

Abstract
The highly tunable properties of poly(ethylene glycol) (PEG)-based hydrogel systems permit their use in a wide array of regenerative medicine and drug delivery applications. One of the most valuable properties of PEG hydrogels is their intrinsic resistance to protein adsorption and cell adhesion, as it allows for a controlled introduction of desired bioactive factors including proteins, peptides, and drugs. Acrylate-PEG-N-hydroxysuccinimide (Acr-PEG-NHS) is widely utilized as a PEG linker to functionalize bioactive factors with photocrosslinkable groups. This enables their facile incorporation into PEG hydrogel networks or the use of PEGylation strategies for drug delivery. However, PEG linkers can sterically block integrin binding sites on functionalized proteins and reduce cell-material interactions. In this study we demonstrate that reducing the density of PEG linkers on protein backbones during functionalization results in significantly improved cell adhesion and spreading to bioactive hydrogels. However, this reduction in functionalization density also increases protein loss from the matrix over time due to ester hydrolysis of the Acr-PEG-NHS linkers. To address this, a novel PEG linker, acrylamide-PEG-isocyanate (Aam-PEG-I), with enhanced hydrolytic stability was synthesized. It was found that decreasing functionalization density with Aam-PEG-I resulted in comparable increases in cell adhesion and spreading to Acr-PEG-NHS systems while maintaining protein and bioactivity levels within the hydrogel network over a significantly longer time frame. Thus, Aam-PEG-I provides a new option for protein functionalization for use in a wide range of applications that improves initial and sustained cell-material interactions to enhance control of bioactivity.
Keywords: Poly(ethylene glycol), hydrogels, protein functionalization, biostability, cell-material interactions
INTRODUCTION
The ability to tune the chemical, mechanical, and biological properties of poly(ethylene glycol) (PEG)-based hydrogels has enabled their extensive use in regenerative medicine and drug delivery applications.1–4 One of the most unique and advantageous characteristics of PEG hydrogels is their inherent resistance to protein adsorption and cell adhesion.5 This not only results in an attenuated immune response but also allows for a controlled introduction of bioactivity into PEG hydrogel scaffolds.6–9 While many methods exist for incorporation of bioactive agents into hydrogels, covalent attachment generally provides greater stability and control over biomolecule presentation and release.10–11 A widely utilized method to covalently incorporate proteins or peptides into PEG hydrogels is functionalization with acrylate-PEG-N-hydroxysuccinimide (Acr-PEG-NHS).12 Many growth factors and proteins contain lysines in their backbones that provide a primary amine for functionalization. Thus, the NHS-lysine ε-amino group reaction can be utilized to functionalize bioactive agents with crosslinkable acrylate groups.9,12–14 Following functionalization, proteins can be immobilized in PEG hydrogels during photopolymerization without detriment to bioactivity. This provides scaffolds that deliver specific bioactive cues for tissue regeneration and healing.6,9,15–16 Functionalization of therapeutic proteins with PEG linkers can also be used in drug delivery systems to increase circulation times.17
The PEG linkers on functionalized proteins serve to covalently stabilize them into synthetic networks; however, they can also physically block and/or increase steric hindrance around integrin binding sites and interfere with cell-protein interactions, Figure 1.18–19 In proteins and growth factors with multiple amine functionalization sites, the density of Acr-PEG-NHS can be easily varied in the traditional functionalization scheme to provide bioactive factors with a range of functionalization densities. A protein that is functionalized with fewer linkers could potentially have improved cell interactions through allowing increased access to integrin binding sites, Figure 1. However, Van Den Bulcke et al. demonstrated that gelatin hydrogels with lower functionalization levels had decreased moduli compared to those with higher functionalization levels. This indicates that a reduction in crosslinking occurs as protein functionalization density is decreased due to reduced crosslinking sites on the protein.20 From these findings, one can also assume that lowering the functionalization density would decrease initial incorporation and retention of proteins in a bioactive hydrogel network. Therefore, there is an evident trade-off between reduced steric interactions that promote enhanced initial cell interactions and sufficient protein retention to support sustained cell interactions. These are both critical factors in the development of bioactive scaffolds for regenerative medicine and PEGylated drugs with improved circulation times. However, the effect of functionalization density on both initial cell interactions and protein retention over time is poorly characterized in current literature.
