

Abstract
The presence of circulating tumor cells (CTCs) is believed to lead to the formation of secondary tumors via an adhesion cascade involving interaction between adhesion receptors of endothelial cells and ligands on CTCs. Many CTCs express sialylated carbohydrate ligands on their surfaces that adhere to selectin protein found on inflamed endothelial cells. We have investigated the feasibility of using immobilized selectin proteins as a targeting mechanism for CTCs under flow. Herein, targeted liposomal doxorubicin (L-DXR) was functionalized with recombinant human E-selectin (ES) and polyethylene glycol (PEG) to target and kill cancer cells under shear flow, both when immobilized along a microtube device or sheared in a cone-and-plate viscometer in a dilute suspension. Healthy circulating cells such as red blood cells were not targeted by this mechanism and were left to freely circulate, and minimal leukocyte death was observed. Halloysite nanotube (HNT)-coated microtube devices immobilized with nanoscale liposomes significantly enhanced the targeting, capture, and killing of cancer cells. This work demonstrates that E-selectin functionalized L-DXR, sheared in suspension or immobilized onto microtube devices, provides a novel approach to selectively target and deliver chemotherapeutics to CTCs in the bloodstream.
Keywords: Circulating tumor cells, Doxorubicin, E-selectin, Halloysite nanotubes, Liposomes, Targeted delivery
1. Introduction
Cancer is one of the leading causes of death, as approximately 90% of human cancer deaths are attributed to cancer metastasis [1,2]. The process of metastasis can involve the presence of circulating tumor cells (CTCs), which are believed to undergo an adhesion cascade similar to leukocytes. This process consists of a discrete sequence of steps involving tethering, rolling, and firm adhesion between cells and the inflamed endothelial lining of the vasculature [3,4]. Cells from the primary tumor detach, invade the surrounding tissue, and intravasate into the blood or lymphatic circulation as CTCs [2,3,5,6]. Following intravasation, CTCs interact with the vasculature by selectin-mediated rolling and cell arrest, where a fraction of cells are presumed to extravasate into the tissue of a distant organ and proliferate to form a secondary tumor [2–4]. Although metastasis is an inefficient process with only about 0.01% of CTCs surviving the pathway [7,8], the development of metastases remains as the primary cause of cancer-related mortality.
Doxorubicin (DXR), an adriamyacin anthracycline antibiotic, is a common chemotherapy agent that has been used in treating a variety of cancers, including metastatic breast cancer, Kaposi’s sarcoma, acute leukemia, Hodgkin’s Disease and other lymphomas and sarcomas [9]. The drug’s potent antineoplastic ability stems from its function in DNA-intercalation, inhibition of topo-isomerase II, and formation of free radicals [9–11]. However, the non-specific activity of DXR can lead to systemic toxicity, tissue necrosis, neutropenia, and side effects including cardiomyopathy, myelosuppression, alopecia, mucositis, nausea and vomiting [9,12,13].
To increase delivery efficiency and reduce toxicity of doxorubicin, a number of delivery systems such as polymers, dendrimers, liposomes, and nanoparticles have been developed [12,14]. In particular, liposomal doxorubicin (L-DXR) has been shown to improve the drug’s efficacy by altering its pharmacokinetics, greatly increasing its circulation time in blood, reducing drug accumulation in organs, and dampening toxic side effects [15–19]. Furthermore, the addition of polyethylene glycol (PEG) coupled to the liposomal surface provides steric stabilization, reducing drug clearance, protein interactions, and opsonization by the reticulo-endothelial system [20–22]. Known as “stealth” liposomes, PEG-conjugated nanoparticles further extend the circulation time of the drug, which due to enhanced permeability and retention effects, aids in tumor uptake [16–19,22–24]. The success of liposomal doxorubicin has been shown in pre-clinical and clinical studies as evidenced by Doxil®, a commercially available L-DXR formulation that has been approved by the FDA for use in treating Kaposi’s sarcoma, and Myocet®, a non-PEGylated L-DXR for the treatment of metastatic breast cancer [25].
While PEGylated L-DXR is effective in treating tumors, the drug vehicle remains limited in its ability to target specific cells. Many CTCs express sialylated carbohydrate ligands on their surface, which adhere to selectin proteins along the inflamed endothelium during metastasis [26–30]. Targeting of CTCs in the bloodstream via selectin bonds could reduce the probability of metastasis. In this study, targeted L-DXR particles were developed by functionalizing the liposome surface with a combination of PEG and recombinant human E-selectin. We then investigated the targeting, capture, and killing of model CTCs using targeted L-DXR (1) immobilized within a microtube device while exposed to physiologically relevant shear stresses, and (2) in a dilute suspension under shear flow in a cone-and-plate viscometer. Naturally-occurring halloysite nanotubes (HNT), which have recently been shown to enhance cell capture under shear flow, were used as a nanostructured surface coating within microtubes to enhance targeted DXR delivery [31].
2. Materials and methods
2.1. Cell lines and cell culture
Colorectal adenocarcinoma cell line COLO 205 (ATCC # CCL-222) and acute promyelocytic leukemic cell line KG-1a (ATCC # CCL-246.1) were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI 1640 cell culture medium with 10% (v/v) fetal bovine serum and 1% (v/v) PenStrep, all purchased from Invitrogen (Grand Island, NY, USA), and incubated under humidified conditions at 37 °C and 5% CO2 (complete media). COLO 205 cells were not allowed to exceed 90% confluence, and KG-1a cells were maintained at less than 1×106 cells/mL.
2.2. Liposomal-doxorubicin preparation
Multilamellar liposomes (Fig. 1) were prepared using a thin lipid film hydration method [32,33] with 125 mM ammonium sulfate (Sigma-Aldrich, St. Louis, MO, USA) followed by 10 freeze–thaw cycles and then extrusion as previously described [34] to prepare empty liposomes (EL). To prepare L-DXR, DXR HCL (Sigma-Aldrich) was encapsulated within ELs using the ammonium sulfate remote loading method as described previously [33] at a doxorubicin-to-lipid ratio of 0.2:1 (w/w). Non-encapsulated DXR was removed using gel-exclusion chromatography with Sephadex G-50 (Sigma-Aldrich). The liposomal doxorubicin concentration was determined by spectrophotometry (λ=490 nm), and the loading efficiency of doxorubicin was determined to be >95%.
Fig. 1.
