

Abstract
Significance: Alzheimer disease (AD) is an age-related neurodegenerative disease. AD is characterized by progressive cognitive impairment. One of the main histopathological hallmarks of AD brain is the presence of senile plaques (SPs) and another is elevated oxidative stress. The main component of SPs is amyloid beta-peptide (Aβ) that is derived from the proteolytic cleavage of amyloid precursor protein. Recent Advances: Recent studies are consistent with the notion that methionine present at 35 position of Aβ is critical to Aβ-induced oxidative stress and neurotoxicity. Further, we also discuss the signatures of oxidatively modified brain proteins, identified using redox proteomics approaches, during the progression of AD. Critical Issues: The exact relationships of the specifically oxidatively modified proteins in AD pathogenesis require additional investigation. Future Directions: Further studies are needed to address whether the therapies directed toward brain oxidative stress and oxidatively modified key brain proteins might help delay or prevent the progression of AD. Antioxid. Redox Signal. 19, 823–835.
Introduction
Alzheimer disease (AD) is an age-related neurodegenerative disorder that affects a large and ever-growing population of Americans 65 years of age or older, a number that current estimates place at ∼5.1 million and that may grow to nearly 20 million by the year 2050 due to an aging Baby Boomer population (63). Pathologically, AD is characterized by a loss of synapses, an increase in the number of extracellular amyloid beta-peptide (Aβ)–rich senile plaques (SPs) formed from the amyloidogenic processing of amyloid precursor protein (APP) (discussed later), and an increase in intracellular neurofibrillary tangles (NFTs) composed of aggregated hyperphosphorylated Tau, a microtubule stabilizing protein (81). In the last decade or so, strong evidence has been put forth linking AD to an increase in oxidative stress due in part to both the increased production of reactive oxygen and nitrogen species (ROS and RNS, respectively) and a loss of function of many antioxidant defense enzymes (3,13,20,21,84,94,95,110,153,162).
Alzheimer Diagnosis and Staging
AD is clinically characterized by a decline in episodic memory that is often mistaken for normal cognitive deficiencies due to aging. Because the pathology remains hidden within the brain tissue, clinical diagnosis during the early stages remains inherently error prone and subjective, though advances have been made to aid a physician in making a correct diagnosis (100,167). To date, physicians have access to tools designed to help with diagnosis such as the Mini Mental State Evaluation (MMSE), which is used to track a patient's cognitive prowess (a 30-point scale is used with >25 being normal and <25 as probable AD), and also imaging alternatives such as magnetic resonance imaging (MRI) and positron emission tomography (PET) scans, which visualize both the potential hippocampal, sulci, and gyri degeneration and decreased glucose utilization (42,48,71,150).
Progression of typical AD can be stratified into four main stages: preclinical AD (PCAD), mild cognitive impairment (MCI), early AD (EAD), and late-stage AD (LAD). PCAD is defined as the potential stage of AD in which the patient presents as a fully functional individual in cognitive exams such as MMSE, yet the growing pathology within the brain tissue is present, but likely unknown precluding early death from a non-neurodegenerative means (141,144,166). MCI has been described as being the transition stage between normal cognition and EAD, and is subdivided into both amnestic MCI (aMCI) and non-amnestic MCI, the former of the two presenting with memory deficits and maintains a 10%–15% conversion rate per year to AD (123,124,150). Pathologically, each stage differs in that both amyloid plaques and NFTs increase in distribution and density from MCI to LAD, though non-demented individuals have also been known to possess both types of pathology while maintaining normal cognition (159). Through use of imaging techniques such as MRI, all stages of clinical AD described demonstrate varying degrees of degeneration, with MCI presenting relatively small degeneration affecting the hippocampus, sulci, and gyri, whereas a larger degree affects the same brain regions in LAD accompanied by additional atrophy of the frontal lobe and ventricular widening in EAD and LAD (45,47,71). Additionally, research conducted using PET scans concluded that regional glucose utilization within the brain, including the temporal lobe, was significantly reduced in AD patients, a trend that was shown to remain for possible PCAD and MCI patients, indicating a severe energy deficiency, as glucose is known to be the predominant source of energy for the brain (34,35,42,66,125).
APP Processing
The main component of SPs is a 4-kDa protein called Aβ (61,99). Aβ is generated by the proteolytic cleavage of APP, a type I transmembrane protein suggested to play an important role in neurite outgrowth, neuronal protein trafficking, signal transduction, calcium metabolism, and others. (176). There are three major alternate splicing variants with APP770, APP751, and APP695.
In the amyloidogenic pathway, APP is cleaved by β-secretase (also referred to as β-site APP-cleaving enzyme) (163) at position 671, resulting in the release of a large N-terminal derivative called β-secretase-cleaved soluble APP (β-sAPP) (Fig. 1). The β-sAPP differ from α-secretase-cleaved soluble APP (α-sAPP) (produced from non-amyloidogenic processing, Fig. 2) by lacking the Aβ(1–16) regions at its C-terminus, but it has been reported to function as a death receptor 6 ligand and also mediate axonal pruning and neuronal cell death (114). The toxicity of C-terminal fragment (CTF) may possibly be mediated by the end products of γ- and/or caspase-cleavage including APP intracellular domain (AICD), C31, and Jcasp. Caspases can cleave APP at position Asp664 resulting in the formation of 31-amino-acid peptide of APP referred to as C31. C31 has been shown to induce cytotoxicity. Further, cleavage by γ-secretase generates JCasp (52,119); however, JCasp has been reported to play a minor role in cytotoxicity. In a transgenic mouse, mutation at caspase cleavage site in APP prevented AD-associated changes suggesting that caspase cleavage of APP might be crucial for Aβ-mediated neurotoxicity. In the next step, the 99-amino-acid CTF of APP (C99) is cleaved by the γ-secretase complex releasing free peptides ranging from 38 to 43 amino acids referred to as Aβ, P83 fragment, and AICD (Fig. 2). Hence, the γ- cleavage is critical for the amount and type of Aβ produced.
FIG. 1.

A general depiction of amyloidogenic processing. Amyloid precursor protein (APP) is cleaved by β-secretase followed by γ-secretase within the bilayer to produce a fragment of amyloid-beta peptide (Aβ), sAPPβ, and AICD. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 2.

A general depiction of non-amyloidogenic processing. APP is cleaved by α-secretase followed by γ-secretase within the bilayer to produce a fragment of P3, sAPPα, and APP intracellular domain (AICD). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.
Aβ40 represents the most abundant form of Aβ in the brain, while Aβ42 shows a significant increase with certain forms of AD (112). Aβ42 has two extra hydrophobic amino acids compared to Aβ40, which promotes greater fibrillar formation in Aβ42 and is known to be more toxic (Fig. 3). The evidence of Aβ toxicity was provided by molecular pathology, human genetics, and discoveries from cell biology (13,16,38,169). The increased hydrophobicity of Aβ42 possibly allows this peptide to integrate within the lipid bilayer initiating the process of cell damage. Schmidt et al. using mass-per-length measurements and electron cryomicroscopy with 3-dimensional reconstruction on an Aβ(1–42) amyloid fibril morphology showed that the Aβ(1–42) fibril morphology has only one protofilament, in contrast to Aβ(1–40) fibril forms two protofilaments. Further, Aβ(1–42) showed pairs of β-sheets at the cores of the two protofilaments making up a fibril (135).
FIG. 3.

Amino acid sequence of beta-amyloid peptides. Red color indicates the two additional hydrophobic amino acids that are present in beta-amyloid (1–42), which is critical for higher aggregation rate of beta-amyloid (1–42), and its associated neurotoxicity (please see text for more details). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Once Aβ is produced, individual amyloid peptides (Aβ42 in particular) aggregate to form small assemblies of dimers, trimers, oligomers, protofibrils, and large insoluble fibrils. Studies showed poor correlation between plaque load and cognitive function (113). Recently, the role of Aβ has been amended to suggest that soluble Aβ oligomers are the more toxic species. Further research has indicated that the soluble oligomers and not the plaques correlate well with cognitive decline (44,53,54,117,165,168). Moreover, Aβ levels and temporal NFT density have been shown to be elevated to a higher degree in LAD when compared with MCI and EAD, which are likewise elevated compared with control (9,11,58,108,159). The relationship between Aβ-containing SPs and NFT formation has been debated, but recently Jin et al. reported that with the addition of soluble Aβ dimers, tau became hyperphosphorylated before cytoarchitectural disruption was observed, followed by subsequent neuritic degeneration. Interestingly, this process was exacerbated with the overexpression of human tau and prevented with the knockdown of human tau (74). Soluble Aβ has also been shown to modulate the pro-survival PI3K/AKT-GSK3β pathway, inhibiting various neurotrophin effects including that of α-sAPP (73). These lines of evidence provide insight into the progression of AD and a potential causal relationship between two known pathological hallmarks of this disease.
