial cells led to reductions in the overall calcium content of 12-day-old mice. This appeared to be, in part, an inverse function of milk PTHrP content, for disruption of the PTHrP gene in MECs using the same BLG-Cre transgene revealed a dose-dependent, inverse relationship between neonatal ash calcium content and milk PTHrP content. Previous studies have shown that manipulating dietary calcium intake in lactating mice results in opposite changes in milk calcium and PTHrP content (20, 22, 52). Increased dietary calcium was associated with increased milk calcium content, decreased milk PTHrP levels and increased neonatal bone volume and density (20, 52). In these studies, it appeared that some of the effects were due to the differences in neonatal calcium intake and/or circulating calcium levels, given that the neonatal skeletal changes were blunted in the setting of global disruption of the CaSR in lactating dams (20). However, milk PTHrP levels were always lower in the mice that had higher milk calcium, consistent with the possibility that the changes in neonatal bone in response to altered maternal calcium intake might have been caused, in part, by simultaneous changes in milk PTHrP. We believe it likely that the loss of the mammary gland CaSR caused a reduced accrual of neonatal calcium due both to the reduction in milk calcium itself and the increase in milk PTHrP.
At this point the mechanisms and the rationale for the effects of milk PTHrP on neonatal calcium content are unclear. However, one hypothesis is that the mammary CaSR acts to entrain maternal and neonatal calcium and bone metabolism to adapt as a unit to alterations in the availability of calcium in the environment. The increase in milk PTHrP content might help avoid hypocalcemia in the neonates when milk calcium content drops. This hypothesis will require many further experiments for its validation, but there are precedents for milk being a vehicle to adjust neonatal physiology. For example, milk TGF-β can affect neonatal immunity, milk cortisol levels can affect the development of neonatal stress responses and behavior, and alterations in milk nutrients can affect lifelong metabolic programming in the offspring of obese or diabetic mothers (53–59). Our studies suggest that milk PTHrP may also represent a metabolic signal from mother to offspring.
In summary, disruption of the CaSR on breast epithelial cells affects maternal calcium and bone metabolism, milk calcium and PTHrP content, and neonatal calcium accrual. By stimulating MEC calcium transport and by suppressing MEC PTHrP production, this receptor coordinates breast calcium uptake with maternal calcium metabolism during lactation. In addition, we found that the CaSR-mediated regulation of PTHrP and calcium secretion into milk appears to coordinate maternal and neonatal calcium economies. These genetic experiments confirm that the lactating breast becomes a calcium-sensing organ that regulates maternal and neonatal calcium and bone homeostasis.
Acknowledgments
We are indebted to Drs Arthur Broadus and Dolores Shoback for guidance and useful conversations about these experiments.
This work was supported by the following grants: National Institutes of Health Grants DK55501, DK077565, and CA153702 (to J.W.); a Veterans Affairs Merit award, a Veterans Affairs Program Project Award and National Institutes of Health Grants AR050023, DK054793, and AR055924 (to D.B.), and National Institutes of Health Grant AG21353 (to W.C.). Infrastructure for these studies was also provided by Yale core facilities supported by Grants P30DK045735 and P30AR46032.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BLG-Cre
- β-lactoglobulin-Cre
- BMD
- bone mineral density
- BV/TV
- bone volume/total volume
- CaSR
- calcium-sensing receptor
- CaSR-cKO
- BLG-Cre;CaSRlox/lox
- CT
- computed tomographic
- CTX
- cross-linked C-telopeptides of type I collagen
- DEXA
- dual-energy x-ray absorptiometry
- HMCt
- high-mineral-density bone
- LMCt
- low-mineral-density bone
- MEC
- mammary epithelial cell
- OS/BS
- osteoid surface/bone surface
- PMCA2
- plasma membrane calcium ATPase 2b
- P1NP
- type 1 procollagen N terminal
- PTHR1
- PTH receptor 1
- WT
- wild-type.