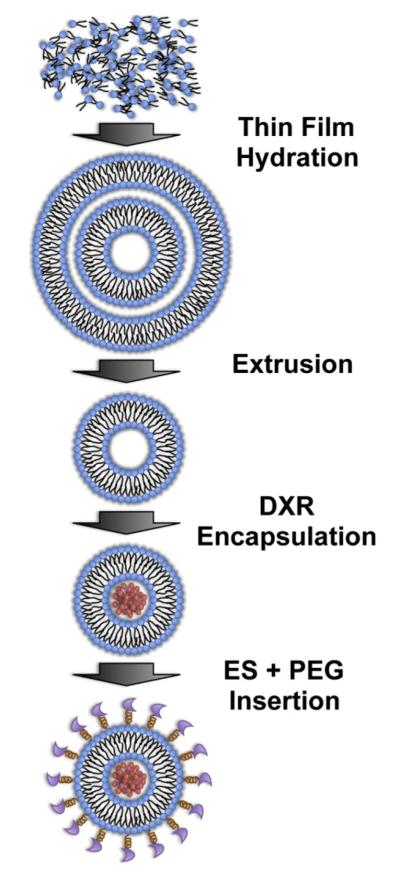
Schematic of E-selectin-targeted PEGylated liposome synthesis.
2.3. Preparation of targeted liposomes
Recombinant human E-selectin/Fc chimera (rhE/Fc) (R&D Systems, Minneapolis, MN) was conjugated to 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-Maleimide 2000 (DSPE-PEG2000 maleimide) (Avanti Polar Lipids, Alabaster, AL, USA) via thiolation, and conjuguates were covalently attached to diluted EL or L-DXR as previously described [34] to make two forms of targeted liposomes, E-selectin-PEG-conjugated liposomal doxorubicin (ES-PEG L-DXR) and E-selectin-PEG-conjugated empty liposomes (ES-PEG EL). The volume ratio of EL or L-DXR in phosphate-buffered saline (PBS) and rhE/Fc-DSPE-PEG2000 maleimide to the diluted liposomes was 1:1 and 1:12, respectively. All liposomes were stored at 4 °C for no more than one week until usage.
Liposomes were freshly prepared and diluted with phosphate-buffered saline (PBS), and the mean particle diameter and zeta potential were measured by dynamic light scattering using a Malvern Zetasizer nano ZS (Malvern Instruments Ltd., Worcestershire, UK), according to the manufacturer’s protocols. To confirm that E-selectin was successfully conjugated to the liposome surface, 10 μL of fluorescently tagged liposomes was mixed with 490 μL COLO 205 or KG-1a cells (106 cells/ mL) and sheared in a cone and plate viscometer at 2 dyn/cm2 for 10 min. All cells were analyzed for fluorescent liposomes conjugated to the surface using an Accuri C6 flow cytometer (Accuri Cytometers, Inc., Ann Arbor, MI, USA). Adhesion of fluorescent liposomes to the cell surface was confirmed via confocal microscopy.
2.4. Doxorubicin encapsulation and leakage studies
A 5 μL sample of L-DXR was treated with or without 20 μL of 10% Triton X-100. Samples of EL, L-DXR, PEG L-DXR, and ES-PEG L-DXR (30 μL) were used for the leakage study. PBS was added to all solutions to achieve a total volume of 200 μL. Fluorescence intensity of DXR in 200 μL solutions was measured in fluorescence units (fu) using a spectrophotometer (SmartSpec Plus; Bio-Rad Laboratories, Philadelphia, PA, USA). To assess DXR leakage, fluorescence readings were recorded over a span of 39 days.
2.5. Static experiments
COLO 205 and KG-1a cells were cultured in complete media at a concentration of 1×105 cells/mL and 3×106 cells/mL, respectively, on 24-well plates (Becton Dickinson, Franklin Lakes, NJ, USA). Cells were treated with ES-PEG L-DXR, naked L-DXR, soluble DXR (20 μM), or naked EL at volumes of 0, 0.001, 0.01, 0.1, 0.5, 1, 2, and 5 μL for 18 h. Adherent COLO 205 cells were then treated with Accutase (Sigma-Aldrich) for 5 min at 37 °C to remove cells from the surface. Both COLO 205 and KG-1a cells were then washed with 1× PBS at 1000 rpm in a refrigerated centrifuge (Allegra XX-22R Centrifuge; Beckman Coulter, Brea, CA, USA) and resuspended in fresh media. After 4 days, cell viability was evaluated on a hemocytometer (Hausser Scientific, Horcham, PA, USA) using trypan blue exclusion dye (Lonza, Wilkersville, MD, USA). The percentage of dead cells was determined by comparing the number of viable cells for treated and untreated cells. For treatment of 0.5 μL liposomes on COLO 205 cells, cell viability was assessed each day for 4 days. Brightfield images were taken of COLO 205 cells at day 4 using an inverted microscope (IX-81; Olympus America Inc., Melville, NY, USA) coupled to a CCD camera (Hitachi) and processed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
2.6. Preparation of cells for capture experiments
COLO 205 cells were treated with Accutase for 5 min at 37 °C before handling. KG-1a and COLO 205 cells were washed with 1× PBS at 1000 rpm and resuspended in flow buffer at a concentration of 1×106 cells/mL. The assay buffer consisted of PBS saturated with Ca2 + and Mg2 +, which activated recombinant human E-selectin receptors to facilitate cell capture. Prior to performing capture experiments, 95% cell viability was confirmed by trypan blue assay.
To prepare blood cells for capture experiments, Human peripheral blood was obtained via venipuncture from healthy blood donors and collected into sterile sodium heparin-containing tubes (BD Biosciences) after informed consent. Mononuclear cells (MNCs) and red blood cells (RBCs) were isolated by centrifugation at 480×g for 50 min at 23 °C in a Marathon 8 K centrifuge (Fisher Scientific, Pittsburgh, PA) using 1-Step™ Polymorphs (Accurate Chemical & Scientific Corporation, Westbury, NY). This centrifugation method creates a density gradient to separate blood into visible layers of plasma, mononuclear cells, neutrophils, and erythrocytes and platelets. MNCs and RBCs were extracted separately and washed in Mg2 + and Ca2 + free HBSS to remove any remaining Polymorphs. The MNC sample was further purified from remaining red blood cells in the suspension by hypotonic lysis. RBC and MNC samples were resuspended at a concentration of 1.0×106 cells/mL in HBSS containing 0.5% HSA, 2 mM Ca2+, 1 mM Mg2+, and 10 mM HEPES (Invitrogen), buffered to pH 7.4.