Genetic Evidences for Aβ Toxicity
The importance of APP and consequently Aβ in AD pathogenesis has emanated from genetic evidence of patients with familial AD (FAD) and Down syndrome (DS). After the cloning of the APP gene, a mutation causing FAD (autosomal dominant) was found at codon 717, close to the C-terminus of the Aβ domain of APP (55). Today, there are at least seven known APP mutations causing FAD (56,138). Interestingly, all APP mutations are located in or near the Aβ region of APP, close to the secretase sites. To date, 32 mutations in APP have been reported, and based on their locations they are grouped into three main classes: the Swedish mutation, located adjacent to the β-cleavage site of APP; London mutations, Flemish mutation, located near the γ-site of APP; the Arctic, Dutch, and the Iowa mutations, located within the Aβ sequence itself. All the APP mutations are found to alter the proteolytic processing of APP, resulting in either increased production of total Aβ or a selective increase in the 42-amino-acid form of Aβ (56,138). In addition to mutations of APP, 177 mutations in presenilin 1 (PS1) (392 families) and 14 mutations in PS2 (23 families) has been identified in FAD, which further support the role of altered APP metabolism in AD pathogenesis. The evidence of involvement of Aβ(1–42) in AD pathogenesis is largely derived by observation of increased Aβ load and increased oxidative stress in FAD. Individuals with FAD mutations consistently show increases in the ratio of Aβ42/40, suggesting that elevated levels of Aβ42 is critical for AD pathogenesis (72,134).
DS patients have three copies of chromosome 21, and the APP gene is present on this chromosome; hence, if these patients live long enough they develop neuropathological features indistinguishable from AD. Further, DS patients had increased accumulation of intracellular Aβ preceding extracellular plaque formation, and the level of intraneuronal Aβ decreases as the extracellular Aβ plaques accumulate (59,109). Further, DS brain also has elevated oxidative stress (32,78,120,121).
Aβ and ROS/RNS in AD
In AD brain, increased levels of Aβ were found in the affected regions; however, Aβ42 is also the predominating form of Aβ in SPs (106), while shorter Aβ proteins predominate in both vascular amyloid and in cerebral spinal fluid (CSF) (106,164). Further, AD CSF showed reduced levels of Aβ42 compared with Aβ40 suggesting that the deposition of the protein in SPs in brain leads to reduced levels of Aβ in the CSF. In AD plasma, the levels of Aβ is controversial, one study found an increase in plasma Aβ42 (102). Most of the studies did not find any change in plasma Aβ42 between AD patients and age-matched controls (70,106). Current data do not provide clear-cut evidence that Aβ protein in plasma/CSF reflects the amount of Aβ deposited in the brain or that plasma/CSF Aβ42 has a potential as a biomarker for AD. Further studies are needed to develop biomarkers for AD diagnosis and therapeutic efficacy.
Several lines of evidence indicate that Aβ induces oxidative stress. Oxidative stress that occurs within the bilayer, hypothesized in the Aβ-induced oxidative stress hypothesis in which Aβ1–42 inserts as oligomers into the bilayer and serves as a source of ROS, has been shown to initiate lipid peroxidation (Figs. 4 and 5) (16,17,93,94,101). For a comprehensive review on oxidative/nitrosative stress in the cell, the reader is referred to the following articles (28,29,151).
FIG. 4.

Schematic illustration of HNE-modified protein. Upon formation of a radical centered allylic carbon on a fatty acid chain, the lipid may interact with molecular O2 that freely diffuses through the bilayer because of its lack of dipole moment, to initiate the lipid peroxidation process that eventually, by way of a proposed Hock cleavage, generates an α/β unsaturated reactive aldehyde [e.g., 4-hydroxy-nonenal (HNE), malondialdehyde, and acrolein]. Membrane-bound proteins may then, by way of nucleophilic side chains such as Cys, Lys, and His, covalently bind the aldehyde that alters the structure and function of the target protein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 5.

Some consequences of elevated ROS and RNS. Reactive oxygen species (ROS) leaked from mitochondria (e.g., O2−•) interact with nitric oxide (NO) produced by nitric oxide synthase (NOS) to produce reactive nitrogen species such as ONOO−, which covalently modify proteins. O2−• can also directly oxidize proteins, lipids, and carbohydrates. O2−• may also be dismutated to H2O2 by superoxide dismutase (SOD) enzymes in an attempt to mitigate O2−• induced damage. However, hydrogen peroxide (H2O2) in the presence of Fe2+ or Cu+ undergoes Fenton chemistry to produce the reactive ROS •OH and −OH, which also cause protein, nucleic acid, and carbohydrate oxidation. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Oxidative Stress at Different Stages of AD
Oxidative stress and its effects have been found as early as MCI in the progression toward AD. Studies conducted in our laboratory and others have found that oxidative stress markers for protein oxidation/nitration, such as protein carbonyls and 3-nitro-tyrosine, are elevated in brains from subjects with aMCI (6–8,25,83). More recently, it has been shown that the phosphorylation profile of proteins such as heme-oxygenase-1 and biliverdin reductase A have been altered in MCI and AD indicating the possibility of aberrant signaling in at least this one critical antioxidant pathway. Increased levels of 8-OHdG, 8-OHG, 5-hydroxycytosine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine, and 4,6-diamino-5-formamidopyrimidine, all markers of nucleic acid oxidation, were found in both mitochondrial DNA and nuclear DNA indicating nucleic acid oxidation in MCI (105,171). 8-OHdG (also found elevated in CSF of AD patients), 8-OHA, and 5-OHU were found in AD brain regions demonstrating that though DNA is more protected from oxidation than RNA, oxidation still occurs (2,50,104,150).
Significant RNA oxidation has been shown to exist in AD, as has been found in the earlier stages of the disease. A high percentage (30%–70%) of mRNA in the frontal cortex was shown to be oxidized in AD brain (139). In EAD brain, 8-OHG was found to be elevated in the cytoplasm of AD hippocampus, frontal, and occipital neocortex, which correlated with the β-amyloid load (89,115,116,140). Ribosomal RNA oxidation was observed in the superior middle gyri and inferior parietal lobule (IPL) of AD brain (43). 8-OHG levels decreased with increased Aβ and NFT levels, a finding that suggests that at the early stages of AD, oxidative damage to RNA may be an early event in AD progression (115).
Increased protein-bound 4-hydroxy-nonenal (HNE) and free HNE, TBARS, and MDA were found, and a higher isoprostane (F2isoP) level in plasma, urine, and CSF in MCI when compared with healthy controls (83,96,173). There have been high levels of free and protein-bound HNE found in AD brain (16,23,86,88,97,103,122). In addition to lipid peroxidation, protein carbonyls were found to be increased in regions of the brain heavily associated with AD, including the hippocampus and parietal cortex, while leaving the cerebellum relatively untouched (64). Moreover, another index of protein oxidation, protein nitration, was also found to be increased in the CSF and AD brain in regions such as the IPL, neocortical regions, and the hippocampus (7,31,65,143,155). Increased protein nitration and protein-bound HNE were found in brains of subjects with EAD (130,131). Inversely correlated to the increase in oxidation observed was the activity of antioxidant systems (both enzymatic and nonenzymatic) found by Sultana et al. and Guidi et al. while no changes in total protein levels were observed, which may be a result of, and contribute to, the observed increase in free radicals during the progression of AD (57,153).
