

Abstract
In this study, we characterized the full-length genome sequences of seven hepatitis C virus (HCV) isolates belonging to genotype 1. These represent the first complete genomes for HCV subtypes 1e, 1h, 1l, plus one novel variant that qualifies for a new but unassigned subtype. The genomes were characterized using 19–22 overlapping fragments. Each was 9400–9439 nt long and contained a single ORF encoding 3019–3020 amino acids. All viruses were isolated in the sera of seven patients residing in, or originating from, Cameroon. Predicted amino acid sequences were inspected and unique patterns of variation were noted. Phylogenetic analysis using full-length sequences provided evidence for nine genotype 1 subtypes, four of which are described for the first time here. Subsequent phylogenetic analysis of 141 partial NS5B sequences further differentiated 13 subtypes (1a–1m) and six additional unclassified lineages within genotype 1. As a result of this study, there are now seven HCV genotype 1 subtypes (1a–1c, 1e, 1g, 1h, 1l) and two unclassified genotype 1 lineages with full-length genomes characterized. Further analysis of 228 genotype 1 sequences from the HCV database with known countries is consistent with an African origin for genotype 1, and with the hypothesis of subsequent dissemination of some subtypes to Asia, Europe and the Americas.
Introduction
Hepatitis C virus (HCV) is one of the major causative agents for chronic hepatitis, cirrhosis and hepatocellular carcinoma (Farci et al., 1991; Liang & Heller, 2004; Nishioka, 1991). As reported by the WHO, HCV infects about 2.2 % of the world’s population, with over a million new cases occurring each year. Furthermore, 27 % of these infected individuals eventually progress to liver cirrhosis among whom 25 % finally develop hepatocellular carcinomas (Alter, 2007). HCV is a positive-sense RNA virus that exhibits extensive genetic heterogeneity and a high level of resistance to antiviral drugs in vivo and in vitro. As such, HCV genetic variation poses a huge problem for global public health (Robinson et al., 2011).
To standardize HCV genetic diversity, consensus proposals for a unified system of HCV nomenclature were published after the 11th International Meeting for HCV and Its Related Viruses (Simmonds et al., 2005). It has been recommended that HCV isolates be classified into six major genotypes defined by phylogenetic analysis. Each genotype contains a variable number of subtypes that are related but genetically and epidemiologically distinct. New subtypes are defined as confirmed or provisional, depending on the availability of complete or partial genetic sequences, or defined as unassigned if less than three examples of a new subtype have been reported. Recently, panels of complete HCV genome sequences have been provided for HCV genotypes 2, 3, 4 and 6 in a series of our previous studies (Li et al., 2006, 2009a, b, 2012; Lu et al., 2006, 2007a, b, 2008, 2013; Wang et al., 2009; Xia et al., 2008). However, similar information is still in short supply for genotype 1. As noted in the HCV nomenclature guidelines (Simmonds et al., 2005), three subtypes (1a, 1b and 1c) of HCV-1 have been confirmed with full-length genomic sequences, while nine subtypes (1d–1l) remained provisionally assigned due to the availability of only partial sequences. Although since then a complete genomic sequence has been reported for subtype 1g (Bracho et al., 2008), the other eight subtypes remain to be sequenced in their entirety.
Evolutionary studies have shown that different HCV genotypes originated in specific geographical regions, such as genotypes 3 and 6 in South and South East Asia (Fu et al., 2012; Pybus et al., 2009), genotype 2 in West Africa (Markov et al., 2009), and genotypes 1 and 4 in Central Africa (Njouom et al., 2007, 2012). Cameroon is located in Central Africa and a diverse range of subtypes of HCV-1, HCV-2 and HCV-4 have been reported from the country. Previous studies have reported Cameroonian isolates of subtypes 1b, 1e, 1h and 1l (Ndjomou et al., 2003; Njouom et al., 2003a, b, 2007; Pasquier et al., 2005); three samples from these studies remained and were used here to generate full genome sequences. In addition, during routine diagnostic HCV screening and genotyping services provided by a company in the UK, four unique HCV-1 isolates were found among patients originating from Cameroon; these represent subtypes 1e and 1l, and a novel HCV-1 variant. The full-length sequences of these four isolates are also provided here. It is hoped that this valuable genomic data will add to our current understanding of HCV classification and nomenclature, and facilitate future studies of HCV molecular epidemiology, evolution and genetics.
Results
Genome sequences and organization
Full-length genome sequences were characterized for seven HCV genotype 1 isolates: 136142, 148636, 160526, 166212, EBW9, EBW424 and EBW443, each with 19–22 overlapping fragments (Fig. S1, available in JGV Online). These genomes were 9420–9439 nt in length, starting from the extreme 5′UTR end through to the variable region of the 3′UTR (Table 1). Each had a single ORF of length 9039–9063 nt. The 5′UTRs were 340–342 nt long, while the 3′UTR lengths varied from 32 to 40 nt. The sizes of the other eight protein genes were consistent with those of the H77 strain (Table 1) with the exception of the E2 (364–371 aa) and NS5A (448–449 aa) genes.
Table 1. Patient and sequence length (nucleotides/amino acids) information for the seven genotype 1 isolates.
Bold entries indicate regions of variable length.
| Sequence name | Age | Sex | Origin | Full | ORF | 5′UTR | Core | E1 | E2 | P7 | NS2 | NS3 | NS4A | NS4B | NS5A | NS5B | 3′UTR |
| H77* | – | – | – | 9646 | 9036/3011 | 341 | 573/191 | 576/192 | 1089/363 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 269 |
| 1e_148636 | ? | ? | ? | 9422 | 9048/3016 | 340 | 573/191 | 576/192 | 1101/367 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 34 |
| 1h_EBW9 | 80 | F | Ebolowa_Cameroon | 9420 | 9039/3013 | 341 | 573/191 | 576/192 | 1092/364 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 40 |
| 1h_EBW443 | 69 | F | Ebolowa_Cameroon | 9420 | 9039/3013 | 341 | 573/191 | 576/192 | 1092/364 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 40 |
| 1l_EBW424 | 77 | M | Ebolowa_Cameroon | 9433 | 9057/3019 | 342 | 573/191 | 576/192 | 1110/370 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 34 |
| 1l_136142 | ? | ? | ? | 9430 | 9054/3018 | 342 | 573/191 | 576/192 | 1107/369 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1775/591 | 34 |
| 1l_166212 | ? | ? | ? | 9428 | 9054/3018 | 342 | 573/191 | 576/192 | 1107/369 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1344/448 | 1776/591 | 32 |
| 1?_160526 | ? | ? | ? | 9439 | 9063/3021 | 341 | 573/191 | 576/192 | 1113/371 | 189/63 | 651/217 | 1893/631 | 162/54 | 783/261 | 1347/449 | 1776/591 | 35 |
The H77 genome (GenBank accession no. NC_004102) is included for comparison.