2.7. Preparation of microtube surfaces
Microrenathane tubing (Braintree Scientific, Braintree, MA, USA) of 300 μm inner diameter was cut to 55 cm in length and washed with 75% ethanol. HNT-coated surfaces were prepared as previously described [31]. Briefly, HNT solution (6.6% by weight) (NaturalNano Rochester, NY, USA) was treated via sonication and then filtration. Microtubes were then washed with distilled water followed by a 5-min incubation with 2:8 poly-L-lysine (0.1% w/v in water) (Sigma-Aldrich) and a 3-min incubation with treated HNTs. Excess HNTs were removed with distilled water and microtubes were incubated overnight at RT. Immobilization of E-selectin targeted liposomes (ES-PEG L-DXR or ES-PEG EL) onto the surface of both HNT-coated and smooth tubes was achieved by incubation with liposomes for 2.5 hours. Surfaces were then blocked for 1 hour with 5% bovine serum albumin (BSA) (Sigma-Aldrich), in PBS (v/v) for smooth tubes and 5% milk protein in PBS for HNT-coated tubes. Blotting grade blocker nonfat dry milk was obtained from Bio-Rad Laboratories (Hercules, CA, USA). All incubations were preceded and followed by PBS washes. Control tubes were incubated with 5% BSA or 5% milk protein for 3.5 h. To activate immobilized E-selectin proteins, microtubes were incubated with calcium-enriched flow buffer prior to beginning cell capture experiments. All steps were performed at RT or 4 °C.
2.8. Cell capture experiments
Microtubes were fastened onto the stage of an Olympus IX81 motorized inverted microscope following surface functionalization. Perfusion of cells suspended in flow buffer through microtubes was controlled via a motorized syringe pump (KDS 230; IITC Life Science, Woodland Hills, CA, USA). Cells were initially perfused through microtubes at a rate of 0.008 mL/min (wall shear stress of 0.5 dyn/ cm2) for 15 min, and then 0.04 mL/min (wall shear stress of 2.5 dyn/cm2) for 2 h. Captured cells were collected by perfusion of flow buffer and air embolism at 0.1 mL/min (wall shear stress≥10 - dyn/cm2). Cells were cultured in 24-well plates in complete media and analyzed for cell viability at day 4 via trypan blue exclusion.
2.9. Cone-and-plate shear assay
COLO 205 and KG-1a cells were washed and resuspended in 490 μL of Hank’s buffered salt solution (HBSS) (Invitrogen) saturated with Ca2 + (HBSS+) at a concentration of 1×106 cells/mL. Samples of ES-PEG L-DXR, naked L-DXR, or ES-PEG EL (10 μL) were then added to the cell suspension to produce a total volume of 500 μL. Dilute suspensions of both cancer cells and liposomes were then sheared in a LVDV-II+Pro cone-and-plate viscometer (Brookfield Engineering Laboratories, Inc., Middleboro, MA, USA) for 2 h at 2 dyn/cm2. Collected cells were placed in culture and analyzed for cell viability at day 4 via trypan blue exclusion.
2.10. Fluorescence microscopy
To assess cellular internalization of L-DXR and ES-PEG L-DXR in static experiments, KG-1a and COLO 205 cells were incubated with 0.5 μL of L-DXR for 2 h at 37 °C and then washed twice with PBS to remove free liposomes from the cell suspension. To determine successful immobilization of ES-PEG L-DXR onto the inner surface of the microtube, surfaces were prepared as previously described and visualized prior to performing capture experiments. Images of intrinsic DXR fluorescence were taken using an Olympus IX81 microscope.
To determine cellular adhesion and uptake of liposomes following shearing and capture experiments, cells were treated with fluorescent ES-PEG EL for both experiments. Fluorescent liposomes were constructed by replacing ovine wool cholesterol with BODIPY-cholesterol (Avanti Polar Lipids). Cell nuclei were visualized by staining with 2 μL of 10 mg/mL trihydrochloride trihydrate (Hoechst 33342) (Invitrogen, Carlsbad, CA, USA) for 15 min. Cells were washed twice with PBS and then placed on a glass slide and visualized with either a Zeiss 710 Spectral Confocal Microscope System (Carl Zeiss MicroImaging GmbH, Jena, Germany), or a Leica TCS SP2 system (Leica Microsystems Heidelberg GmbH, Mannheim, Germany) at 65× magnification with FITC and DAPI filters. Both individual and Z-stack images were taken and processed using Zen 2009 light edition software (Carl Zeiss MicroImaging GmbH) or LCS software (Leica Microsystems Heidelberg GmbH).
2.11. Statistical analysis
Results were reported as mean±standard error of the mean and statistical significance was calculated using GraphPad Prism Software (San Diego, CA, USA). Paired two-tailed P-value t-tests were performed for static and cell capture experiments, and unpaired t-tests were performed for cone-and-plate experiments. P-values less than 0.05 were considered significant.
3. Results
3.1. Characterization of targeted liposomes
The physical characteristics of the liposomes are summarized in Table 1. The diameters of L-DXR and PEG L-DXR were similar, while ES-PEG L-DXR was 35–40 nm larger in diameter than non-targeting liposomes. The zeta potential of L-DXR was reduced by PEG surface modification from −24.56 mV to −8.49 mV, compared to the zeta potential of naked liposomes. Attachment of ES-PEG showed a slight increase in the zeta potential compared to PEG modified liposomes, but remained lower than the naked liposome zeta potential.
Table 1.
Characterization of liposomes
| L-DXR | PEG L-DXR | ES-PEG L-DXR | |
|---|---|---|---|
| Particle size (nm) | 106.3+/−5.9 | 111.5+/−6.4 | 143.7+/−11.2 |
| Zeta potential (mV) | −24.56+/−6.34 | −8.49+/−5.85 | −13.12+/−6.28 |
Data are mean+/−standard deviation of three independent measurements.
To assess whether functional E-selectin was successfully conjugated to the surface, fluorescently tagged liposomes were sheared with COLO 205 and KG-1a cells in a cone-and-plate viscometer for 10 min at a shear stress of 2 dyn/cm2. Liposomes lacking E-selectin showed negligible adhesion to the cells, due to the lack of E-selectin interactions with E-selectin ligands on the cell surface. Cells sheared with ES-PEG L-DXR showed nearly 99.9% adherence to fluorescent liposomes (Fig. 2A). A nearly identical percentage of cells adhered to ESPEG EL, suggesting that the fluorescence was due to E-selectin adhesion, rather than fluorescence due to DXR drug release. The 10 min shear duration was sufficient for complete adhesion to all cell surfaces with negligible time for significant liposome internalization to occur (Fig. 2B).
Fig. 2.

(A) Percent of COLO 205 and KG-1a cells with liposomes adhered to the surface. Data presented as mean±SEM (n=3). (B) Representative COLO 205 cell (blue = cell nuclei) with fluorescent ES-PEG liposomes (green) adhered to the surface. Scale bar=10 μm.