Figure 1.

Schematic representation of hypothesized effects of protein functionalization density on integrin binding. A decreased number of PEG linkers on the protein backbone (low functionalization density) could reduce steric blocking and improve integrin binding to functionalized proteins.
In the present studies, we utilized Acr-PEG-NHS to functionalize collagen and a Streptococcal collagen-like (Scl2-1) protein for incorporation into PEG hydrogels.6 Scl2-1 is based upon a collagen-mimetic protein that is derived from group A Streptococcus. It contains the Gly-Xaa-Yaa (GXY) motifs of collagen but lacks hydroxyproline and therefore provides a stable triple helix without the need for costly post-translational modification.21–22 This enables recombinant expression in E. coli, thereby eliminating batch-to-batch variability concerns associated with native collagen and cost concerns with solid-phase synthesis of extracellular matrix peptides.23 Additionally, Scl2-1 proteins act a biological blank slate and are resistant to cell adhesion and platelet aggregation, even in the presence of serum.6,24 This allows for controlled introduction of receptor binding motifs via site-directed mutagenesis to induce specific cell interactions. Scl2-1 proteins represent a valuable biomaterial system that can be utilized to promote controlled bioactivity in regenerative medicine applications. For these studies, the GFPGER sequence recognized by α1β1 and α2β1 integrins was inserted into Scl2-1 to produce a modified protein, Scl2-2, that promotes endothelial cell (EC) adhesion.25–26 The impact of reducing Acr-PEG-NHS functionalization density on EC adhesion and protein maintenance over time was analyzed in both PEG-collagen and PEG-Scl2-2 hydrogel systems to determine the versatility of these techniques. Additionally, hydrolytic stability of the PEG matrix and the protein linker was enhanced in an effort to improve collagen and Scl2-2 retention, Figure 2. The resulting protein levels in the hydrogel matrix and the corollary effects on bioactivity were analyzed over a swelling time frame of 6 weeks to determine a functionalization scheme that increases initial and sustained cell-material interactions.27
Figure 2.
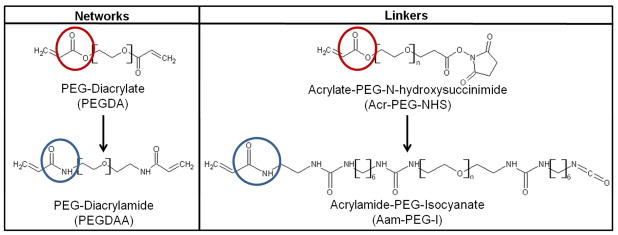
Comparison of network macromers and PEG functionalization linkers used to tune retention of collagen and Scl2-2. Ester linkages in PEGDA and Acr-PEG-NHS (red) were replaced with amides in PEGDAA and Aam-PEG-I (blue) to increase hydrolytic stability.
MATERIALS AND METHODS
Materials
All chemicals and materials were purchased from Sigma Aldrich (St. Louis, MO) and used as received unless otherwise noted. Acr-PEG-NHS (3.5 kDa) and PEG diamine (3.4 kDa) were purchased from Jenkem Technology USA (Allen, TX).
Synthesis of PEGDA and PEGDAA
Poly(ethylene glycol) diacrylate (PEGDA) was synthesized as previously described.28 Briefly, acryloyl chloride was added dropwise to a solution of PEG (3.4 kDa) diol and triethylamine (TEA) in anhydrous dichloromethane (DCM) under nitrogen. The molar ratios of PEG diol, TEA, and acryloyl chloride were 1:2:4, respectively. After 24 hours of stirring, the reaction solution was washed with 8 molar equivalents of 2M potassium bicarbonate and dried with anhydrous sodium sulfate. The final product was precipitated in cold diethyl ether, filtered, and dried under vacuum.