References
- 1. Kovacs CS. Calcium and bone metabolism during pregnancy and lactation. J Mamm Gland Biol Neoplasia. 2005;10(2):105–118 [DOI] [PubMed] [Google Scholar]
- 2. Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev. 1997;18(6):832–872 [DOI] [PubMed] [Google Scholar]
- 3. VanHouten J. Maternal calcium and bone metabolism during lactation. Curr Opin Endocrinol Diabetes. 2005;12:477–482 [Google Scholar]
- 4. Wysolmerski JJ. Interactions between breast, bone, and brain regulate mineral and skeletal metabolism during lactation. Ann NY Acad Sci. 2010;1192:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. VanHouten JN, Dann P, Stewart AF, et al. Mammary-specific deletion of parathyroid hormone-related protein preserves bone mass during lactation. J Clin Invest. 2003;112(9):1429–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. VanHouten JN, Wysolmerski JJ. Low estrogen and high parathyroid hormone-related peptide levels contribute to accelerated bone resorption and bone loss in lactating mice. Endocrinology. 2003;144(12):5521–5529 [DOI] [PubMed] [Google Scholar]
- 7. Sowers MF, Hollis BW, Shapiro B, et al. Elevated parathyroid hormone-related peptide associated with lactation and bone density loss. JAMA. 1996;276(7):549–554 [PubMed] [Google Scholar]
- 8. Ardeshirpour L, Brian S, Dann P, VanHouten J, Wysolmerski J. Increased PTHrP and decreased estrogens alter bone turnover but do not reproduce the full effects of lactation on the skeleton. Endocrinology. 2010;151(12):5591–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366(6455):575–580 [DOI] [PubMed] [Google Scholar]
- 10. Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81(1):239–297 [DOI] [PubMed] [Google Scholar]
- 11. Chakravarti B, Chattopadhyay N, Brown EM. Signaling through the extracellular calcium-sensing receptor (CaSR). Adv Exp Med Biol. 2012;740:103–142 [DOI] [PubMed] [Google Scholar]
- 12. Ho C, Conner DA, Pollak MR, et al. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet. 1995;11(4):389–394 [DOI] [PubMed] [Google Scholar]
- 13. Kantham L, Quinn SJ, Egbuna OI, et al. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab. 2009;297(4):E915–E923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Loupy A, Ramakrishnan SK, Wootla B, et al. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest. 2012;122(9):3355–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pearce SH, Trump D, Wooding C, et al. Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest. 1995;96(6):2683–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pollak MR, Brown EM, Chou YH, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75(7):1297–1303 [DOI] [PubMed] [Google Scholar]
- 17. Pollak MR, Brown EM, Estep HL, et al. Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet. 1994;8(3):303–307 [DOI] [PubMed] [Google Scholar]
- 18. Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Science Signaling. 2008;1(35):ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dvorak-Ewell MM, Chen TH, Liang N, et al. Osteoblast extracellular Ca2+ -sensing receptor regulates bone development, mineralization, and turnover. J Bone Miner Res. 2011;26(12):2935–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shu L, Ji J, Zhu Q, et al. The calcium-sensing receptor mediates bone turnover induced by dietary calcium and parathyroid hormone in neonates. J Bone Miner Res. 2011;26(5):1057–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng I, Klingensmith ME, Chattopadhyay N, et al. Identification and localization of the extracellular calcium-sensing receptor in human breast. J Clin Endocrinol Metab. 1998;83(2):703–707 [DOI] [PubMed] [Google Scholar]
- 22. VanHouten J, Dann P, McGeoch G, et al. The calcium-sensing receptor regulates mammary gland parathyroid hormone-related protein production and calcium transport. J Clin Invest. 2004;113(4):598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. VanHouten JN, Neville MC, Wysolmerski JJ. The calcium-sensing receptor regulates plasma membrane calcium adenosine triphosphatase isoform 2 activity in mammary epithelial cells: a mechanism for calcium-regulated calcium transport into milk. Endocrinology. 2007;148(12):5943–5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ardeshirpour L, Dann P, Pollak M, Wysolmerski J, VanHouten J. The calcium-sensing receptor regulates PTHrP production and calcium transport in the lactating mammary gland. Bone. 2006;38(6):787–793 [DOI] [PubMed] [Google Scholar]