3.2. Encapsulation with minimal leakage of DXR from liposomes
To evaluate encapsulation efficacy of DXR, the fluorescence intensity of intrinsically fluorescent DXR in the presence or absence of Triton X-100 was measured, and showed that DXR was successfully encapsulated into liposomes (Fig. 3A). The fluorescence intensity of both samples was compared to 1 μM soluble DXR in PBS, which revealed 3-fold greater fluorescence intensity compared to the sample without Triton X-100. The leakage profile of DXR from L-DXR demonstrates minimal release of the drug over time, regardless of E-selectin and/or PEG conjugation to the liposome surface (Fig. 3B).
Fig. 3.

(A) Fluorescence intensity of soluble DXR (1 μM) and 5 μL L-DXR in the presence or absence of 10% Triton X-100. Results expressed in fluorescence units (fu). (B) Leakage profile of 30 μL liposomes over time as measured by fluorescence intensity. Data presented as mean±SEM (n=3).
3.3. Cellular uptake and cytotoxicity of L-DXR
COLO 205 and KG-1a cancer cell lines were treated with 0.001–5 μL of L-DXR, EL, soluble DXR, or ES-PEG L-DXR for 18 h and assessed for cell viability at day 4. The dose response curves for both KG-1a (Fig. 4A) and COLO 205 (Fig. 4B) cells showed a significant decrease in cell viability with all treatment doses of L-DXR and ES-PEG L-DXR when compared to untreated cells. Fluorescence from DXR was visible within the cell, indicating liposome internalization (Fig. 5). Conjugation of ES-PEG to the liposome surface did not decrease the effectiveness of the liposome formulation, as treatment of cancer cells with L-DXR and ES-PEG showed no significant difference in static conditions, and maximum efficacy was observed at 0.5 μL. Neither KG-1a (Fig. 4A) nor COLO 205 (Fig. 4B) cells were affected by treatment with EL. The growth curve of COLO 205 cells treated with 0.5 μL of EL over 4 days was not significantly different from that of untreated cells (Fig. 6), further implying that liposomes themselves do not negatively affect cell viability. The difference in treatment responses is readily seen in brightfield images taken at day 4 (Fig. 7).
Fig. 4.

Liposome uptake and cytotoxic potential on cancer cells under static conditions. KG-1a cells (A) and COLO 205 cells (B) were placed in static conditions for 4 days and evaluated for viability using trypan-blue. (A,B) Dose response curves relating the volume of L-DXR, EL, ES-PEG L-DXR or soluble DXR treatment and viability of cells. All data are presented as mean±SEM (n=3). *** denotes P<0.001.
Fig. 5.
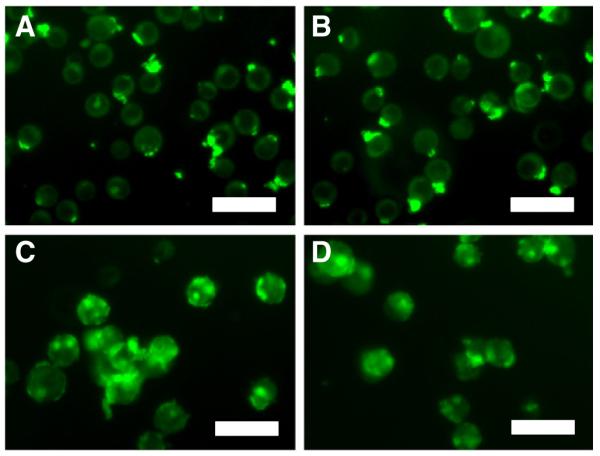
Fluorescent images of L-DXR internalization by KG-1a cells (A,B) and COLO 205 cells (C, D) after treatment with L-DXR (A,C) and ES-PEG L-DXR (B,D). Scale bar=30 μm.
Fig. 6.
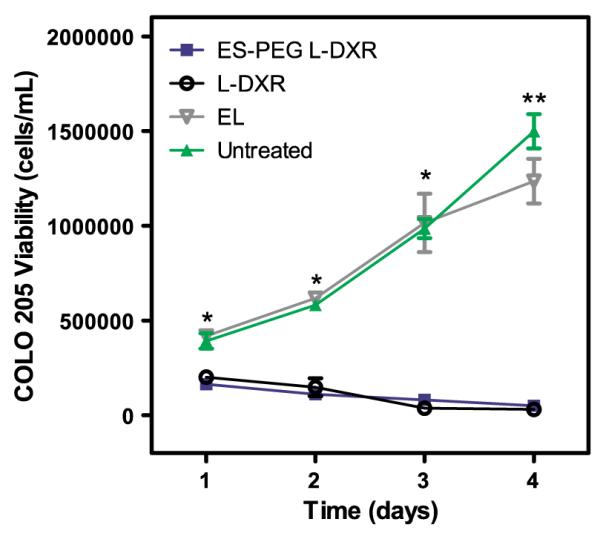
Viability of COLO 205 cells over 4 days after treatment with 0.5 μL of ES-PEG L-DXR, naked L-DXR, or naked EL. All data presented as mean±SEM (n=3). * denotes P<0.05, and ** denotes P<0.01.
Fig. 7.
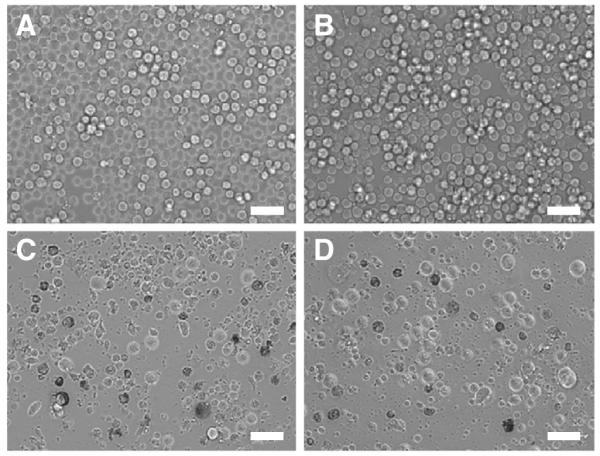
Brightfield images of COLO 205 cells (A) untreated or treated with 0.5 uL of (B) EL, (C) naked L-DXR, or (D) ES-PEG L-DXR taken 4 days after treatment. Scale bar=50 μm.