Redox Proteomics Studies of MCI, EAD, and AD
Redox proteomics is a method of identification of oxidatively modified proteins pioneered by our laboratory that employs redox-specific antibodies, two-dimensional polyacrylamide gel electrophoresis, and tandem-mass spectrometry (MS/MS) with the identification of specific proteins based on their tryptic peptide amino acid sequence after interrogation of protein databases such as SwissProt (41,67,68). Our laboratory has identified proteins in MCI, EAD, and AD brain that are vital to cellular function as being oxidatively modified and dysfunctional; however, for the sake of this review only a select few will be discussed. For a discussion of oxidatively modified proteins discovered by our laboratory using redox proteomics, the reader is referred to articles cited here (4,24,27,41,150).
Sultana et al. found that the important protein regulator Pin1 is oxidized and activity decreased (148). Recently, there has been much interest in the area of regulation via the phosphorylation specific peptidyl-prolyl cis-trans isomerase (PPIase), Pin1, and its role in neuronal cell cycle checkpoints and cellular phosphorylation status in diseases such as AD and cancer (5,14,46,82). Pin1 recognizes the specific motif of phosphorylated serine or threonine on the amino-terminal side of an adjacent proline (pSer/Thr-Pro) and catalyzes the isomerization of the peptide bond (90,128). This regulation has been shown to be important in the phosphorylation status of both APP and Tau, and some kinases and phosphatases that act on those target proteins, giving Pin1 both a direct and indirect regulation of two key pathological hallmarks of AD (14,85,87,92).
Another link between the stages of AD is the presence of oxidatively modified proteins important to cellular energy production (150). Three enzymes, α-enolase, adenosine-triphosphate-synthase, and lactate dehydrogenase were implicated as being oxidatively modified in brains of subjects with aMCI and AD, while α-enolase in particular was found to modified in EAD as well (25,30,31,122,129,149,152,155,156). Additionally, enolase was identified by redox proteomics as oxidatively modified in brains of subjects with FAD (19).
The activity of α-enolase as a glycolytic protein is well understood. Consequently, the oxidative modification and subsequent loss of activity may significantly hinder energy production (150). α-Enolase however, possesses nonglycolytic activities in signaling pathways important to cell survival and in Aβ clearance (22). Evidence also suggests that α-enolase may be a neurotrophic factor, play a role in hypoxic stress regulation, and have transcription factor capabilities (1,62,147,158).
The examples of Pin1 and α-enolase were selected to demonstrate the power of redox proteomics in identifying specific links in cell signaling pathways that are damaged and dysfunctional as opposed to global tissue oxidation, and free radical induced oxidative stress of enzymatic proteins with multifunctional roles that may have far-reaching effects. In using redox proteomics, researchers may identify proteins that are more susceptible to oxidative modification and from this information garner insight regarding the progression and possibly even potential treatment for diseases such as AD.
Role of Methionine in Aβ-Induced Oxidative Stress
Studies from our laboratory and others showed Met-35 of Aβ peptides is critical for Aβ-associated toxicity and oxidative stress (15,160,174,175). Met can undergo two-electron oxidation to form methionine sulfoxide (MetSOx) (127,137). Oxidation of Met to the sulfoxide might play an important role in the regulation of protein function or cellular defense mechanism (145). Further, the presence of methionine sulfoxide reductase (MSR), which catalyzes the conversion of MetSOx to Met (91,98,146), suggests that MSR might play an antioxidant role. Interestingly, in AD brain the activity of MSR is less, and a significant fraction of SP-resident Aβ peptide has Met in the form of MetSOx (112), suggesting that Met oxidation might play an important role AD progression and pathogenesis (49). However, in vitro studies showed that Aβ with MetSOx is less toxic at a shorter incubation time (160), this could possibly be related to altered production of toxic Aβ oligomers (75,107).
In addition, Aβ-resident Met in the lipid bilayer can undergo one-electron oxidation forming sulfuranyl free radical [MetS+]. Since, Aβ is generated from cleavage of APP, a transmembrane protein as discussed above, we proposed that Aβ once produced can insert as small oligomers into the lipid bilayer adopting an α-helical conformation (9,175). According to the α-helix conformation rule of i+4 rule, that is, every fourth amino acid interacts; hence, the Met-35 S-atom would interact with carbonyl oxygen of Ile-31 (79,80,137) (Fig. 6). Since oxygen of Ile31 is more electronegative than sulfur it will pull the electron density toward it, making the S-atom in Met-35 more vulnerable to one-electron oxidation to form sulfuranyl free radical [MetS+] on Met (79,136,160) (Fig. 7). The substitution of Ile-31 by proline, an α-helix breaker, abrogates the oxidative stress and neurotoxicity associated with Aβ(1–42) (80), suggesting that the secondary structure of Aβ(1–42) contributes to reactivity of the neurotoxic peptide. However, until now the source of oxidant that triggers this event largely remains unknown. It is proposed that either molecular oxygen or Cu2+ might be key in the oxidation of Met to the sulfuranyl radical. In the absence of oxygen, Aβs cannot lead to free radical production (161). Prior studies showed that Aβ(1–42) has Cu/Zn SOD-like properties (39), and that amyloid plaques had high levels of copper (33). In vitro studies showed that Aβ(1–42) can promote the reduction of peptide-bound Cu2+ to Cu+ and form hydrogen peroxide (H2O2). Further Cu+, can react with the H2O2 to form highly reactive, hydroxyl free radicals (69,76). Further, chelation of copper by clioquinol (CQ, 5-chloro-7-iodoquinolin-8-ol), hydroxyquinoline antibiotic that has nanomolar affinity for Cu2+ (118), reduced the formation of H2O2 by Aβ (12,142,172). In vivo studies showed that oral administration of the clioquinol in Tg2576 mice reduced amyloid levels. Further, Phase 2 clinical trial showed that CQ slowed the rate of cognitive decline and reduced the plasma Aβ42 levels in moderately severe AD patients (132). The importance of copper in Aβ-induced toxicity is suggested by a study where Met35 was substituted by Val that showed to increase the toxicity (36), suggesting that this substitution might lead to a change in the conformation of Aβ from α-helix to a mixture of α-helical and β-sheet conformations, thereby increasing the binding of Cu+2 and subsequently its associated toxicity. Further, substitution of His 6,13,14 in Aβ(1–42) by Tyr, which binds Cu2+ with less affinity than His, showed that it did not affect the oxidative stress and neurotoxicity further emphasizing the importance of Met-35 in the Aβ-induced toxicity and oxidative stress (10,160). Further research is needed to understand the role of copper in Aβ.
FIG. 6.

A pictorial representation of Aβ oligomerization and insertion into the bilayer. When inserted into the bilayer, Aβ forms an α-helix that allows the peptide backbone carbonyl of Ile-31 to come within Van der Waals distance of the sulfur atom on Met-35, as explained by the i+4 rule of α-helicies. This interaction allows for the formation of a sulfuranyl radical that leads to a catalytic lipid peroxidation process. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 7.

A proposed mechanism for the Aβ-induced free radical stress hypothesis. As shown, the electron density surrounding the sulfur atom of Met-35 is pulled away by the more electronegative oxygen of the carbonyl located on the peptide backbone at the position of Ile-31. As discussed, the carbonyl is within Van der Waals distance to the sulfur, which primes the lone pair on the sulfur for one-electron oxidation, forming the sulfuranyl radical. Because this occurs within the bilayer, unsaturated lipids are present, allowing for an allylic hydrogen atom abstraction by the sulfuranyl radical to eventually form a reduced Met-35 that recycles back upon deprotonation to the starting conditions for another cycle. The carbon centered radical may then go on to undergo peroxidation to create reactive aldehydes or may directly interact with another protein or lipid in a radical propagation step. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Once MetS+ radical is generated it can abstract allylic H atoms from the acyl chains of unsaturated fatty acids in the lipid bilayer to initiate the process of lipid peroxidation (60), and consequently affect the lipid bilayer. The products of oxidation further diffuse through the membrane affecting other cellular compartments, greatly amplifying the effect of the original Aβ-centered free radical, eventually leading to cell loss and AD. Consistent with this model, we substituted Gly at residue 37 of Aβ(1–42) by aspartic acid. The effect of this negatively charged amino acid was to remove the Met-35 residue from the bilayer, and no oxidative stress was observed in neuronal cultures (93). Vitamin E, a chain-breaking antioxidant blocks the chain reaction in the mechanism of lipid peroxidation, preventing oxidative stress to neurons (80). However, clinical trials conducted using vitamin E for the most part did not show beneficial effects in AD, which could be due to experimental design (77).