- 25. Lippuner K, Zehnder HJ, Casez JP, Takkinen R, Jaeger P. PTH-related protein is released into the mother's bloodstream during location: evidence for beneficial effects on maternal calcium-phosphate metabolism. J Bone Miner Res. 1996;11(10):1394–1399 [DOI] [PubMed] [Google Scholar]
- 26. Selbert S, Bentley DJ, Melton DW, et al. Efficient BLG-Cre mediated gene deletion in the mammary gland. Transgen Res. 1998;7(5):387–396 [DOI] [PubMed] [Google Scholar]
- 27. Applied Biosystems Relative quantitation of gene expression: ABI Prism 7700 sequence detection system. User bulletin number 2. Foster City, CA: Applied Biosystems; 1997 [Google Scholar]
- 28. Bai M, Trivedi S, Brown EM. Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J Biol Chem. 1998;273(36):23605–23610 [DOI] [PubMed] [Google Scholar]
- 29. Chattopadhyay N, Baum M, Bai M, et al. Ontogeny of the extracellular calcium-sensing receptor in rat kidney. Am J Physiol. 1996;271(3 Pt 2):F736–F743 [DOI] [PubMed] [Google Scholar]
- 30. Ward DT, Brown EM, Harris HW. Disulfide bonds in the extracellular calcium-polyvalent cation-sensing receptor correlate with dimer formation and its response to divalent cations in vitro. J Biol Chem. 1998;273(23):14476–14483 [DOI] [PubMed] [Google Scholar]
- 31. Pidasheva S, Grant M, Canaff L, Ercan O, Kumar U, Hendy GN. Calcium-sensing receptor dimerizes in the endoplasmic reticulum: biochemical and biophysical characterization of CASR mutants retained intracellularly. Hum Mol Genet. 2006;15(14):2200–2209 [DOI] [PubMed] [Google Scholar]
- 32. Cifuentes M, Albala C, Rojas C. Calcium-sensing receptor expression in human adipocytes. Endocrinology. 2005;146(5):2176–2179 [DOI] [PubMed] [Google Scholar]
- 33. Broadus AE, Mahaffey JE, Bartter FC, Neer RM. Nephrogenous cyclic adenosine monophosphate as a parathyroid function test. J Clin Invest. 1977;60(4):771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reinhardt TA, Lippolis JD, Shull GE, Horst RL. Null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2 impairs calcium transport into milk. J Biol Chem. 2004;279(41):42369–42373 [DOI] [PubMed] [Google Scholar]
- 35. Boras-Granic K, VanHouten J, Hiremath M, Wysolmerski J. Parathyroid hormone-related protein is not required for normal ductal or alveolar development in the post-natal mammary gland. PloS One. 2011;6(11):e27278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thiede MA, Rodan GA. Expression of a calcium-mobilizing parathyroid hormone-like peptide in lactating mammary tissue. Science. 1988;242(4876):278–280 [DOI] [PubMed] [Google Scholar]
- 37. Wysolmerski JJ, Philbrick WM, Dunbar ME, Lanske B, Kronenberg H, Broadus AE. Rescue of the parathyroid hormone-related protein knockout mouse demonstrates that parathyroid hormone-related protein is essential for mammary gland development. Development. 1998;125(7):1285–1294 [DOI] [PubMed] [Google Scholar]
- 38. Anderson JJ, Garner SC, Mar MH, Boass A, Toverud SU, Parikh I. The ovariectomized, lactating rat as an experimental model for osteopenia: calcium metabolism and bone changes. Bone Mineral. 1990;11(1):43–53 [DOI] [PubMed] [Google Scholar]
- 39. Kameda T, Mano H, Yamada Y, et al. Calcium-sensing receptor in mature osteoclasts, which are bone resorbing cells. Biochem Biophys Res Commun. 1998;245(2):419–422 [DOI] [PubMed] [Google Scholar]
- 40. Kanatani M, Sugimoto T, Kanzawa M, Yano S, Chihara K. High extracellular calcium inhibits osteoclast-like cell formation by directly acting on the calcium-sensing receptor existing in osteoclast precursor cells. Biochem Biophys Res Commun. 1999;261(1):144–148 [DOI] [PubMed] [Google Scholar]