Phylogeny of full-length genome sequences
A maximum-likelihood (ML) tree was estimated using 52 full-length HCV genome sequences (Fig. 1). The phylogeny exhibited seven major branches, representing HCV genotypes 1–7, each supported by a maximum bootstrap support of 100 %. The most recent HCV classification and nomenclature proposal (Simmonds et al., 2005) reported that full-length HCV genome sequences had been confirmed for four HCV-1 subtypes: 1a, 1b, 1c and 1g. However, in the Los Alamos HCV database we identified an additional 14 full-length sequences that were classified as HCV genotype 1 but carried no subtype designations. Inclusion of these 14 sequences in Fig. 1 revealed that 13 of them belonged to subtype 1a while the remaining genome (accession number AJ851228) represented an unclassified variant. Thus, prior to this study, full-length HCV genome sequences have only been characterized for four subtypes: 1a, 1b, 1c, 1g, and one unclassified strain.
Fig. 1.

An ML phylogenetic tree estimated using full-length genomic sequences of HCV. Reference sequences from confirmed subtypes of genotypes 1, 2, 3, 4, 5, 6 and 7 are included, together with the seven new isolates from this study (black circles). Each genotype is denoted at the right-hand side of the tree. Within genotype 1, all subtypes and equivalents are also indicated. Isolates are named using the following format: subtype_sampling country_sampling date (if available)_isolate name_accession number. The symbol ‘?’ indicates that a subtype has not been assigned. Bootstrap supports are shown at internal branches and the scale bar represents 0.10 nt substitutions per site. Country codes: CA, Canada; CM, Cameroon; EG, Egypt; ES, Spain; HEC, 〈?〉; HK, 〈?〉; ID, Indonesia; IN, India; JP, 〈?〉; MD, 〈?〉; TH, 〈?〉; UK; US, USA; VN, Vietnam.
The seven full-length genomes determined here represented three subtypes and a novel unclassified lineage within HCV genotype 1 (new genomes indicated by black circles in Fig. 1). Isolates EBW9 and EBW443 formed a cluster of two taxa, designated subtype 1h, and isolate 160526 was placed as a single branch corresponding to a new subtype equivalent. Adjacent to the latter lineage, a tight cluster was formed by the isolates EBW424, 136142 and 166212, designated subtype 1l. The isolate 148636, which represents subtype 1e, was also represented in this tree by a single branch. Thus, the three subtypes, 1e, 1h and 1l, were confirmed for the first time in this study by full-length genome sequencing. Similarly, the 160526 sequence should be considered to be a new but currently unclassified subtype equivalent.
Phylogeny of partial NS5B region sequences
A segment of the NS5B region corresponding to nucleotides 8276–8615 of the H77 reference genome, has been found reliably to differentiate HCV genotypes and subtypes in most cases (Murphy et al., 2007). To investigate the full genetic diversity of HCV genotype 1, 141 HCV-1 sequences of this genome region were analysed, each representing an individual isolate and including the seven from this study. The resulting ML tree shows 19 major lineages within genotype 1 (Fig. 2). Among those, 13 have been assigned to HCV subtypes 1a–1m (Jeannel et al., 1998; Simmonds et al., 2005), while six are currently unassigned. The latter were indicated in Fig. 2 as subtype 1(I) to subtype 1(VI). Of the seven isolates from this study, six (148636, EBW9, EBW443, EBW424, 136142 and 166212) represent three of the 13 assigned subtypes, while the seventh isolate (160526) corresponds to one of the six unassigned lineages and is designated in the tree as subtype 1(VI). Specifically, isolate 148636 grouped with a cluster of 37 isolates: 35 from Cameroon (Ndjomou et al., 2003; Njouom et al., 2003b; Pasquier et al., 2005) and two from Canada (Murphy et al., 2007). Both EBW9 and EBW443 were classified into a cluster of 25 isolates: 22 from Cameroon (Ndjomou et al., 2003; Pasquier et al., 2005), two from Canada (Murphy et al., 2007) and one from France (Laperche et al., 2005). EBW424, 136142 and 166212 grouped with a cluster of 25 isolates, all from Cameroon (Ndjomou et al., 2003; Njouom et al., 2003b, 2007; Pasquier et al., 2005).
Fig. 2.