The earliest study using transgenic Caenorhabditis elegans expressing human Aβ(1–42) showed increased oxidation that correlated with the phenotypic expression (e.g., paralysis) of the worm (170,175), which was confirmed by others (44). However, when the Met-35 was substituted by Cys no oxidative stress was found, but the deposition of modified Aβ(1–42) was not altered (175). Consistent with the role of Met, an in vitro study demonstrated that when the sulfur atom of methionine in Aβ(1–42) was substituted by a methylene moiety [Aβ(1–42)M35NLE] that has the same side chain length and hydrophobicity as Met (175), Aβ loses its associated free radical formation, oxidative stress, and toxicity (37,40,111). In contrast, some studies suggested that the 33–35 region of Aβ (25–35) is critical for the aggregation and neurotoxic properties of Aβ peptide, but substitution of Met by norleucine did not reduce the toxicity associated with this peptide (126). However, the chemistry of C-terminal Met is entirely different than Met within the peptide chain.
A recent study from our laboratory used for the first time an in vivo mammalian model to show that Aβ-resident Met-35 is critical to oxidative stress and neurotoxicity (18). In this study the PDAPP mouse, with Swedish and Indiana familial mutations of APP, has a third mutation introduced: substitution of leucine in APP at M631, corresponding to Met-35 of Aβ(1–42) (18). These mice were referred to as PDAPPM631L mice. In contrast, to the brain from PDAPP mice, which demonstrate oxidative stress, brain from PDAPPM631L mice showed no in vivo oxidative stress. In addition, punctate deposits of Aβ(1–42) were found in the latter brain compared to frank amyloid deposits in the brain of PDAPP mice, suggesting that Aβ(1–42)-resident Met not only affects in vivo oxidative stress but also affects plaque formation. Interestingly, Met substitution in Aβ(1–42) did not rescue spatial learning and memory deficits at 6 months of age as assessed by the Morris water maze test. Given that APP is processed to produce toxic sAPPβ and other toxic fragments of APP, this result may not be surprising. Other, more sensitive cognitive tests are needed to better understand the effect of the loss of Met on learning and memory. Proteomics analysis on brain from PDAPPM631L mice showed reduced oxidation of key proteins that are critical in regulating cellular pathways such as energy metabolism, cellular defense, protein degradation, and pH regulation compared to PDAPP mice (133,157). The decreased oxidation in general and reduced oxidation of key proteins like Pin1 (Pin1, discussed earlier) might play an important role in preventing AD pathogenesis (157).
Conclusion
The overproduction and accumulation of Aβ are key to the progression and pathogenesis of AD. Hence, the use of treatments to reduce Aβ formation or the downstream oxidative stress associated with Aβ could be helpful in preventing, treating, or delaying the progression of AD (51). There are various approaches that could be potential candidates to reduce Aβ levels: inhibiting Aβ production (by inhibiting secretase enzymes) or increasing the clearance of Aβ or using compounds that bind Aβ to impair aggregation (Fig. 8). Dissolving the extant SP may not be a good idea to combat this devastating disease, since oligomeric Aβ, the likely main toxic species of this peptide would be elevated by simple equilibrium considerations. Studies are in progress in our laboratory and others to further delineate the mechanism of Aβ-associated toxicity and develop a regimen to treat, slow, or hopefully one day prevent AD.
FIG. 8.
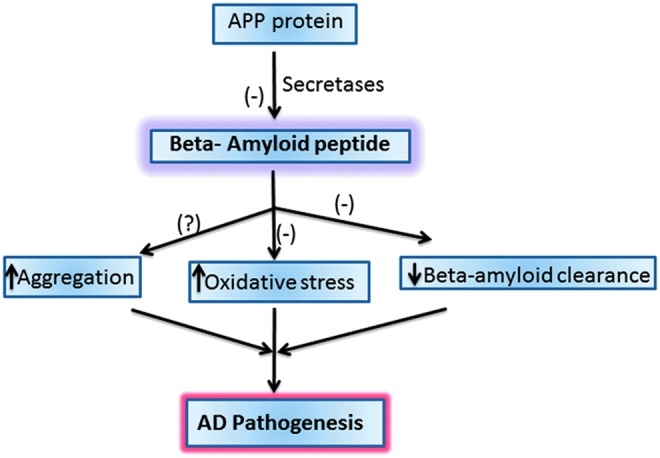
Potential therapeutic targets for AD. There are various potential targets to prevent Alzheimer disease (AD) progression and pathogenesis that include inhibiting the beta-amyloid formation or increasing its clearance from the brain or inhibiting the oxidative stress induced by beta-amyloid peptide. (-) indicates potential targets to combat AD. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Abbreviations Used
- Aβ
amyloid-beta peptide
- α-sAPP
α-secretase-cleaved soluble APP
- β-sAPP
β-secretase-cleaved soluble APP
- AD
Alzheimer disease
- AICD
APP intracellular domain
- aMCI
amnestic MCI
- APP
amyloid precursor protein
- CQ
5-chloro-7-iodoquinolin-8-ol
- CSF
cerebral spinal fluid
- CTF
C-terminal fragment
- DS
Down syndrome
- EAD
early AD
- F2isoP
isoprostane
- FAD
familial AD
- H2O2
hydrogen peroxide
- HNE
4-hydroxy-nonenal
- IPL
inferior parietal lobule
- LAD
late-stage AD
- MCI
mild cognitive impairment
- MetSOx
methionine sulfoxide
- MMSE
Mini Mental State Evaluation
- MRI
magnetic resonance imaging
- MSR
methionine sulfoxide reductase
- NFTs
neurofibrillay tangles
- NO•
nitric oxide
- NOS
nitric oxide synthase
- O2•−
superoxide radical anion
- •OH
hydroxyl radical
- ONOO−
peroxynitrite anion
- PCAD
preclinical AD
- PET
positron emission tomography
- Pin1
peptidyl-prolyl cis-trans isomerase 1
- PS
presenilin
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- SP
senile plaques
Acknowledgment
This research was supported by NIH grants to D.A.B. [AG-05119].