3.4. Microtubes coated with ES-PEG L-DXR capture and kill cancer cells under flow
Previously, we demonstrated that E-selectin could be absorbed onto the inner surface of microrenathane tubes and cause rolling adhesion of cells expressing E-selectin ligand [31,35]. Microrenathane tubes, which were previously demonstrated as a vascular shunt prototype in rats to successfully capture CD34+ hematopoietic stem and progenitor cells from the circulation [36], as well as for use in multiple in vitro studies examining leukocyte adhesion [37,38] and cancer cell adhesion [35,39] along the inflamed endothelium, were used as a model of human vasculature in our experiments. To ensure that ES-PEG L-DXR was successfully immobilized onto the inner surface of microtubes, fluorescence microscopy was used to detect the intrinsic fluorescence of DXR from liposomes in the coated tubes. Tubes coated with ES-PEG L-DXR displayed a uniform amount of fluorescence throughout the tube (Fig. 8B), while control tubes showed minimal auto-fluorescence (Fig. 8A). Untreated control tubes were coated with BSA to prevent non-specific binding. After targeted liposomes were immobilized onto the surface of the microtube, KG-1a or COLO 205 cells were perfused through coated microtubes at physiological shear stresses to mimic the behavior of CTCs in the vasculature. COLO 205 and KG-1a were chosen as model CTCs for these experiments because they are cancer cell lines that express sialylated carbohydrate ligands and have been shown to exhibit rolling adhesion along immobilized selectin proteins [31,35], characteristics that leukocytes use for the initial steps in diapedesis [4,28,40–42]. Both KG-1a cells (not shown) and COLO 205 cells were captured by ESPEG liposomes (Fig. 8C, Supplemental video 1). No rolling, tethering, or adhesion was observed in control tubes, indicating that cells were specifically captured by ES-PEG liposomes in microtubes (Fig. 8D, Supplemental video 2).
Fig. 8.

Fluorescent images of a (A) control tube and (B) tube coated with ES-PEG L-DXR. Video frames of COLO 205 cells perfused through a (C) ES-PEG L-DXR coated tube and (D) BSA-coated control tube. Scale bar = 100 μm.
After perfusion, captured cells were collected and placed in culture. Of captured KG-1a cells, there was a significant reduction in cell viability of approximately 20% for cells captured with ES-PEG L-DXR compared to untreated cells and those captured with ES-PEG EL (Fig. 9A). Viability of COLO 205 cells was also reduced by ~30% when captured and treated with ES-PEG L-DXR (Fig. 9A). For both cell lines, cell viability was not significantly affected when captured with ES-PEG EL.
Fig. 9.

(A) Comparison of viability of COLO 205 and KG-1a cells following perfusion through control tubes (BSA-coated or 20 μM DXR) or tubes coated with ES-PEG L-DXR or ES-PEG EL. Viability of KG-1a and COLO 205 cells were taken at day 4 (n=3). (B) Percent of nonadhered, flowing RBCs recovered following perfusion through device. (C) Viability of MNCs following perfusion through control tubes or tubes coated with liposomes. Data presented as mean± SEM. * denotes P<0.05. NS denotes not significant.
The cytotoxic effects of the targeting mechanism on healthy blood cells was assessed using red blood cells and mononuclear white blood cells from healthy human donors. RBCs are not targeted by this mechanism, as they did not display significant adhesion to ESPEG liposome coated surfaces compared to BSA coated surfaces (Fig. 9B). MNCs only experienced ~16% decrease in cell viability after perfusion through the device (Fig. 9C).
3.5. ES-PEG L-DXR enhances cancer cell death in a dilute suspension under flow
To assess the effectiveness of targeted liposomes in suspension under flow, KG-1a and COLO 205 cells were exposed to liposomes in a dilute suspension and sheared in a cone-and-plate viscometer for 2 h. The cone-and-plate assay mimics targeted liposomes freely circulating in the bloodstream to target CTCs, as an injectable, alternative method to liposomes immobilized along a microtube platform. Confocal microscopy images taken of KG-1a (Fig. 10A) and COLO 205 (Fig. 10B) cells illustrate the adhesion and internalization of targeted liposomes to cells following shearing experiments. Green and blue fluorescence represents liposomes and the cell nuclei, respectively.
Fig. 10.
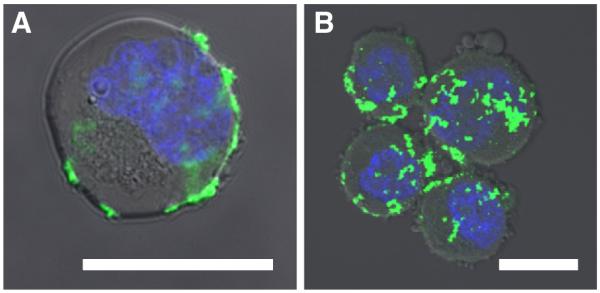
Overlay of brightfield and fluorescence microscopy images taken of KG-1a (A) and COLO 205 (B) cells after 2 h of shearing in a dilute suspension. Scale bars=10 μm. Green fluorescence labels liposomes and the blue fluorescence labels DNA.
KG-1a cells treated with ES-PEG L-DXR experienced a nearly 75% decrease in cell viability compared to EL and untreated controls (Fig. 11). When compared to naked L-DXR, ES-PEG L-DXR also significantly reduced cell viability by nearly 65%, indicating targeted liposomes are more effective than naked liposomes in adhering to and killing cancer cells expressing E-selectin ligands. Similarly, viability of COLO 205 cells treated with ES-PEG L-DXR experienced a 90% decrease in cell viability compared to untreated cells. Taken together, these results suggest that increased interaction between cells and liposomes are necessary for improved targeted delivery of liposomal doxorubicin.
Fig. 11.
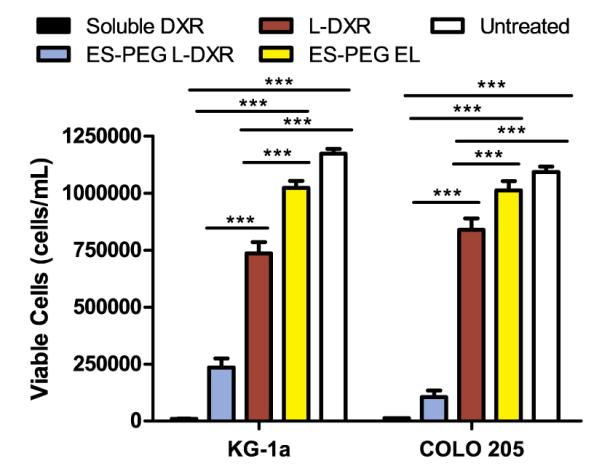
Viability of KG-1a and COLO 205 cells at day 4 following treatment with targeted L-DXR under shear in a dilute suspension. Data presented as mean±SEM (n=3). *** denotes P<0.001.