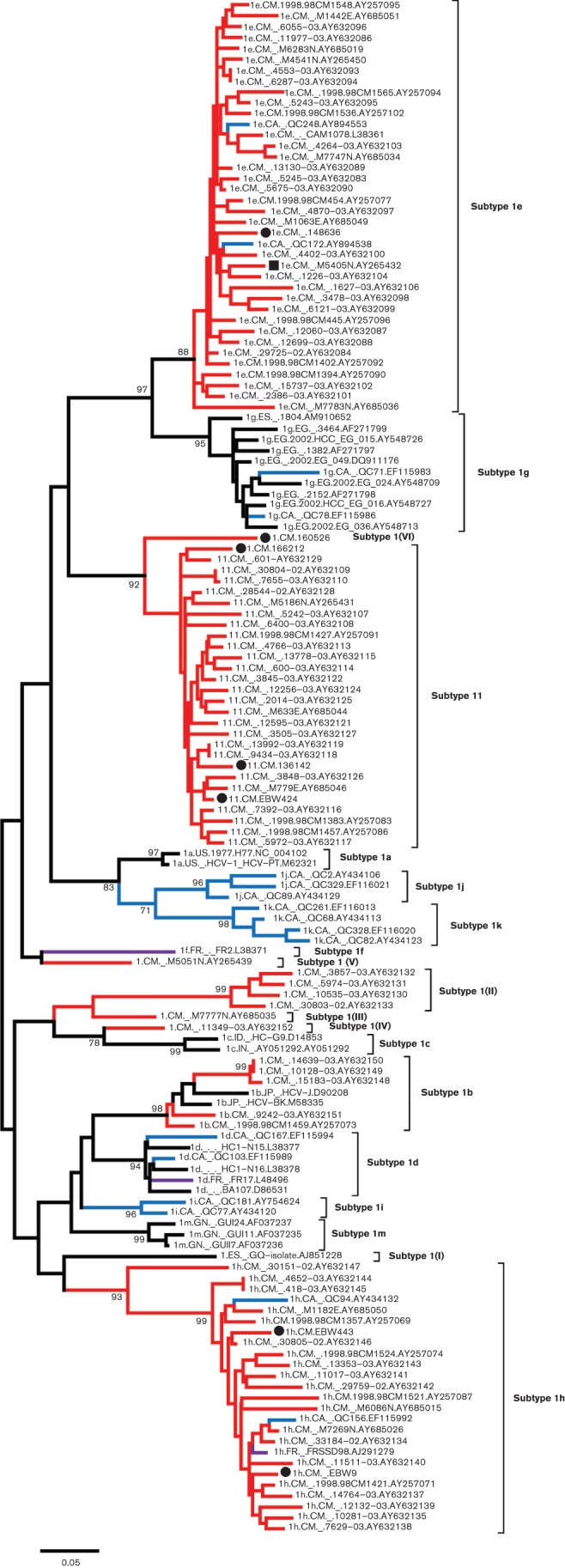
An ML phylogenetic tree estimated using 141 HCV-1 partial NS5B sequences (positions 8276–8615 in the H77 reference genome). Each subtype or subtype equivalent is denoted at the right-hand side of the tree. Isolates from Cameroon are represented with red branches, and isolates from Canada are represented with blue branches, while isolates from France are indicated with purple branches. The seven isolates determined in this study are indicated by black circles. A black square indicates an isolate previously classified as subtype 1a, but which in this study was shown to belong to subtype 1e. For other details, see legend of Fig. 1.
HCV genotype 1 distribution globally and in Cameroon
At the time of study, the Los Alamos HCV database provided 228 sequences that were classified as subtypes 1c to 1m and for which the country of sampling was stated. Using this information, we summarized the geographical distribution of HCV-1 strains (after excluding subtypes 1a and 1b, which are found worldwide). As shown in Table 2, subtypes 1c and 1g were the most widely distributed subtypes; they have been identified in seven and eight countries, respectively. This was followed by subtypes 1d, 1e and 1h, which have been reported from four, three and three countries, respectively. In contrast, the other six subtypes appeared to be restricted; they have only been found in one or two countries. The greatest number of subtypes was found in Canada followed by France and Cameroon. On a continental scale, North America showed the largest genetic diversity of HCV-1 isolates, representing eight subtypes (Table 3). This was followed by Africa, which showed seven subtypes, and by Europe, which showed six subtypes. Only one or two subtypes were reported from other continents. The continent for which the largest number of HCV-1 isolates has been reported was Africa (47.4 %), followed by Asia (37.3 %) and America (8.33 %). Although these results may be interpreted as suggesting that HCV-1 originated in North America, it is much more likely that this genotype originated in Africa. Many of the recently reported divergent HCV isolates sampled in Canada are known to be from recent African immigrants to that country (Li et al., 2012).
Table 2. Geographical distribution of subtypes of HCV genotype 1.
| HCV | No. of isolates | Percentage of | |
| subtype* | Countries with subtypes detected† | (n = 228)‡ | isolates |
| 1c | CA, DE, GB, ID, IN, PK, ZA | 87 | 38.3 |
| 1d | CA, FR, NL, TN | 6 | 2.64 |
| 1e | CA, CM, VN | 43 | 18.9 |
| 1f | FR | 1 | 0.441 |
| 1g | CA, DE, EG, ES, LB, NL, SD, VE | 26 | 11.5 |
| 1h | CA, CM, FR | 24 | 10.6 |
| 1i | CA, FR | 3 | 1.32 |
| 1j | CA§ | 3 | 1.32 |
| 1k | CA§ | 4 | 1.76 |
| 1l | CM | 28 | 12.3 |
| 1m | GN | 3 | 1.32 |
| Total | 19 countries | 228 | 100 |
Subtypes 1a and 1b are not listed in this table.
ISO 3166-1 two-letter country codes: CA, Canada; CM, Cameroon; DE, Germany; EG, Egypt; ES, Spain; FR, France; GB, Great Britain; GN, Guinea; ID, Indonesia; IN, India; LB, Lebanon; NL, Netherlands; PK, Pakistan; SD, Sudan; TN, Tunisia; VE, VN, Vietnam; ZA, South Africa.
228 isolates represented by 385 sequences at time of study (see main text).
Patient originally from Haiti.
Table 3. Genotype 1 subtype distribution among continents.
| Continent | No. of subtypes (name of subtypes)* | No. of isolates (n = 228) | Percentage of isolates |
| Africa | 7 (1c, 1d, 1e, 1g, 1h, 1l, 1m) | 108 | 47.4 |
| Asia | 2(1c, 1e) | 85 | 37.3 |
| East Mediterranean | 1(1g) | 2 | 0.877 |
| Europe | 6 (1c, 1d, 1f, 1g, 1h, 1i) | 13 | 5.70 |
| North America | 8 (1c, 1d, 1e, 1g, 1h, 1i, 1j, 1k) | 19 | 8.33 |
| South America | 1 (1g) | 1 | 0.439 |
| Total | 11(1c–1m) | 228 | 100 |
All HCV-1 subtypes except for 1a and 1b were retrieved (see main text).
Because six isolates from this study were classified into subtypes 1e, 1h and 1l, all available sequences belonging to these three subtypes were analysed separately. As shown in Fig. 3(a), these sequences originated most frequently in Cameroon (87/96), while only a few (9/96) were sampled from other countries (e.g. France, Canada and Vietnam; all these locations are linked to colonial-era France). The phylogeny in Fig. 2 suggests that isolates from these three countries were interspersed among those from Cameroon, implying that subtypes 1e, 1h and 1l had origins in Cameroon and were later brought to other continents, most likely by French colonial travellers. This may be also the case for subtype 1d, although no isolates were entirely sequenced in the present study. As summarized in Table 2 and Fig. 2, 1d isolates have been isolated not only in Tunisia, a former French colonial country in Africa, but also in France and The Netherlands in Europe.