References
- 1.Aaronson RM. Graven KK. Tucci M. McDonald RJ. Farber HW. Non-neuronal enolase is an endothelial hypoxic stress protein. J Biol Chem. 1995;270:27752–27757. doi: 10.1074/jbc.270.46.27752. [DOI] [PubMed] [Google Scholar]
- 2.Abe T. Tohgi H. Isobe C. Murata T. Sato C. Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer's disease. J Neurosci Res. 2002;70:447–450. doi: 10.1002/jnr.10349. [DOI] [PubMed] [Google Scholar]
- 3.Aksenov MY. Tucker HM. Nair P. Aksenova MV. Butterfield DA. Estus S. Markesbery WR. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer's disease. J Mol Neurosci. 1998;11:151–164. doi: 10.1385/JMN:11:2:151. [DOI] [PubMed] [Google Scholar]
- 4.Aluise CD. Robinson RAS. Cai JA. Pierce WM. Markesbery WR. Butterfield DA. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer's disease: insights into memory loss in MCI. J Alzheimers Dis. 2011;23:257–269. doi: 10.3233/JAD-2010-101083. [DOI] [PubMed] [Google Scholar]
- 5.Balastik M. Lim J. Pastorino L. Lu KP. Pin1 in Alzheimer's disease: multiple substrates, one regulatory mechanism? Biochim Biophys Acta. 2007;1772:422–429. doi: 10.1016/j.bbadis.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barone E. Di Domenico F. Cenini G. Sultana R. Cini C. Preziosi P. Perluigi M. Mancuso C. Butterfield DA. Biliverdin reductase—a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011;1812:480–487. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barone E. Di Domenico F. Cenini G. Sultana R. Coccia R. Preziosi P. Perluigi M. Mancuso C. Butterfield DA. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. J Alzheimers Dis. 2011;25:623–633. doi: 10.3233/JAD-2011-110092. [DOI] [PubMed] [Google Scholar]
- 8.Barone E. Di Domenico F. Sultana R. Coccia R. Mancuso C. Perluigi M. Butterfield DA. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med. 2012;52:2292–2301. doi: 10.1016/j.freeradbiomed.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyd-Kimball D. Castegna A. Sultana R. Poon HF. Petroze R. Lynn BC. Klein JB. Butterfield DA. Proteomic identification of proteins oxidized by Abeta(1–42) in synaptosomes: implications for Alzheimer's disease. Brain Res. 2005;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 10.Boyd-Kimball D. Mohmmad Abdul H. Reed T. Sultana R. Butterfield DA. Role of phenylalanine 20 in Alzheimer's amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol. 2004;17:1743–1749. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- 11.Boyd-Kimball D. Poon HF. Lynn BC. Cai J. Pierce WM., Jr. Klein JB. Ferguson J. Link CD. Butterfield DA. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1–42): implications for Alzheimer's disease. Neurobiol Aging. 2006;27:1239–1249. doi: 10.1016/j.neurobiolaging.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Bush AI. Drug development based on the metals hypothesis of Alzheimer's disease. J Alzheimers Dis. 2008;15:223–240. doi: 10.3233/jad-2008-15208. [DOI] [PubMed] [Google Scholar]
- 13.Butterfield DA. beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chem Res Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 14.Butterfield DA. Abdul HM. Opii W. Newman SF. Joshi G. Ansari MA. Sultana R. Pin1 in Alzheimer's disease. J Neurochem. 2006;98:1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- 15.Butterfield DA. Boyd-Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Butterfield DA. Castegna A. Lauderback CM. Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 17.Butterfield DA. Drake J. Pocernich C. Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield DA. Galvan V. Lange MB. Tang H. Sowell RA. Spilman P. Fombonne J. Gorostiza O. Zhang J. Sultana R. Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48:136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterfield DA. Gnjec A. Poon HF. Castegna A. Pierce WM. Klein JB. Martins RN. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer's disease: an initial assessment. J Alzheimers Dis. 2006;10:391–397. doi: 10.3233/jad-2006-10407. [DOI] [PubMed] [Google Scholar]
- 20.Butterfield DA. Howard B. Yatin S. Koppal T. Drake J. Hensley K. Aksenov M. Aksenova M. Subramaniam R. Varadarajan S. Harris-White ME. Pedigo NW., Jr. Carney JM. Elevated oxidative stress in models of normal brain aging and Alzheimer's disease. Life Sci. 1999;65:1883–1892. doi: 10.1016/s0024-3205(99)00442-7. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield DA. Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide 1–42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 22.Butterfield DA. Lange ML. Multifunctional roles of enolase in Alzheimer's disease brain: beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butterfield DA. Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield DA. Perluigi M. Reed T. Muharib T. Hughes CP. Robinson RA. Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butterfield DA. Poon HF. Clair DS. Keller JN. Pierce WM. Klein JB. Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 26. This reference has been deleted.
- 27.Butterfield DA. Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer's disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis. 2007;12:61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 28.Calabrese V. Cornelius C. Mancuso C. Lentile R. Stella AM. Butterfield DA. Redox homeostasis and cellular stress response in aging and neurodegeneration. Methods Mol Biol. 2010;610:285–308. doi: 10.1007/978-1-60327-029-8_17. [DOI] [PubMed] [Google Scholar]
- 29.Calabrese V. Cornelius C. Rizzarelli E. Owen JB. Dinkova-Kostova AT. Butterfield DA. Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal. 2009;11:2717–2739. doi: 10.1089/ars.2009.2721. [DOI] [PubMed] [Google Scholar]
- 30.Castegna A. Aksenov M. Thongboonkerd V. Klein JB. Pierce WM. Booze R. Markesbery WR. Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 31.Castegna A. Thongboonkerd V. Klein JB. Lynn B. Markesbery WR. Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 32.Cenini G. Dowling AL. Beckett TL. Barone E. Mancuso C. Murphy MP. Levine H., 3rd Lott IT. Schmitt FA. Butterfield DA. Head E. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822:130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerpa WF. Barria MI. Chacon MA. Suazo M. Gonzalez M. Opazo C. Bush AI. Inestrosa NC. The N-terminal copper-binding domain of the amyloid precursor protein protects against Cu2+ neurotoxicity in vivo. FASEB J. 2004;18:1701–1703. doi: 10.1096/fj.03-1349fje. [DOI] [PubMed] [Google Scholar]
- 34.Chetelat G. Desgranges B. de la Sayette V. Viader F. Eustache F. Baron JC. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer's disease? Neurology. 2003;60:1374–1377. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- 35.Chetelat G. Eustache F. Viader F. De La Sayette V. Pelerin A. Mezenge F. Hannequin D. Dupuy B. Baron JC. Desgranges B. FDG-PET measurement is more accurate than neuropsychological assessments to predict global cognitive deterioration in patients with mild cognitive impairment. Neurocase. 2005;11:14–25. doi: 10.1080/13554790490896938. [DOI] [PubMed] [Google Scholar]
- 36.Ciccotosto GD. Tew D. Curtain CC. Smith D. Carrington D. Masters CL. Bush AI. Cherny RA. Cappai R. Barnham KJ. Enhanced toxicity and cellular binding of a modified amyloid beta peptide with a methionine to valine substitution. J Biol Chem. 2004;279:42528–42534. doi: 10.1074/jbc.M406465200. [DOI] [PubMed] [Google Scholar]
- 37.Clementi ME. Pezzotti M. Orsini F. Sampaolese B. Mezzogori D. Grassi C. Giardina B. Misiti F. Alzheimer's amyloid beta-peptide (1–42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun. 2006;342:206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- 38.Copani A. Koh JY. Cotman CW. Beta-amyloid increases neuronal susceptibility to injury by glucose deprivation. Neuroreport. 1991;2:763–765. doi: 10.1097/00001756-199112000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Curtain CC. Ali F. Volitakis I. Cherny RA. Norton RS. Beyreuther K. Barrow CJ. Masters CL. Bush AI. Barnham KJ. Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 40.Dai XL. Sun YX. Jiang ZF. Attenuated cytotoxicity but enhanced betafibril of a mutant amyloid beta-peptide with a methionine to cysteine substitution. FEBS Lett. 2007;581:1269–1274. doi: 10.1016/j.febslet.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 41.Dalle-Donne I. Scaloni A. Butterfield DA. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases. Hoboken, NJ: John Wiley and Sons; 2006. [DOI] [PubMed] [Google Scholar]
- 42.de Leon MJ. George AE. Ferris SH. Rosenbloom S. Christman DR. Gentes CI. Reisberg B. Kricheff II. Wolf AP. Regional correlation of PET and CT in senile dementia of the Alzheimer type. AJNR Am J Neuroradiol. 1983;4:553–556. [PMC free article] [PubMed] [Google Scholar]