3.6. HNTs promote the delivery and cytotoxic potential of immobilized ES-PEG L-DXR to cancer cells under flow
A nanostructured surface coating of HNTs was applied to the microtube surfaces to enhance the adhesion of cancer cells to E-selectin liposomes, and thus improve their uptake into cancer cells (Fig. 12). For COLO 205 and KG-1a cells treated with ES-PEG L-DXR coated on HNT-coated microtubes, cell viability decreased by over 87% and nearly 35% respectively, when compared to that on smooth tubes and decreased by approximately 96% and 45% respectively, when compared to control HNT-coated microtubes (Fig. 13A,B). For both cell lines, cell viability was not negatively affected by control HNT-coated microtubes and either smooth or HNT-coated tubes treated with ES-PEG EL. Confocal microscopy images taken of COLO 205 cells following perfusion over targeted liposomes coated on smooth (Fig. 14A) and HNT-coated (Fig. 14B) tubes illustrate the location of liposomes on the cell surface.
Fig. 12.
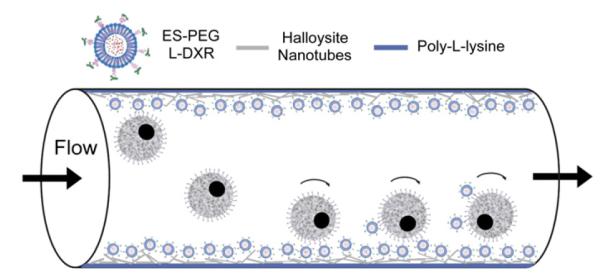
Schematic of halloysite nanotube (HNT)-coated microtube device with immobilized targeted L-DXR to enhance capture and killing of tumor cells under flow.
Fig. 13.

Comparison of smooth and HNT-coated tube with immobilized liposomes on cell viability of (A) KG-1a and (B) COLO 205 after perfusion through microtubes. Cell viability of KG-1a and COLO 205 cells were taken at day 4 (n=3), respectively. Data presented as mean±SEM. * denotes P<0.05, ** denotes P<0.01.
Fig. 14.
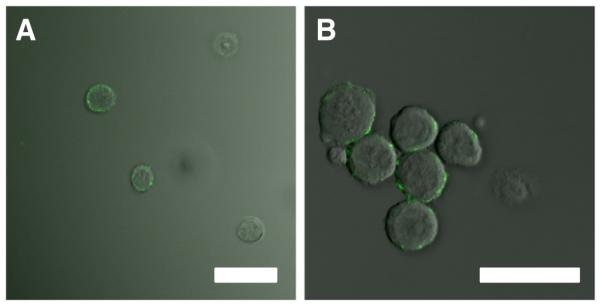
Overlay of brightfield and fluorescence microscopy images of COLO 205 cells following perfusion through a (A) smooth tube and (B) HNT-coated tube incubated with fluorescently-tagged ES-PEG EL. Scale bar=30 μm.
4. Discussion
The capture and killing of cancer cells was successfully achieved using a combination of E-selectin functionalized L-DXR immobilized along the inner surface of a blood-compatible microfluidic device. Results on the use of L-DXR in our study suggest that liposomes were satisfactory for cytotoxic evaluation and effective in killing cancer cells. Over the course of several weeks, L-DXR maintained its retention of the drug, even without the addition of PEG (Fig. 3). This was consistent with previous results [33,43] and can be attributed to two factors: the lipid composition and method of DXR loading. The cholesterol formulation for L-DXR exhibits resistance to strong shear forces, which aid in withstanding forces applied during the extrusion process [33]; thus, providing liposomal stability when encapsulating DXR. Remote loading of DXR was facilitated using an ammonium sulfate gradient, which lies between the basic outer phase and the acidic inner phase of the liposome, and drives DXR inside and ammonium ions out of the vesicle [44]. DXR precipitates as it is driven into liposomes, enhancing drug retention [22].
In treating model CTCs, L-DXR nanoparticles demonstrated great potential in their cytotoxicity to cancer cells. With over 95% of cells killed, a treatment dosage 0.5 μL of liposomal doxorubicin was shown to have a maximum therapeutic efficacy in both KG-1a and COLO 205 cancer cell lines (Fig. 4A,B), despite physiological differences between the cell lineages. This high cell death rate can be attributed to DXR alone. For both cell lines, empty liposomes tended to have either little or no effect following internalization of the liposomes, which occurs within 2 hours of treatment for KG-1a and COLO 205 cells (Fig. 5). Moreover, free DXR and liposomes were removed from the media with multiple washes, suggesting that DXR released from L-DXR inside the cells was responsible for triggering cell death.
Cytotoxic potential to CTCs was shown to be equally or more effective with the functionalization of L-DXR using E-selectin. E-selectin, an adhesion molecule that mediates tumor cell dissemination [3,27,28,30,45], was chosen to decorate the surface of liposomes for target-specificity and DXR delivery to cancer cells expressing E-selectin ligands. The addition of PEG was used to provide stability and charge neutralization of liposomes [34]. To incorporate both E-selectin and PEG to the surface of the liposomes, E-selectin was attached to PEG via a maleimide group [46]. Using COLO 205 and KG-1a cells as model CTCs, static experiments confirmed that ES-PEG L-DXR was no less effective than L-DXR, making them a good candidate for targeting under flow conditions. Cell capture experiments demonstrated that CTCs could be captured and killed under flow conditions, while leukocytes only experienced a minimal decrease in cell viability. While both CTCs and leukocytes express ligands for E-selectin on their surface, metastatic cancer cells typically have very high expression of E-selectin ligands [47–50]. Thus, more liposomes are likely to be adhering to cancer cells than leukocytes, which could explain the differences in cell death. Red blood cells do not possess E-selectin ligands, and thus experienced negligible adhesion using this novel targeting mechanism.
Under shear flow in a cone-and-plate viscometer, improved cytotoxicity of targeted liposomes was evident from a substantial increase in cell death compared to delivery using liposome-coated microtubes. The dynamic environment of the viscometer mimics sustained flow through a closed system, and in contrast to static conditions, allows for an increased number of collisions between the cells and nanoparticles, thus delivering more L-DXR to the cells. Although naked L-DXR contributes considerably to cell death, the bond between E-selectin and its ligands expressed by cells is strong and remains so even after several washes, as evidenced by a light red pellet following centrifugation of the sample (not shown). This further supports the conclusion that the target-specificity of ES-PEG L-DXR is a strong improvement over naked L-DXR and in general, confirms the demonstrated improvement of ligand-targeted liposomes described in previous studies [45,51]. However, in the bloodstream, suspension liposomes could have less opportunity to adhere to CTCs in the complex milieu of blood, which would necessitate high concentrations of chemotherapeutics that could cause undesired side effects. An immobilized platform within the microtube would adhesively interact only with cells that circulate along the near-wall region of blood vessels. Here, margination elevates the local concentration of leukocytes and CTC above those in the systemic circulation by several-fold, allowing more opportunities for an immobilized platform to interact with CTCs. The nanostructured coating of HNTs along the microtube surface enhanced the CTC capture and killing of the microtube platform to similar levels as the cone and plate assay, providing potential that an immobilized platform of targeted chemotherapeutic can be used to capture and kill CTCs, and possibly decrease the probability of metastasis.