Fig. 3.

Distribution of genotype 1 isolates in different countries and in Cameroon. (a) Histogram showing the frequencies of subtype 1e, 1h and 1l in four countries (CM, Cameroon; CA, Canada; FR, France; VN, Vietnam; ?, unknown country). The number of isolates is shown above each column while the country of origin is indicated below. (b) Histogram showing the frequencies of genotype 1 subtypes sampled in Cameroon. The number of isolates is shown above each column.
We also investigated all available HCV-1 sequences from Cameroon. Among these, 15 (representing 13 individual isolates) had no subtype designations. Analysis of the NS5B region sequences from Cameroon (solid diamonds in Fig. 2) revealed that one of them was classified into subtype 1e, two into 1l and three into 1b; the remaining seven formed four independent clusters labelled as subtype 1(II), subtype 1(III), subtype 1(IV) and subtype 1(V), respectively. Similarly, sequences that had subtype designations were also classified. Except for 1e, 1h and 1l, only one isolate of 1a (Njouom et al., 2003b) and five isolates of 1b were from Cameroon. However, after reanalysis, the isolate classified as subtype 1a was found to group into subtype 1e (represented by a solid square in Fig. 2). Thus to date no subtype 1a isolate has been identified in Cameroon. In summary, HCV-1 isolates sampled in Cameroon (102 in total) represented four of the 13 assigned subtypes and five of the six unassigned lineages (Figs 2 and 3b).
Specific variations in the E2 and NS5A regions
The number of amino acids in the E2 and NS5A regions varied from 364 to 371 and from 448 to 449, respectively, among the seven isolates determined in this study (Table 1). Within these two regions, many genetic differences were observed when compared with the H77 strain.
E2 region.
Generally, the E2 protein of HCV has 11 potential glycosylation sites (Slater-Handshy et al., 2004). Compared with the H77 strain, the seven sequences from this study only showed six (N2, N4, N6, N7, N10 and N11) of these 11 sites conserved, while the other five sites (N1, N3, N5, N8 and N9) were variable. The N5 site is sited between two conserved amino acids (P471 and R483 according to H77 numbering). Within this site, many mutations were observed: the 148636 sequence showed an insertion of four amino acids, and the EBW424, 166212, 136142 and 160526 sequences each showed an insertion of five amino acids. However, such insertions were not observed in either EBW9 or EBW443. As shown in Fig. 4 for the E2 protein, amino acid insertions occurred at four positions: E384–T385, H384–K395, H445–K446 and G573–V574.
Fig. 4.
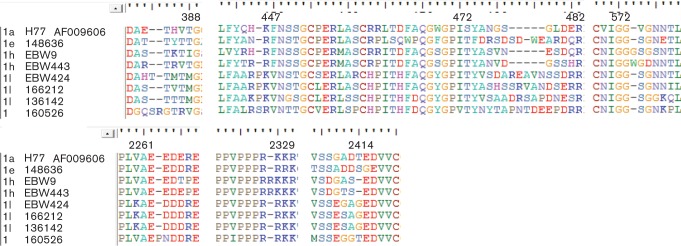
Analysis of amino acid sequences in the E2 and NS5A regions. Amino acids are shown using standard IUPAC codes and bars indicate indels. Isolate names are listed to the left of the alignment while amino acid positions (relative to the H77 reference genome) are indicated above.
NS5A region.
A variety of insertions and deletions were observed in the NS5A region among the new HCV-1 sequences reported in this study (Fig. 4). They were located in the subgenomic region of protein kinase region (PKR)-BD and Domain III. The PKR-BD of the 160526 isolate had an insertion of one amino acid at position 2262–2263; the Domain III of subtype 1h (EBW9 and EBW443) had one insertion at position 2328–2329 and one deletion at position 2414.
Similarity plotting
To exclude the possibility of viral recombination, pairwise nucleotide similarity curves were plotted along HCV genomes using the RDP3 software. Upon comparison of the seven isolates from this study with each other, and with the 45 reference sequences shown in Fig. 1 that represent various HCV genotypes and subtypes, no such evidence was detected (data not shown). Similarity plotting also showed that three hypervariable regions (HVR1, HVR2 and V3) were positioned as previously described (Simmonds, 2004).
Discussion
In this study, full-length genome sequences of seven genotype 1 isolates of HCV were characterized (isolates EBW9, EBW424, EBW443, 136142, 148636, 160526 and 166212). Based on the consensus criteria for HCV classification and nomenclature (Simmonds et al., 2005), isolate 148636 should be classified as subtype 1e, both EBW9 and EBW443 as subtype 1h, EBW424, 136142 and 166212 as subtype 1l, while isolate 160526 may represent a novel as yet unassigned subtype. These classifications are supported not only by analysis of full-length genome sequences but also by analysis of partial Core, E1 and NS5B regions (Figs 1, 2 and S2). Previous nomenclature guidelines (Simmonds et al., 2005) regarded subtypes 1e, 1h and 1l as being provisional subtypes. With the seven full-length genome sequences characterized here, the definition of these three subtypes is now confirmed. However, a formal designation cannot currently be made for the potential new subtype represented by isolate 160526, because no other closely related isolates have been identified; isolate 160526 therefore remains an unassigned subtype equivalent. Within genotype 1, full-length genome sequences are still missing for subtypes 1d, 1f, 1i, 1j, 1k and 1m; this situation should be rectified in future studies in order to further develop HCV nomenclature, which provides important background information for effective HCV prevention and treatment.