- 43.Ding QX. Markesbery WR. Cecarini V. Keller JN. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer's disease. Neurochem Res. 2006;31:705–710. doi: 10.1007/s11064-006-9071-5. [DOI] [PubMed] [Google Scholar]
- 44.Drake J. Link CD. Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 45.Drayer BP. Heyman A. Wilkinson W. Barrett L. Weinberg T. Early-onset Alzheimer's disease: an analysis of CT findings. Ann Neurol. 1985;17:407–410. doi: 10.1002/ana.410170420. [DOI] [PubMed] [Google Scholar]
- 46.Driver JA. Lu KP. Pin1: a new genetic link between Alzheimer's disease, cancer and aging. Curr Aging Sci. 2010;3:158–165. doi: 10.2174/1874609811003030158. [DOI] [PubMed] [Google Scholar]
- 47.Farrow TF. Thiyagesh SN. Wilkinson ID. Parks RW. Ingram L. Woodruff PW. Fronto-temporal-lobe atrophy in early-stage Alzheimer's disease identified using an improved detection methodology. Psychiatry Res. 2007;155:11–19. doi: 10.1016/j.pscychresns.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 48.Folstein MF. Folstein SE. McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 49.Gabbita SP. Aksenov MY. Lovell MA. Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer's disease brain. J Neurochem. 1999;73:1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 50.Gabbita SP. Lovell MA. Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 51.Ganjei JK. Targeting amyloid precursor protein secretases: Alzheimer's disease and beyond. Drug News Perspect. 2010;23:573–584. doi: 10.1358/dnp.2010.23.9.1507297. [DOI] [PubMed] [Google Scholar]
- 52.Gervais FG. Xu D. Robertson GS. Vaillancourt JP. Zhu Y. Huang J. LeBlanc A. Smith D. Rigby M. Shearman MS. Clarke EE. Zheng H. Van Der Ploeg LH. Ruffolo SC. Thornberry NA. Xanthoudakis S. Zamboni RJ. Roy S. Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 53.Geula C. Mesulam MM. Saroff DM. Wu CK. Relationship between plaques, tangles, and loss of cortical cholinergic fibers in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:63–75. doi: 10.1097/00005072-199801000-00008. [DOI] [PubMed] [Google Scholar]
- 54.Glabe CC. Amyloid accumulation and pathogenesis of Alzheimer's disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 55.Goate A. Segregation of a missense mutation in the amyloid beta-protein precursor gene with familial Alzheimer's disease. J Alzheimers Dis. 2006;9:341–347. doi: 10.3233/jad-2006-9s338. [DOI] [PubMed] [Google Scholar]
- 56.Goate A. Hardy J. Twenty years of Alzheimer's disease-causing mutations. J Neurochem. 2012;120(Suppl 1):3–8. doi: 10.1111/j.1471-4159.2011.07575.x. [DOI] [PubMed] [Google Scholar]
- 57.Guidi I. Galimberti D. Lonati S. Novembrino C. Bamonti F. Tiriticco M. Fenoglio C. Venturelli E. Baron P. Bresolin N. Scarpini E. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2006;27:262–269. doi: 10.1016/j.neurobiolaging.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 58.Guillozet AL. Weintraub S. Mash DC. Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 59.Gyure KA. Durham R. Stewart WF. Smialek JE. Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- 60.Halliwell B. Gutteridge JM. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992;307:108–112. doi: 10.1016/0014-5793(92)80911-y. [DOI] [PubMed] [Google Scholar]
- 61.Hardy J. Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 62.Hattori T. Takei N. Mizuno Y. Kato K. Kohsaka S. Neurotrophic and neuroprotective effects of neuron-specific enolase on cultured neurons from embryonic rat-brain. Neurosci Res. 1995;21:191–198. doi: 10.1016/0168-0102(94)00849-b. [DOI] [PubMed] [Google Scholar]
- 63.Hebert LE. Scherr PA. Bienias JL. Bennett DA. Evans DA. Alzheimer disease in the US population—prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 64.Hensley K. Hall N. Subramaniam R. Cole P. Harris M. Aksenov M. Aksenova M. Gabbita SP. Wu JF. Carney JM. Lovell M. Markesbery WR. Butterfield DA. Brain regional correspondence between Alzheimers-disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 65.Hensley K. Maidt ML. Yu ZQ. Sang H. Markesbery WR. Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herholz K. Salmon E. Perani D. Baron JC. Holthoff V. Frolich L. Schonknecht P. Ito K. Mielke R. Kalbe E. Zundorf G. Delbeuck X. Pelati O. Anchisi D. Fazio F. Kerrouche N. Desgranges B. Eustache F. Beuthien-Baumann B. Menzel C. Schroder J. Kato T. Arahata Y. Henze M. Heiss WD. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 2002;17:302–316. doi: 10.1006/nimg.2002.1208. [DOI] [PubMed] [Google Scholar]
- 67.Hoogland C. Mostaguir K. Sanchez JC. Hochstrasser DF. Appel RD. SWISS-2DPAGE, ten years later. Proteomics. 2004;4:2352–2356. doi: 10.1002/pmic.200300830. [DOI] [PubMed] [Google Scholar]
- 68.Hoogland C. Sanchez JC. Tonella L. Binz PA. Bairoch A. Hochstrasser DF. Appel RD. The 1999 SWISS-2DPAGE database update. Nucleic Acids Res. 2000;28:286–288. doi: 10.1093/nar/28.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang X. Atwood CS. Hartshorn MA. Multhaup G. Goldstein LE. Scarpa RC. Cuajungco MP. Gray DN. Lim J. Moir RD. Tanzi RE. Bush AI. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 70.Ida N. Hartmann T. Pantel J. Schroder J. Zerfass R. Forstl H. Sandbrink R. Masters CL. Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 71.Jack CR., Jr. Petersen RC. Xu YC. O'Brien PC. Smith GE. Ivnik RJ. Boeve BF. Waring SC. Tangalos EG. Kokmen E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52:1397–1403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jarrett JT. Berger EP. Lansbury PT., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 73.Jimenez S. Torres M. Vizuete M. Sanchez-Varo R. Sanchez-Mejias E. Trujillo-Estrada L. Carmona-Cuenca I. Caballero C. Ruano D. Gutierrez A. Vitorica J. Age-dependent accumulation of soluble amyloid beta (Abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J Biol Chem. 2011;286:18414–18425. doi: 10.1074/jbc.M110.209718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin M. Shepardson N. Yang T. Chen G. Walsh D. Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johansson AS. Bergquist J. Volbracht C. Paivio A. Leist M. Lannfelt L. Westlind-Danielsson A. Attenuated amyloid-beta aggregation and neurotoxicity owing to methionine oxidation. Neuroreport. 2007;18:559–563. doi: 10.1097/WNR.0b013e3280b07c21. [DOI] [PubMed] [Google Scholar]
- 76.Jomova K. Vondrakova D. Lawson M. Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- 77.Joshi YB. Pratico D. Vitamin E in aging, dementia, and Alzheimer's disease. Biofactors. 2012;38:90–97. doi: 10.1002/biof.195. [DOI] [PubMed] [Google Scholar]
- 78.Jovanovic SV. Clements D. MacLeod K. Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radic Biol Med. 1998;25:1044–1048. doi: 10.1016/s0891-5849(98)00137-3. [DOI] [PubMed] [Google Scholar]
- 79.Kanski J. Aksenova M. Butterfield DA. The hydrophobic environment of Met35 of Alzheimer's Abeta(1–42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res. 2002;4:219–223. doi: 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- 80.Kanski J. Aksenova M. Schoneich C. Butterfield DA. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide. Free Radic Biol Med. 2002;32:1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 81.Katzman R. Saitoh T. Advances in Alzheimer's disease. FASEB J. 1991;5:278–286. [PubMed] [Google Scholar]
- 82.Keeney JT. Swomley AM. Harris JL. Fiorini A. Mitov MI. Perluigi M. Sultana R. Butterfield DA. Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res. 2012;22:220–230. doi: 10.1007/s12640-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 83.Keller JN. Schmitt FA. Scheff SW. Ding Q. Chen Q. Butterfield DA. Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 84.Koppal T. Drake J. Yatin S. Jordan B. Varadarajan S. Bettenhausen L. Butterfield DA. Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer's disease. J Neurochem. 1999;72:310–317. doi: 10.1046/j.1471-4159.1999.0720310.x. [DOI] [PubMed] [Google Scholar]