5. Conclusion
In this study, we demonstrated the efficacy of targeting, capturing, and killing model CTCs using a novel combination of E-selectin functionalized L-DXR and a biomimetic microtube device. Targeted ES-PEG L-DXR was shown to increase cancer cell killing in both static and dynamic environments. The cone-and-plate shear experiments demonstrated the effectiveness of target-specific nanoparticles in solution, which can aid in the development of drugs intended for systemic delivery. Furthermore, by immobilizing liposomes onto HNT-coated microtubes, capture and kill rates of cells in flow were significantly increased. One advantage of this device for capturing and killing of CTCs is that by immobilizing liposomes onto the microtube surface, the potential distribution of targeted L-DXR into circulation is reduced and a lower dose of the drug is necessary. The proposed device demonstrates potential application of HNT-coated microtubes for reducing the probability of metastasis, and suggests new strategies for enhancing targeted delivery of chemotherapeutics to CTCs.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health grant no. CA143876. The authors acknowledge Jeff Mattison for work with blood sample collection and donor recruitment.
Footnotes
Supplementary materials related to this article can be found on-line at doi:10.1016/j.jconrel.2012.02.018.
References
- [1].Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat. Rev. Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- [2].Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- [3].Chambers AF, MacDonald IC, Schmidt EE, Koop S, Morris VL, Khokha R, Groom AC. Steps in tumor metastasis: new concepts from intravital videomicroscopy. Cancer Metastasis Rev. 1995;14:279–301. doi: 10.1007/BF00690599. [DOI] [PubMed] [Google Scholar]
- [4].Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- [6].Brechot P. Paterlini, Benali NL. Ciruclating tumor cells (CTC) detection: clinical impact and future directions. Cancer Lett. 2007;253:180–204. doi: 10.1016/j.canlet.2006.12.014. [DOI] [PubMed] [Google Scholar]
- [7].Chambers AF, Naumov GN, Vantyghem SA, Tuck AB. Molecular biology of breast cancer metastasis, clinical implications of experimental studies on metastasic inefficiency. Breast Cancer Res. 2000;2:400–407. doi: 10.1186/bcr86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].MacDonald IC, Groom AC, Chambers AF. Cancer spread and micrometastasis development: quantitative approaches for in vivo models. Bioessays. 2002;24:885–893. doi: 10.1002/bies.10156. [DOI] [PubMed] [Google Scholar]
- [9].Young RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N. Engl. J. Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- [10].Bouma J, Beijnen JH, Bult A, Underberg WJ. Anthracycline antitumor agent: a review of physicochemical, analytical, and stability properties. Pharm. Weekbl. Sci. 1986;8:109–133. doi: 10.1007/BF02086146. [DOI] [PubMed] [Google Scholar]
- [11].Osheroff N, Corbett AH, Robinson MJ. Mechanism of action of topoisomerase II-targeted antineoplastic drugs. Adv. Pharmacol. 1994;29:105–126. doi: 10.1016/s1054-3589(08)61134-5. [DOI] [PubMed] [Google Scholar]
- [12].Reddy LH. Drug delivery to tumours: recent strategies. J. Pharm. Pharmacol. 2005;57:1231–1242. doi: 10.1211/jpp.57.10.0001. [DOI] [PubMed] [Google Scholar]
- [13].Fritze A, Hens F, Kimpfler A, Schubert R, Suss R. Peschka. Remote loading of doxorubicin intro liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta. 2006;1758:1633–1640. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]
- [14].Saad M, Garbuzenko OB, Ber E, Chandna P, Khandare JJ, Pozharov VP, Minko T. Receptor targeted polymers, dendrimers, liposomes: which nanocarrier is the most efficient for tumor-specific treatment and imaging? J. Control. Release. 2008;130:107–114. doi: 10.1016/j.jconrel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Herman EH, Rahman A, Ferrans VJ, Vick JA, Schein PS. Prevention of chronic doxorubicin cardiotoxicity in beagles by liposomal encapsulation. Cancer Res. 1983;43:5427–5432. [PubMed] [Google Scholar]
- [16].Krishna R, McIntosh N, Riggs RW, Mayer LD. Doxorubicin encapsulated in sterically stablized liposomes exhibits renal and biliary clearance properties that are independent of Valspodar (PSC 833) under conditions that significantly inhibit nonencapsulated drug excretion. Clin. Cancer Res. 1999;5:2939–2947. [PubMed] [Google Scholar]
- [17].Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19:424–436. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- [18].Saltzman WM. Drug Delivery: Engineering Principles for Drug Therapy. Oxford University Press Inc.; New York: 2004. [Google Scholar]
- [19].Soundararajan A, Bao A, Phillips WT, Perez R, Goins BA. [(186)Re]Liposomal doxorubicin (Doxil): in vitro stability, pharmacokinetics, imaging and biodistribution in a head and neck squamous cell carcinoma xenograft model. Nucl. Med. Biol. 2009;36:515–524. doi: 10.1016/j.nucmedbio.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lasic DD. Doxorubicin in sterically stabilized liposomes. Nature. 1996;380:561–562. doi: 10.1038/380561a0. [DOI] [PubMed] [Google Scholar]
- [21].Silvander M. Steric stabilization of liposomes — a review. Prog. Colloid Polym. Sci. 2002;120:35–40. [Google Scholar]
- [22].Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin: a review of animal and human studies. Clin. Pharmacokinet. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- [23].Carrion C, de Madariaga MA, Domingo JC. In vitro cytotoxic study of immunoliposomal doxorubicin targeted to human CD34(+) leukemic cells. Life Sci. 2004;75:313–328. doi: 10.1016/j.lfs.2003.12.020. [DOI] [PubMed] [Google Scholar]
- [24].Han HD, Lee A, Hwang T, Song CK, Seong H, Hyun J, Shin BC. Enhanced circulation time and antitumor activity of doxorubicin by comblike polymer-incorporated liposomes. J. Control. Release. 2007;120:161–168. doi: 10.1016/j.jconrel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- [25].Ceh B, Winterhalter M, Frederik PM, Vallner JJ, Lasic DD. Stealth® liposomes: from theory to product. Adv. Drug Deliv. Rev. 1997;24:165–177. [Google Scholar]