Previously, a total of 13 subtypes (1a–1m) of HCV genotype 1 have been proposed (Simmonds et al., 2005). Of these, subtypes 1a and 1b are distributed worldwide and for this reason were not analysed in this study. In addition to subtypes 1a–1m, this study proposes the existence of six genotype 1 lineages that are equivalent to new but unassigned subtypes. Each of these lacks a sufficient number of closely related isolates to meet the criteria for proposing new subtypes, although they showed sufficient genetic distances from other subtypes to qualify. Excluding subtypes 1a and 1b, we also summarized the genetic diversity and geographical distribution of the remaining 11 assigned subtypes and the six unassigned lineages. Genotype 1 isolates most often originated in Cameroon. It has been previously noted that HCV-1 has been historically endemic in Africa, particularly in Central-West Africa (Simmonds et al., 2005). Of the 108 isolates classified into subtypes 1c–1m that were reported as being African in origin, we found isolates of subtypes 1e, 1h and 1l from Cameroon accounted for more than 80 % (87/108) (Fig. 3a) (Ndjomou et al., 2003; Njouom et al., 2003b; Pasquier et al., 2005). This could be taken as evidence suggesting that these three subtypes originated in Cameroon and that from Cameroon they subsequently disseminated to other areas. These subtypes were possibly amplified in Cameroon by unsafe medical interventions, such as the use of intravenous antimalarial drugs and contaminated blood transfusions that are considered to be major historical risk factors for HCV infection among people in Cameroon (Pépin & Labbé, 2008; Pépin et al., 2010). Remarkably, among these three subtypes, four isolates have been detected in Quebec, Canada (Murphy et al., 2007), three in Vietnam (GenBank numbers are AB306393, AB306376 and AB301765), and one in France (Laperche et al., 2005). Historically, these three countries/regions were linked to the former French colonial empire. Similar links among geographical regions for various strains of HCV genotype 2 have been proposed to result from the actions of former European colonial powers (Li et al., 2012; Markov et al., 2012). Such scenarios could have also occurred for a variety of HCV-1 lineages and the currently observed worldwide circulation of subtypes 1a and 1b may thus reflect such consequences. Among the six unassigned lineages of genotype 1, five have also been discovered in Cameroon, namely lineages 1(II), 1(III), 1(IV), 1(V) and 1(VI). This may further suggest Cameroon as the area where HCV-1 originated, although more comprehensive sampling of HCV from other countries of West and Central Africa will be necessary before this hypothesis can be confirmed.
Methods
Subjects and specimens.
Three serum samples, EBW9, EBW443 and EBW424, were collected during 2000–2003 among patients living in Cameroon (Pasquier et al., 2005). Detailed information of the age and gender of these three patients and dates of sampling are shown in Table 1. In addition, four samples (148636, 136142, 166212 and 160526) were provided by Micropathology Ltd in the UK, who identified them as atypical HCV genotype 1 sequences during routine service provision to a range of primary healthcare providers. These samples were obtained from patients who were believed to have acquired their infections in Cameroon.
PCR amplification and sequencing.
Full-length HCV genome sequences were each determined from 140 µl of serum sample using the approaches we have recently described (Li et al., 2012). Briefly, RNA extraction (Qiagen Viral RNA extraction kit, Qiagen) and cDNA synthesis (RevertAid First Strand cDNA Synthesis kit, Fermentas) were performed following the manufacturer’s guidelines; overlapping fragments spanning the full HCV genome were amplified using conventional nested or semi-nested PCR with the primers listed in Table S1 (only degenerate primers are shown). Standard procedures were adopted to avoid possible carryover contamination (Kwok & Higuchi, 1989). At least one negative control, one positive control, and a blank composed of water were included in each of the following steps: RNA extraction, reverse transcription, and the first and second rounds of PCR. After PCR, the amplicons were purified using QIAquick PCR purification kit (Qiagen) according to the manufacturer’s protocol. To obtain consensus sequences that reflected the possible heterogeneity of the viral population within each individual, purified amplicons were sequenced directly in both directions. Some fragments amplified using degenerate primers failed to yield clear sequencing chromatograms. These fragments were cloned into the pGEM-T Easy Vector (Promega), of which four clones each were picked and sequenced. All sequencing reactions were conducted using ABI Prism BigDye 3.0 Terminators with an appropriate primer and resolved on an ABI Prism 3500 genetic analyser (PE Applied Biosystems). Any errors in base calling were corrected using the SeqMan program and the edited sequences inspected for functional ORFs using the EditSeq program. Both programs are part of the Lasergene 8.1 package (dnastar). Finally, the obtained sequences were aligned using BioEdit (Hall, 1999), in which any further manual adjustments and corrections were made.
In the Los Alamos HCV database (Kuiken et al., 2005), full-length HCV genome sequences are available for four HCV-1 subtypes: 1a, 1b, 1c and 1g. An alignment was constructed that comprised nine sequences (GenBank accession numbers AF009606, AJ278830, EU260395, AB249644, EF407502, AM910652, AY051292, AY651061, D14853) representing these four subtypes. This was done in order to design degenerate primers in conserved regions, which was critical to the success of whole genome amplification and was assisted using the BioEdit and PrimerSelect programs of the Lasergene 8.1 package.
To amplify the extreme 5′ ends in a semi-nested PCR, the upstream primer used was H77-5end (30 bp), while the downstream primers were 4-367R or 4-342R (Table S1). This strategy was used for all seven HCV-1 isolates since the extreme 5′ ends of their 5′UTRs are highly conserved. To amplify the 3′ ends, a semi-nested PCR or nested RACE PCR was utilized in which the upstream primers were specific to the 3′ end of the NS5B region, while the downstream primers used were poly(A), NUP or 1-9411R (see Table S1). The primer 1-9411R was designed based on the conserved 3′UTR region upstream to the poly(A) tail.
All PCRs were conducted with an initial denaturation at 95 °C for 3 min, followed by 35 cycles each consisting of 95 °C for 30 s, 55 °C for 30 s and 72 °C for a variable time according to the fragment sizes (about 1 min/kb). The final cycle of extension was at 72 °C for 7–10 min. All PCRs utilized the FastStart Taq DNA polymerase system (Roche).
Phylogenetic analyses and inspection of genome sequences.
The seven full-length HCV genomic sequences obtained were annotated according to the standard nucleotide numbering scheme of the H77 genome, from the extreme 5′ end to the 3′X tail (Kuiken & Simmonds, 2009). To classify subtypes more clearly we retrieved all HCV-1 sequences available in the Los Alamos HCV database that have lengths of greater than 9000 nt. We identified 543 such sequences for subtype 1a, 478 for 1b, three for 1c, one for 1g, and 14 representing unclassified genotype 1 isolates (database accessed on June 25, 2012). From these, two each belonging to subtypes 1a and 1b, all those belonging to subtypes 1c and 1g, as well as the 14 unclassified sequences (13 of which were found to belong to subtype 1a) were retained; these were subjected to phylogenetic analysis together with reference genomes representing genotypes 2, 3, 4, 5, 6 (Simmonds et al., 2005) and 7 (Murphy et al., 2007). Together with the seven sequences obtained in this study, a total of 52 sequences were considered.