- 85.Landrieu I. Smet-Nocca C. Amniai L. Louis JV. Wieruszeski JM. Goris J. Janssens V. Lippens G. Molecular implication of PP2A and Pin1 in the Alzheimer's disease specific hyperphosphorylation of Tau. PLoS One. 2011;6:e21521. doi: 10.1371/journal.pone.0021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lauderback CM. Hackett JM. Huang FF. Keller JN. Szweda LI. Markesbery WR. Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of A beta 1–42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 87.Lim J. Balastik M. Lee TH. Nakamura K. Liou YC. Sun A. Finn G. Pastorino L. Lee VM. Lu KP. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest. 2008;118:1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lovell MA. Ehmann WD. Mattson MP. Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 89.Lovell MA. Markesbery WR. Oxidatively modified RNA in mild cognitive impairment. Neurobiol Dis. 2008;29:169–175. doi: 10.1016/j.nbd.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu PJ. Wulf G. Zhou XZ. Davies P. Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 91.Luo S. Levine RL. Methionine in proteins defends against oxidative stress. FASEB J. 2009;23:464–472. doi: 10.1096/fj.08-118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma SL. Pastorino L. Zhou XZ. Lu KP. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J Biol Chem. 2012;287:6969–6973. doi: 10.1074/jbc.C111.298596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mark RJ. Lovell MA. Markesbery WR. Uchida K. Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 94.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 95.Markesbery WR. Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9:133–146. doi: 10.1111/j.1750-3639.1999.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Markesbery WR. Kryscio RJ. Lovell MA. Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 97.Markesbery WR. Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 98.Mary J. Vougier S. Picot CR. Perichon M. Petropoulos I. Friguet B. Enzymatic reactions involved in the repair of oxidized proteins. Exp Gerontol. 2004;39:1117–1123. doi: 10.1016/j.exger.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 99.Masters CL. Simms G. Weinman NA. Multhaup G. McDonald BL. Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mattila J. Koikkalainen J. Virkki A. Simonsen A. van Gils M. Waldemar G. Soininen H. Lotjonen J. A disease state fingerprint for evaluation of Alzheimer's disease. J Alzheimers Dis. 2011;27:163–176. doi: 10.3233/JAD-2011-110365. [DOI] [PubMed] [Google Scholar]
- 101.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 102.Mayeux R. Tang MX. Jacobs DM. Manly J. Bell K. Merchant C. Small SA. Stern Y. Wisniewski HM. Mehta PD. Plasma amyloid beta-peptide 1–42 and incipient Alzheimer's disease. Ann Neurol. 1999;46:412–416. doi: 10.1002/1531-8249(199909)46:3<412::aid-ana19>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 103.McGrath LT. McGleenon BM. Brennan S. McColl D. McIlroy S. Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM-Monthly J Associaf Phys. 2001;94:485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 104.Mecocci P. MacGarvey U. Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 105.Migliore L. Fontana I. Trippi F. Colognato R. Coppede F. Tognoni G. Nucciarone B. Siciliano G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol Aging. 2005;26:567–573. doi: 10.1016/j.neurobiolaging.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 106.Miller DL. Papayannopoulos IA. Styles J. Bobin SA. Lin YY. Biemann K. Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 107.Misiti F. Clementi ME. Giardina B. Oxidation of methionine 35 reduces toxicity of the amyloid beta-peptide(1–42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochem Int. 2010;56:597–602. doi: 10.1016/j.neuint.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 108.Mohmmad Abdul H. Sultana R. Keller JN. St Clair DK. Markesbery WR. Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1–42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 109.Mori C. Spooner ET. Wisniewsk KE. Wisniewski TM. Yamaguch H. Saido TC. Tolan DR. Selkoe DJ. Lemere CA. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- 110.Munch G. Schinzel R. Loske C. Wong A. Durany N. Li JJ. Vlassara H. Smith MA. Perry G. Riederer P. Alzheimer's disease—synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105:439–461. doi: 10.1007/s007020050069. [DOI] [PubMed] [Google Scholar]
- 111.Murray MM. Bernstein SL. Nyugen V. Condron MM. Teplow DB. Bowers MT. Amyloid beta protein: Abeta40 inhibits Abeta42 oligomerization. J Am Chem Soc. 2009;131:6316–6317. doi: 10.1021/ja8092604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Naslund J. Schierhorn A. Hellman U. Lannfelt L. Roses AD. Tjernberg LO. Silberring J. Gandy SE. Winblad B. Greengard P, et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A. 1994;91:8378–8382. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson PT. Alafuzoff I. Bigio EH. Bouras C. Braak H. Cairns NJ. Castellani RJ. Crain BJ. Davies P. Del Tredici K. Duyckaerts C. Frosch MP. Haroutunian V. Hof PR. Hulette CM. Hyman BT. Iwatsubo T. Jellinger KA. Jicha GA. Kovari E. Kukull WA. Leverenz JB. Love S. Mackenzie IR. Mann DM. Masliah E. McKee AC. Montine TJ. Morris JC. Schneider JA. Sonnen JA. Thal DR. Trojanowski JQ. Troncoso JC. Wisniewski T. Woltjer RL. Beach TG. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nikolaev A. McLaughlin T. O'Leary DD. Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Nunomura A. Perry G. Aliev G. Hirai K. Takeda A. Balraj EK. Jones PK. Ghanbari H. Wataya T. Shimohama S. Chiba S. Atwood CS. Petersen RB. Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 116.Nunomura A. Perry G. Pappolla MA. Wade R. Hirai K. Chiba S. Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oda T. Wals P. Osterburg HH. Johnson SA. Pasinetti GM. Morgan TE. Rozovsky I. Stine WB. Snyder SW. Holzman TF, et al. Clusterin (apoJ) alters the aggregation of amyloid beta-peptide (A beta 1–42) and forms slowly sedimenting A beta complexes that cause oxidative stress. Exp Neurol. 1995;136:22–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 118.Opazo C. Luza S. Villemagne VL. Volitakis I. Rowe C. Barnham KJ. Strozyk D. Masters CL. Cherny RA. Bush AI. Radioiodinated clioquinol as a biomarker for beta-amyloid: Zn complexes in Alzheimer's disease. Aging Cell. 2006;5:69–79. doi: 10.1111/j.1474-9726.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- 119.Park SA. Shaked GM. Bredesen DE. Koo EH. Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem Biophys Res Commun. 2009;388:450–455. doi: 10.1016/j.bbrc.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perluigi M. Butterfield DA. The identification of protein biomarkers for oxidative stress in Down syndrome. Expert Rev Proteomics. 2011;8:427–429. doi: 10.1586/epr.11.36. [DOI] [PubMed] [Google Scholar]
- 121.Perluigi M. Butterfield DA. Oxidative stress and Down syndrome: a route toward Alzheimer-like dementia. Curr Gerontol Geriatr Res. 2012;2012:724904. doi: 10.1155/2012/724904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Perluigi M. Sultana R. Cenini G. Di Domenico F. Memo M. Pierce WM. Coccia R. Butterfield DA. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer's disease: role of lipid peroxidation in Alzheimer's disease pathogenesis. Proteomics Clin Appl. 2009;3:682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Petersen RC. Mild cognitive impairment: transition between aging and Alzheimer's disease. Neurologia. 2000;15:93–101. [PubMed] [Google Scholar]
- 124.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 125.Petrie EC. Cross DJ. Galasko D. Schellenberg GD. Raskind MA. Peskind ER. Minoshima S. Preclinical evidence of Alzheimer changes: convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Arch Neurol. 2009;66:632–637. doi: 10.1001/archneurol.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pike CJ. Walencewicz-Wasserman AJ. Kosmoski J. Cribbs DH. Glabe CG. Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 127.Pogocki D. Schoneich C. Redox properties of Met(35) in neurotoxic beta-amyloid peptide. A molecular modeling study. Chem Res Toxicol. 2002;15:408–418. doi: 10.1021/tx0101550. [DOI] [PubMed] [Google Scholar]
- 128.Ranganathan R. Lu KP. Hunter T. Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 129.Reed T. Perluigi M. Sultana R. Pierce WM. Klein JB. Turner DM. Coccia R. Markesbery WR. Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 130.Reed TT. Pierce WM., Jr. Turner DM. Markesbery WR. Butterfield DA. Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J Cell Mol Med. 2009;13:2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reed TT. Pierce WM. Markesbery WR. Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 132.Ritchie CW. Bush AI. Mackinnon A. Macfarlane S. Mastwyk M. MacGregor L. Kiers L. Cherny R. Li QX. Tammer A. Carrington D. Mavros C. Volitakis I. Xilinas M. Ames D. Davis S. Beyreuther K. Tanzi RE. Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 133.Robinson RA. Lange MB. Sultana R. Galvan V. Fombonne J. Gorostiza O. Zhang J. Warrier G. Cai J. Pierce WM. Bredesen DE. Butterfield DA. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-beta peptide of amyloid precursor protein. Neuroscience. 2011;177:207–222. doi: 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Scheuner D. Eckman C. Jensen M. Song X. Citron M. Suzuki N. Bird TD. Hardy J. Hutton M. Kukull W. Larson E. Levy-Lahad E. Viitanen M. Peskind E. Poorkaj P. Schellenberg G. Tanzi R. Wasco W. Lannfelt L. Selkoe D. Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 135.Schmidt M. Sachse C. Richter W. Xu C. Fandrich M. Grigorieff N. Comparison of Alzheimer Abeta(1–40) and Abeta(1–42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schoneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer's disease. Biochim Biophys Acta. 2005;1703:111–119. doi: 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 137.Schoneich C. Pogocki D. Hug GL. Bobrowski K. Free radical reactions of methionine in peptides: mechanisms relevant to beta-amyloid oxidation and Alzheimer's disease. J Am Chem Soc. 2003;125:13700–13713. doi: 10.1021/ja036733b. [DOI] [PubMed] [Google Scholar]