- [26].Kitayama J, Tsuno N, Sunami E, Osada T, Muto T, Nagawa H. E-selectin can mediate the arrest type of adhesion colon cancer cells under physiological shear flow. Eur. J. Cancer. 2000;36:121–127. doi: 10.1016/s0959-8049(99)00228-2. [DOI] [PubMed] [Google Scholar]
- [27].Krause T, Turner GA. Are selectins involved in metastasis? Clin. Exp. Metastasis. 1999;17:183–192. doi: 10.1023/a:1006626500852. [DOI] [PubMed] [Google Scholar]
- [28].Gout S, Tremblay PL, Huot J. Selectins and selectin ligand in extravasation of cancer cells and organ selectivity of metastasis. Clin. Exp. Metastasis. 2008;25:335–344. doi: 10.1007/s10585-007-9096-4. [DOI] [PubMed] [Google Scholar]
- [29].Kohler S, Ullrich S, Richter U, Schumacher U. E-/P-selectins in colon carcinoma metastasis: first in vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br. J. Cancer. 2010;102:602–609. doi: 10.1038/sj.bjc.6605492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Laubli H, Borsig L. Selectins promote tumor metastasis. Semin. Cancer Biol. 2010;20:169–177. doi: 10.1016/j.semcancer.2010.04.005. [DOI] [PubMed] [Google Scholar]
- [31].Hughes AD, King MR. Use of naturally occurring halloysite nanotubes for enhanced capture of flowing cells. Langmuir. 2010;26:12155–12164. doi: 10.1021/la101179y. [DOI] [PubMed] [Google Scholar]
- [32].Amselem S, Cohen R, Druckmann S, Gabizon A, Goren D, Abra RM, Huang A, New R, Barenholz Y. Preparation and characterization of liposomal doxorubicin for human use. J. Liposome Res. 1992;2:93–123. [Google Scholar]
- [33].Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- [34].Huang Z, King MR. An immobilized nanoparticle-based platform for efficient gene knockdown of targeted cells in the circulation. Gene Ther. 2009;16:1271–1282. doi: 10.1038/gt.2009.76. [DOI] [PubMed] [Google Scholar]
- [35].Rana K, Liesveld JL, King MR. Delivery of apoptotic signal to rolling cancer cells: a novel biomimetic technique using immobilized TRAIL and E-selectin. Biotechnol. Bioeng. 2009;102:1692–1702. doi: 10.1002/bit.22204. [DOI] [PubMed] [Google Scholar]
- [36].Wojciechowski JC, Narasipura SD, Charles N, Mickelsen D, Rana K, Blair ML, King MR. Capture and enrichment of CD34-positive haematopoietic stem and progenitor cells from blood circulation using P-selectin in an implantable device. Br. J. Haematol. 2008;140:673–681. doi: 10.1111/j.1365-2141.2007.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ball CJ, King MR. Role of c-Abl in L-selectin shedding from the neutrophil surface. Blood Cells Mol. Dis. 2011;46:246–251. doi: 10.1016/j.bcmd.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee D, Schultz JB, Knauf PA, King MR. Mechanical shedding of L-selectin from the neutrophil surface during rolling on sialyl lewis X under flow. J. Biol. Chem. 2007;282:4812–4820. doi: 10.1074/jbc.M609994200. [DOI] [PubMed] [Google Scholar]
- [39].Yin X, Rana K, Ponmudi V, King MR. Knockdown of fucosyltransferase III disrupts the adhesion of circulating cancer ce lls to E-selectin without affecting hematopoietic cell adhesion. Carbohydr. Res. 2010;345:2334–2342. doi: 10.1016/j.carres.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM. Interactions between cancer cells and the endothelium in metastasis. J. Pathol. 2000;190:310–329. doi: 10.1002/(SICI)1096-9896(200002)190:3<310::AID-PATH525>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- [41].Kim YJ, Borsig L, Han HL, Varki NM, Varki A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am. J. Pathol. 1999;155:461–472. doi: 10.1016/S0002-9440(10)65142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int. J. Cancer. 2009;125:1298–1305. doi: 10.1002/ijc.24409. [DOI] [PubMed] [Google Scholar]
- [43].Ceh B, Lasic DD. A rigorous theory of remote loading of drugs into liposomes. Langmuir. 1995;11:3356–3368. doi: 10.1006/jcis.1996.4555. [DOI] [PubMed] [Google Scholar]
- [44].Chemin C, Pean JM, Bourgaux C, Pabst G, Wuthrich P, Couvreur P, Ollivon M. Supramolecular organization of S12363-liposomes prepared with two different remote loading processes. Biochim. Biophys. Acta. 2009;1788:926–935. doi: 10.1016/j.bbamem.2008.11.017. [DOI] [PubMed] [Google Scholar]
- [45].Forssen E, Willis M. Ligand-targeted liposomes. Adv. Drug Deliv. Rev. 1998;29:249–271. doi: 10.1016/s0169-409x(97)00083-5. [DOI] [PubMed] [Google Scholar]
- [46].Nallamothu R, Wood GC, Pattillo CB, Scott RC, Kiani MF, Moore BM, Thoma LA. A tumor vasculature targeted liposome delivery system for combretastatin A4: design, characterization and in vitro evaluation. AAPS PharmSciTech. 2006;7:E32. doi: 10.1208/pt070232. [DOI] [PubMed] [Google Scholar]
- [47].Dimitroff CJ, Lechpammer M, Woodward D. Long, Kutok JL. Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelial cells under shear flow is mediated by E-selectin. Cancer Res. 2004;64:5261–5269. doi: 10.1158/0008-5472.CAN-04-0691. [DOI] [PubMed] [Google Scholar]
- [48].Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004;95:377–384. doi: 10.1111/j.1349-7006.2004.tb03219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Magnani JL. The discovery, biology, and drug development of sialyl Lea and sialyl Lex. Arch. Biochem. Biophys. 2004;426:122–131. doi: 10.1016/j.abb.2004.04.008. [DOI] [PubMed] [Google Scholar]
- [50].Ogawa J, Inoue H, Koide S. Expression of α-1,3-fucosyltransferase type IV and VII genes is related to poor prognosis in lung cancer. Cancer Res. 1996;56:325–329. [PubMed] [Google Scholar]
- [51].Tsuruta W, Tsurushima H, Yamamoto T, Suzuki K, Yamazaki N, Matsumura A. Application of liposomes incorporating doxorubicin with sialyl lewis X to prevent stenosis after rat carotid artery injury. Biomaterials. 2009;30:118–125. doi: 10.1016/j.biomaterials.2008.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.