These 52 full-length HCV genomic sequences were aligned using BioEdit and investigated with the MEGA5 sequence editor (Tamura et al., 2011). Particular interest was paid to amino acid variation in the interferon-sensitivity determining region (ISDR) (Enomoto & Sato, 1995), the RNA-activated protein kinase region (PKR), and some other domains in the E2 and NS5A regions (Gale & Katze, 1998; Gale et al., 1998; Taylor et al., 1999).
To explore genetic variation and subtype classifications within HCV genotype 1, an NS5B region dataset was assembled that represented all assigned subtypes and unassigned variants. Only two sequences were selected as representatives of subtypes 1a, 1b and 1c, as these subtypes are well known to be prevalent worldwide and are represented by thousands of sequences in the Los Alamos HCV database. However, for all other subtypes (such as 1d–1m) all available partial NS5B sequences were retrieved from the database. To understand the diversity of HCV-1 strains in Cameroon, all HCV-1 isolates sampled in Cameroon were retrieved regardless of whether they had a subtype assigned or not. In total, the NS5B dataset contained 141 HCV-1 sequences, each approximately 340 nt long and corresponding to nucleotide positions 8276–8615 in the H77 genome.
ML phylogenetic trees were reconstructed for the two sequence datasets using mega5 (Tamura et al., 2011). The most appropriate nucleotide substitution model for phylogenetic analysis was determined using the model selection procedure implemented in the program modeltest (Posada & Crandall, 1998). For the full-length and partial NS5B alignments, the GTR+I+Γ6 model was found to be the best. To assess the statistical robustness of phylogenetic groupings, bootstrap analyses were conducted with 500 replicates. The sequence name, sampling country, subtype and accession number were indicated at the tips of the resulting phylogenies.
To exclude recent virus recombination events (Colina et al., 2004; Kalinina et al., 2002, 2004; Lee et al., 2010; Legrand-Abravanel et al., 2007; Noppornpanth et al., 2006), the rdp3 software (Martin et al., 2010) was run with settings as previously described (Lu et al., 2007a). This analysis was only performed for the full-length alignment.
Geographical distribution of HCV-1 sequences.
In the Los Alamos HCV database a total of 76 912 sequences were classified as HCV-1 (including five recombinants; accessed on 25 June 2012). Among these, subtype 1a had 33 086 sequences; subtype 1b had 40 752; subtypes 1c–1m had 385; and 2689 sequences had no subtype designations. To analyse the geographical distribution of HCV-1 sequences, only subtype 1c–1m were included (because subtypes 1a and 1b are distributed worldwide). When the cloning status of these 385 sequences was examined we were able to distinguish 228 individual isolates. Because the seven new genomes in this study all had origins in Cameroon and all (excluding one) belong to subtypes 1e, 1h and 1l, the 228 individual isolates were further analysed to see if they were sampled in Cameroon, or were classified as subtypes 1e, 1h and 1l.
Acknowledgements
The study described was supported by a grant from the National Institute of Allergy and Infectious Diseases (5 R01 AI080734-03A). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. No potential conflicts of interest have been reported. We thank Ms Brandi Miller for assistance in proofreading and editing this manuscript.
Footnotes
Two supplementary figures and one supplementary table are available with the online version of this paper.
References
- Alter M. J. (2007). Epidemiology of hepatitis C virus infection. World J Gastroenterol 13, 2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracho M. A., Saludes V., Martró E., Bargalló A., González-Candelas F., Ausina V. (2008). Complete genome of a European hepatitis C virus subtype 1g isolate: phylogenetic and genetic analyses. Virol J 5, 72 10.1186/1743-422X-5-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colina R., Casane D., Vasquez S., García-Aguirre L., Chunga A., Romero H., Khan B., Cristina J. (2004). Evidence of intratypic recombination in natural populations of hepatitis C virus. J Gen Virol 85, 31–37 10.1099/vir.0.19472-0 [DOI] [PubMed] [Google Scholar]
- Enomoto N., Sato C. (1995). Clinical relevance of hepatitis C virus quasispecies. J Viral Hepat 2, 267–272 10.1111/j.1365-2893.1995.tb00040.x [DOI] [PubMed] [Google Scholar]
- Farci P., Alter H. J., Wong D., Miller R. H., Shih J. W., Jett B., Purcell R. H. (1991). A long-term study of hepatitis C virus replication in non-A, non-B hepatitis. N Engl J Med 325, 98–104 10.1056/NEJM199107113250205 [DOI] [PubMed] [Google Scholar]
- Fu Y., Qin W., Cao H., Xu R., Tan Y., Lu T., Wang H., Tong W., Rong X. & other authors (2012). HCV 6a prevalence in Guangdong province had the origin from Vietnam and recent dissemination to other regions of China: phylogeographic analyses. PLoS ONE 7, e28006 10.1371/journal.pone.0028006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M., Jr, Katze M. G. (1998). Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther 78, 29–46 10.1016/S0163-7258(97)00165-4 [DOI] [PubMed] [Google Scholar]
- Gale M. J., Jr, Korth M. J., Katze M. G. (1998). Repression of the PKR protein kinase by the hepatitis C virus NS5A protein: a potential mechanism of interferon resistance. Clin Diagn Virol 10, 157–162 10.1016/S0928-0197(98)00034-8 [DOI] [PubMed] [Google Scholar]
- Hall T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41, 95–98 [Google Scholar]