- 138.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 139.Shan X. Lin CLG. Quantification of oxidized RNAs in Alzheimer's disease. Neurobiol Aging. 2006;27:657–662. doi: 10.1016/j.neurobiolaging.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 140.Shan X. Tashiro H. Lin CLG. The identification and characterization of oxidized RNAs in Alzheimer's disease. J Neurosci. 2003;23:4913–4921. doi: 10.1523/JNEUROSCI.23-12-04913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Small BJ. Fratiglioni L. Backman L. Canaries in a coal mine: cognitive markers of preclinical Alzheimer disease. Arch Gen Psychiatry. 2001;58:859–860. doi: 10.1001/archpsyc.58.9.859. [DOI] [PubMed] [Google Scholar]
- 142.Smith DG. Cappai R. Barnham KJ. The redox chemistry of the Alzheimer's disease amyloid beta peptide. Biochim Biophys Acta. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 143.Smith MA. Harris PLR. Sayre LM. Beckman JS. Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sperling RA. Aisen PS. Beckett LA. Bennett DA. Craft S. Fagan AM. Iwatsubo T. Jack CR., Jr. Kaye J. Montine TJ. Park DC. Reiman EM. Rowe CC. Siemers E. Stern Y. Yaffe K. Carrillo MC. Thies B. Morrison-Bogorad M. Wagster MV. Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stadtman ER. Cyclic oxidation and reduction of methionine residues of proteins in antioxidant defense and cellular regulation. Arch Biochem Biophys. 2004;423:2–5. doi: 10.1016/j.abb.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 146.Stadtman ER. Moskovitz J. Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- 147.Subramanian A. Miller DM. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J Biol Chem. 2000;275:5958–5965. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 148.Sultana R. Boyd-Kimball D. Poon HF. Cai J. Pierce WM. Klein JB. Markesbery WR. Zhou XZ. Lu KP. Butterfield DA. Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 149.Sultana R. Boyd-Kimball D. Poon HF. Cai J. Pierce WM. Klein JB. Merchant M. Markesbery WR. Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 150.Sultana R. Butterfield DA. Role of oxidative stress in the progression of Alzheimer's disease. J Alzheimers Dis. 2010;19:341–353. doi: 10.3233/JAD-2010-1222. [DOI] [PubMed] [Google Scholar]
- 151.Sultana R. Mecocci P. Mangialasche F. Cecchetti R. Baglioni M. Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer's disease: insights into the role of oxidative stress in Alzheimer's disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis. 2011;24:77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 152.Sultana R. Perluigi M. Butterfield DA. Redox proteomics identification of oxidatively modified proteins in Alzheimer's disease brain and in vivo and in vitro models of AD centered around Abeta(1–42) J Chromatogr B Analyt Technol Biomed Life Sci. 2006;833:3–11. doi: 10.1016/j.jchromb.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 153.Sultana R. Piroddi M. Galli F. Butterfield D. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem Res. 2008;33:2540–2546. doi: 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- 154. This reference has been deleted.
- 155.Sultana R. Poon HF. Cai J. Pierce WM. Merchant M. Klein JB. Markesbery WR. Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 156.Sultana R. Reed T. Perluigi M. Coccia R. Pierce WM. Butterfield DA. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med. 2007;11:839–851. doi: 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sultana R. Robinson RA. Bader Lange M. Fiorini A. Galvan V. Fombonne J. Baker A. Gorostiza O. Zhang J. Cai J. Pierce WM. Bredesen DE. Butterfield DA. Do proteomics analyses provide insights into reduced oxidative stress in the brain of an Alzheimer disease transgenic mouse model with an M631L amyloid precursor protein substitution and thereby the importance of amyloid-beta-resident methionine 35 in Alzheimer disease pathogenesis? Antioxid Redox Signal. 2012;17:1507–1514. doi: 10.1089/ars.2011.4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Takei N. Kondo J. Nagaike K. Ohsawa K. Kato K. Kohsaka S. Neuronal survival factor from bovine brain is identical to neuron-specific enolase. J Neurochem. 1991;57:1178–1184. doi: 10.1111/j.1471-4159.1991.tb08277.x. [DOI] [PubMed] [Google Scholar]
- 159.Tremblay C. Pilote M. Phivilay A. Emond V. Bennett DA. Calon F. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2007;12:377–390. doi: 10.3233/jad-2007-12411. [DOI] [PubMed] [Google Scholar]
- 160.Varadarajan S. Kanski J. Aksenova M. Lauderback C. Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's A beta(1–42) and A beta(25–35) J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 161.Varadarajan S. Yatin S. Aksenova M. Butterfield DA. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 162.Varadarajan S. Yatin S. Kanski J. Jahanshahi F. Butterfield DA. Methionine residue 35 is important in amyloid beta-peptide-associated free radical oxidative stress. Brain Res Bull. 1999;50:133–141. doi: 10.1016/s0361-9230(99)00093-3. [DOI] [PubMed] [Google Scholar]
- 163.Vassar R. Bennett BD. Babu-Khan S. Kahn S. Mendiaz EA. Denis P. Teplow DB. Ross S. Amarante P. Loeloff R. Luo Y. Fisher S. Fuller J. Edenson S. Lile J. Jarosinski MA. Biere AL. Curran E. Burgess T. Louis JC. Collins F. Treanor J. Rogers G. Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 164.Vigo-Pelfrey C. Lee D. Keim P. Lieberburg I. Schenk DB. Characterization of beta-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 165.Viola KL. Velasco PT. Klein WL. Why Alzheimer's is a disease of memory: the attack on synapses by A beta oligomers (ADDLs) J Nutr Health Aging. 2008;12:51S–57S. doi: 10.1007/BF02982587. [DOI] [PubMed] [Google Scholar]
- 166.Vlassenko AG. Benzinger TL. Morris JC. PET amyloid-beta imaging in preclinical Alzheimer's disease. Biochim Biophys Acta. 2012;1822:370–379. doi: 10.1016/j.bbadis.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Waldemar G. Phung KT. Burns A. Georges J. Hansen FR. Iliffe S. Marking C. Rikkert MO. Selmes J. Stoppe G. Sartorius N. Access to diagnostic evaluation and treatment for dementia in Europe. Int J Geriatr Psychiatry. 2007;22:47–54. doi: 10.1002/gps.1652. [DOI] [PubMed] [Google Scholar]
- 168.Walsh DM. Klyubin I. Fadeeva JV. Rowan MJ. Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 169.Walsh DM. Teplow DB. Alzheimer's disease and the amyloid beta-protein. Prog Mol Biol Transl Sci. 2012;107:101–124. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- 170.Wan L. Nie G. Zhang J. Luo Y. Zhang P. Zhang Z. Zhao B. beta-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med. 2010;50:122–129. doi: 10.1016/j.freeradbiomed.2010.10.707. [DOI] [PubMed] [Google Scholar]
- 171.Wang J. Markesbery WR. Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- 172.White AR. Du T. Laughton KM. Volitakis I. Sharples RA. Xilinas ME. Hoke DE. Holsinger RM. Evin G. Cherny RA. Hill AF. Barnham KJ. Li QX. Bush AI. Masters CL. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006;281:17670–17680. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- 173.Yao Y. Clark CM. Trojanowski JQ. Lee VM. Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58:623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 174.Yatin SM. Varadarajan S. Butterfield DA. Vitamin E prevents Alzheimer's amyloid beta-peptide (1–42)-induced neuronal protein oxidation and reactive oxygen species production. J Alzheimers Dis. 2000;2:123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- 175.Yatin SM. Varadarajan S. Link CD. Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1–42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339–342. [DOI] [PubMed] [Google Scholar]
- 176.Zheng H. Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]