- 41. Lorget F, Kamel S, Mentaverri R, et al. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem Biophys Res Commun. 2000;268(3):899–903 [DOI] [PubMed] [Google Scholar]
- 42. Malgaroli A, Meldolesi J, Zallone AZ, Teti A. Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. J Biol Chem. 1989;264(24):14342–14347 [PubMed] [Google Scholar]
- 43. Marie PJ. The calcium-sensing receptor in bone cells: a potential therapeutic target in osteoporosis. Bone. 2010;46(3):571–576 [DOI] [PubMed] [Google Scholar]
- 44. Mentaverri R, Yano S, Chattopadhyay N, et al. The calcium sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J. 2006;20(14):2562–2564 [DOI] [PubMed] [Google Scholar]
- 45. Miyauchi A, Hruska KA, Greenfield EM, et al. Osteoclast cytosolic calcium, regulated by voltage-gated calcium channels and extracellular calcium, controls podosome assembly and bone resorption. J Cell Biol. 1990;111(6 Pt 1):2543–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Woodrow JP, Sharpe CJ, Fudge NJ, Hoff AO, Gagel RF, Kovacs CS. Calcitonin plays a critical role in regulating skeletal mineral metabolism during lactation. Endocrinology. 2006;147(9):4010–4021 [DOI] [PubMed] [Google Scholar]
- 47. Horwitz MJ, Tedesco MB, Sereika SM, et al. Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)2 vitamin D. J Bone Miner Res. 2005;20(10):1792–1803 [DOI] [PubMed] [Google Scholar]
- 48. Gopalakrishnan R, Suttamanatwong S, Carlson AE, Franceschi RT. Role of matrix Gla protein in parathyroid hormone inhibition of osteoblast mineralization. Cells Tissues Organs. 2005;181(3–4):166–175 [DOI] [PubMed] [Google Scholar]
- 49. Ishizuya T, Yokose S, Hori M, et al. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99(12):2961–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malluche HH, Sherman D, Meyer W, Ritz E, Norman AW, Massry SG. Effects of long-term infusion of physiologic doses of 1–34 PTH on bone. Am J Physiol. 1982;242(2):F197–F201 [DOI] [PubMed] [Google Scholar]
- 51. Yuan Q, Sato T, Densmore M, et al. Deletion of PTH rescues skeletal abnormalities and high osteopontin levels in Klotho−/− mice. PLoS Genet. 2012;8(5):e1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cao G, Gu Z, Ren Y, et al. Parathyroid hormone contributes to regulating milk calcium content and modulates neonatal bone formation cooperatively with calcium. Endocrinology. 2009;150(2):561–569 [DOI] [PubMed] [Google Scholar]
- 53. de Moura EG, Passos MC. Neonatal programming of body weight regulation and energetic metabolism. Biosci Rep. 2005;25(3–4):251–269 [DOI] [PubMed] [Google Scholar]
- 54. Kainonen E, Rautava S, Isolauri E. Immunological programming by breast milk creates an anti-inflammatory cytokine milieu in breast-fed infants compared to formula-fed infants. Br J Nutr. 2012;1–9 [DOI] [PubMed] [Google Scholar]
- 55. Oddy WH, McMahon RJ. Milk-derived or recombinant transforming growth factor-β has effects on immunological outcomes: a review of evidence from animal experimental studies. Clin Exp Allergy. 2011;41(6):783–793 [DOI] [PubMed] [Google Scholar]
- 56. Rautava S, Lu L, Nanthakumar NN, Dubert-Ferrandon A, Walker WA. TGF-β2 induces maturation of immature human intestinal epithelial cells and inhibits inflammatory cytokine responses induced via the NF-κB pathway. J Pediatr Gastroenterol Nutr. 2012;54(5):630–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sullivan EC, Hinde K, Mendoza SP, Capitanio JP. Cortisol concentrations in the milk of rhesus monkey mothers are associated with confident temperament in sons, but not daughters. Dev Psychobiol. 2011;53(1):96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Glynn LM, Davis EP, Schetter CD, Chicz-Demet A, Hobel CJ, Sandman CA. Postnatal maternal cortisol levels predict temperament in healthy breastfed infants. Early Hum Dev. 2007;83(10):675–681 [DOI] [PubMed] [Google Scholar]
- 59. Wahlig JL, Bales ES, Jackman MR, Johnson GC, McManaman JL, Maclean PS. Impact of high-fat diet and obesity on energy balance and fuel utilization during the metabolic challenge of lactation. Obesity. 2012;20(1):65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]