- Jeannel D., Fretz C., Traore Y., Kohdjo N., Bigot A., Pê Gamy E., Jourdan G., Kourouma K., Maertens G. & other authors (1998). Evidence for high genetic diversity and long-term endemicity of hepatitis C virus genotypes 1 and 2 in West Africa. J Med Virol 55, 92–97 10.1002/(SICI)1096-9071(199806)55:2<92::AID-JMV2>3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- Kalinina O., Norder H., Mukomolov S., Magnius L. O. (2002). A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J Virol 76, 4034–4043 10.1128/JVI.76.8.4034-4043.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinina O., Norder H., Magnius L. O. (2004). Full-length open reading frame of a recombinant hepatitis C virus strain from St Petersburg: proposed mechanism for its formation. J Gen Virol 85, 1853–1857 10.1099/vir.0.79984-0 [DOI] [PubMed] [Google Scholar]
- Kuiken C., Simmonds P. (2009). Nomenclature and numbering of the hepatitis C virus. Methods Mol Biol 510, 33–53 10.1007/978-1-59745-394-3_4 [DOI] [PubMed] [Google Scholar]
- Kuiken C., Yusim K., Boykin L., Richardson R. (2005). The Los Alamos hepatitis C sequence database. Bioinformatics 21, 379–384 10.1093/bioinformatics/bth485 [DOI] [PubMed] [Google Scholar]
- Kwok S., Higuchi R. (1989). Avoiding false positives with PCR. Nature 339, 237–238 10.1038/339237a0 [DOI] [PubMed] [Google Scholar]
- Laperche S., Lunel F., Izopet J., Alain S., Dény P., Duverlie G., Gaudy C., Pawlotsky J. M., Plantier J. C. & other authors (2005). Comparison of hepatitis C virus NS5b and 5′ noncoding gene sequencing methods in a multicenter study. J Clin Microbiol 43, 733–739 10.1128/JCM.43.2.733-739.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. R., Yearwood G. D., Guillon G. B., Kurtz L. A., Fischl M., Friel T., Berne C. A., Kardos K. W. (2010). Evaluation of a rapid, point-of-care test device for the diagnosis of hepatitis C infection. J Clin Virol 48, 15–17 10.1016/j.jcv.2010.02.018 [DOI] [PubMed] [Google Scholar]
- Legrand-Abravanel F., Claudinon J., Nicot F., Dubois M., Chapuy-Regaud S., Sandres-Saune K., Pasquier C., Izopet J. (2007). New natural intergenotypic (2/5) recombinant of hepatitis C virus. J Virol 81, 4357–4362 10.1128/JVI.02639-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Fu Y., Lu L., Ji W., Yu J., Hagedorn C. H., Zhang L. (2006). Complete genomic sequences for hepatitis C virus subtypes 6e and 6g isolated from Chinese patients with injection drug use and HIV-1 co-infection. J Med Virol 78, 1061–1069 10.1002/jmv.20663 [DOI] [PubMed] [Google Scholar]
- Li C., Lu L., Wu X., Wang C., Bennett P., Lu T., Murphy D. (2009a). Complete genomic sequences for hepatitis C virus subtypes 4b, 4c, 4d, 4g, 4k, 4l, 4m, 4n, 4o, 4p, 4q, 4r and 4t. J Gen Virol 90, 1820–1826 10.1099/vir.0.010330-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Lu L., Zhang X., Murphy D. (2009b). Entire genome sequences of two new HCV subtypes, 6r and 6s, and characterization of unique HVR1 variation patterns within genotype 6. J Viral Hepat 16, 406–417 10.1111/j.1365-2893.2009.01086.x [DOI] [PubMed] [Google Scholar]
- Li C., Cao H., Lu L., Murphy D. (2012). Full-length sequences of 11 hepatitis C virus genotype 2 isolates representing five subtypes and six unclassified lineages with unique geographical distributions and genetic variation patterns. J Gen Virol 93, 1173–1184 10.1099/vir.0.038315-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T. J., Heller T. (2004). Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 127 (Suppl 1), S62–S71 10.1053/j.gastro.2004.09.017 [DOI] [PubMed] [Google Scholar]
- Lu L., Nakano T., Li C., Fu Y., Miller S., Kuiken C., Robertson B. H., Hagedorn C. H. (2006). Hepatitis C virus complete genome sequences identified from China representing subtypes 6k and 6n and a novel, as yet unassigned subtype within genotype 6. J Gen Virol 87, 629–634 10.1099/vir.0.81400-0 [DOI] [PubMed] [Google Scholar]
- Lu L., Li C., Fu Y., Gao F., Pybus O. G., Abe K., Okamoto H., Hagedorn C. H., Murphy D. (2007a). Complete genomes of hepatitis C virus (HCV) subtypes 6c, 6l, 6o, 6p and 6q: completion of a full panel of genomes for HCV genotype 6. J Gen Virol 88, 1519–1525 10.1099/vir.0.82820-0 [DOI] [PubMed] [Google Scholar]
- Lu L., Li C., Fu Y., Thaikruea L., Thongswat S., Maneekarn N., Apichartpiyakul C., Hotta H., Okamoto H. & other authors (2007b). Complete genomes for hepatitis C virus subtypes 6f, 6i, 6j and 6m: viral genetic diversity among Thai blood donors and infected spouses. J Gen Virol 88, 1505–1518 10.1099/vir.0.82604-0 [DOI] [PubMed] [Google Scholar]
- Lu L., Murphy D., Li C., Liu S., Xia X., Pham P. H., Jin Y., Hagedorn C. H., Abe K. (2008). Complete genomes of three subtype 6t isolates and analysis of many novel hepatitis C virus variants within genotype 6. J Gen Virol 89, 444–452 10.1099/vir.0.83460-0 [DOI] [PubMed] [Google Scholar]
- Lu L., Li C., Yuan J., Lu T., Okamoto H., Murphy D. G. (2013). Full-length genome sequences of five hepatitis C virus isolates representing subtypes 3g, 3h, 3i and 3k, and a unique genotype 3 variant. J Gen Virol 94, 543–548 10.1099/vir.0.83460-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markov P. V., Pepin J., Frost E., Deslandes S., Labbé A. C., Pybus O. G. (2009). Phylogeography and molecular epidemiology of hepatitis C virus genotype 2 in Africa. J Gen Virol 90, 2086–2096 10.1099/vir.0.011569-0 [DOI] [PubMed] [Google Scholar]
- Markov P. V., van de Laar T. J., Thomas X. V., Aronson S. J., Weegink C. J., van den Berk G. E., Prins M., Pybus O. G., Schinkel J. (2012). Colonial history and contemporary transmission shape the genetic diversity of hepatitis C virus genotype 2 in Amsterdam. J Virol 86, 7677–7687 10.1128/JVI.06910-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P., Lemey P., Lott M., Moulton V., Posada D., Lefeuvre P. (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D. G., Willems B., Deschênes M., Hilzenrat N., Mousseau R., Sabbah S. (2007). Use of sequence analysis of the NS5B region for routine genotyping of hepatitis C virus with reference to C/E1 and 5′ untranslated region sequences. J Clin Microbiol 45, 1102–1112 10.1128/JCM.02366-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndjomou J., Pybus O. G., Matz B. (2003). Phylogenetic analysis of hepatitis C virus isolates indicates a unique pattern of endemic infection in Cameroon. J Gen Virol 84, 2333–2341 10.1099/vir.0.19240-0 [DOI] [PubMed] [Google Scholar]
- Nishioka K. (1991). Hepatitis C virus infection in Japan. Gastroenterol Jpn 26 (Suppl 3), 152–155 [DOI] [PubMed] [Google Scholar]
- Njouom R., Pasquier C., Ayouba A., Gessain A., Froment A., Mfoupouendoun J., Pouillot R., Dubois M., Sandres-Sauné K. & other authors (2003a). High rate of hepatitis C virus infection and predominance of genotype 4 among elderly inhabitants of a remote village of the rain forest of South Cameroon. J Med Virol 71, 219–225 10.1002/jmv.10473 [DOI] [PubMed] [Google Scholar]
- Njouom R., Pasquier C., Ayouba A., Sandres-Sauné K., Mfoupouendoun J., Mony Lobe M., Tene G., Thonnon J., Izopet J., Nerrienet E. (2003b). Hepatitis C virus infection among pregnant women in Yaounde, Cameroon: prevalence, viremia, and genotypes. J Med Virol 69, 384–390 10.1002/jmv.10300 [DOI] [PubMed] [Google Scholar]
- Njouom R., Nerrienet E., Dubois M., Lachenal G., Rousset D., Vessière A., Ayouba A., Pasquier C., Pouillot R. (2007). The hepatitis C virus epidemic in Cameroon: genetic evidence for rapid transmission between 1920 and 1960. Infect Genet Evol 7, 361–367 10.1016/j.meegid.2006.10.003 [DOI] [PubMed] [Google Scholar]
- Njouom R., Caron M., Besson G., Ndong-Atome G. R., Makuwa M., Pouillot R., Nkoghé D., Leroy E., Kazanji M. (2012). Phylogeography, risk factors and genetic history of hepatitis C virus in Gabon, Central Africa. PLoS ONE 7, e42002 10.1371/journal.pone.0042002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noppornpanth S., Lien T. X., Poovorawan Y., Smits S. L., Osterhaus A. D., Haagmans B. L. (2006). Identification of a naturally occurring recombinant genotype 2/6 hepatitis C virus. J Virol 80, 7569–7577 10.1128/JVI.00312-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier C., Njouom R., Ayouba A., Dubois M., Sartre M. T., Vessière A., Timba I., Thonnon J., Izopet J., Nerrienet E. (2005). Distribution and heterogeneity of hepatitis C genotypes in hepatitis patients in Cameroon. J Med Virol 77, 390–398 10.1002/jmv.20468 [DOI] [PubMed] [Google Scholar]
- Pépin J., Labbé A. C. (2008). Noble goals, unforeseen consequences: control of tropical diseases in colonial Central Africa and the iatrogenic transmission of blood-borne viruses. Trop Med Int Health 13, 744–753 10.1111/j.1365-3156.2008.02060.x [DOI] [PubMed] [Google Scholar]
- Pépin J., Lavoie M., Pybus O. G., Pouillot R., Foupouapouognigni Y., Rousset D., Labbé A. C., Njouom R. (2010). Risk factors for hepatitis C virus transmission in colonial Cameroon. Clin Infect Dis 51, 768–776 10.1086/656233 [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- Pybus O. G., Barnes E., Taggart R., Lemey P., Markov P. V., Rasachak B., Syhavong B., Phetsouvanah R., Sheridan I. & other authors (2009). Genetic history of hepatitis C virus in East Asia. J Virol 83, 1071–1082 10.1128/JVI.01501-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M., Tian Y., Delaney W. E., IV, Greenstein A. E. (2011). Preexisting drug-resistance mutations reveal unique barriers to resistance for distinct antivirals. Proc Natl Acad Sci U S A 108, 10290–10295 10.1073/pnas.1101515108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. (2004). Genetic diversity and evolution of hepatitis C virus – 15 years on. J Gen Virol 85, 3173–3188 10.1099/vir.0.80401-0 [DOI] [PubMed] [Google Scholar]
- Simmonds P., Bukh J., Combet C., Deléage G., Enomoto N., Feinstone S., Halfon P., Inchauspé G., Kuiken C. & other authors (2005). Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42, 962–973 10.1002/hep.20819 [DOI] [PubMed] [Google Scholar]
- Slater-Handshy T., Droll D. A., Fan X., Di Bisceglie A. M., Chambers T. J. (2004). HCV E2 glycoprotein: mutagenesis of N-linked glycosylation sites and its effects on E2 expression and processing. Virology 319, 36–48 10.1016/j.virol.2003.10.008 [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. R., Shi S. T., Romano P. R., Barber G. N., Lai M. M. (1999). Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 285, 107–110 10.1126/science.285.5424.107 [DOI] [PubMed] [Google Scholar]
- Wang Y., Xia X., Li C., Maneekarn N., Xia W., Zhao W., Feng Y., Kung H. F., Fu Y., Lu L. (2009). A new HCV genotype 6 subtype designated 6v was confirmed with three complete genome sequences. J Clin Virol 44, 195–199 10.1016/j.jcv.2008.12.009 [DOI] [PubMed] [Google Scholar]
- Xia X., Lu L., Tee K. K., Zhao W., Wu J., Yu J., Li X., Lin Y., Mukhtar M. M. & other authors (2008). The unique HCV genotype distribution and the discovery of a novel subtype 6u among IDUs co-infected with HIV-1 in Yunnan, China. J Med Virol 80, 1142–1152 10.1002/jmv.21204 [DOI] [PMC free article] [PubMed] [Google Scholar]