PEG diacrylamide (PEGDAA) was prepared as previously described in a similar method to PEGDA.27 Briefly, acryloyl chloride (4 molar equivalents) was added dropwise to a solution of PEG (3.4 kDa) diamine (1 molar equivalent) and TEA (2 molar equivalents) in anhydrous DCM under nitrogen. The reaction was allowed to proceed for 24 hours, and then it was washed with 2M potassium bicarbonate (8 molar equivalents). Following drying with anhydrous sodium sulfate, the product was precipitated in cold diethyl ether, filtered, and dried under vacuum.
Successful synthesis of PEGDA and PEGDAA was confirmed with Fourier transform infrared (FTIR) and proton nuclear magnetic resonance (1H-NMR) spectroscopy. A Bruker TENSOR 27 spectrometer was used to obtain transmission FTIR spectra of control and functionalized polymers that were solution cast onto KBr pellets. Successful acrylation of PEG diol was indicated by the introduction of an ester peak at 1730 cm−1 and loss of the hydroxyl peak at 3300 cm−1. The introduction of amide peaks at 1640 cm−1 and 1675 cm−1 indicated successful acrylamidation of PEG diamine. A Mercury 300 MHz spectrometer with a TMS/solvent signal as an internal reference was utilized to obtain 1H-NMR spectra of control and functionalized polymers. Greater than 90% conversions of hydroxyl to acrylate endgroups and amine to acrylamide endgroups were observed for all candidate macromers. PEGDA: 1H-NMR (CDCl3): 3.6 ppm (m, -OCH2CH2-), 4.3 ppm (t, -CH2OCO-), 6.1 ppm (dd, -CH=CH2), 5.8 and 6.4 ppm (dd, -CH=CH2). PEGDAA: 1H-NMR (CDCl3): 3.6 ppm (m, -OCH2CH2-); 6.5 ppm (s, -CH2-NH-); 6.4 ppm (m, -CH=CH2); 5.6 and 6.1 ppm (m, -CH=CH2).
Protein Functionalization with Acrylate-PEG-N-hydroxysuccinimide
The Scl2.28 sequence was amplified and purified as previously described.22 Scl2-2 was modified from Scl2-1 through the introduction of the GFPGER sequence during site directed mutagenesis as previously described.22 Scl2-2 and non-gelling rat tail collagen type I (Collagen, Type I solution from rat tail, Sigma Aldrich, St. Louis, MO) were functionalized with photoreactive crosslink sites according to a previously described protocol.12 The backbones of both Scl2-2 and collagen type I contain ~33 lysines that can be utilized in the established NHS-lysine ε-amino group reaction to allow for bioconjugation. The proteins were reacted with Acr-PEG-NHS (MW 3500) in 50 mM sodium bicarbonate buffer (pH 8.5) for 24 hours at room temperature. The molar ratio of Acr-PEG-NHS:NH2 was varied from 0.1:1 to 1:1 to provide proteins with varied functionalization densities (0.1×, 0.5×, and 1×). Dialysis against 0.1M hydrochloric acid was carried out for 24 hours to remove basic byproducts, and additional purification was completed by dialysis against reverse osmosis (RO) water for 24 hours (MWCO = 20,000 Da). Following lyophilization of purified products, FTIR spectroscopy was utilized as described above to confirm functionalization of modified proteins with varied PEG-linker densities.
Synthesis of Acrylamide-PEG-Isocyanate Linker
A solution of PEG (3.4 kDa) diamine in anhydrous DCM was added dropwise to hexane diisocyanate under nitrogen at a 1:2 molar equivalent to synthesize PEG diisocyanate (PEGDI). The reaction was allowed to proceed with stirring for 2 hours. Upon completion of the PEGDI synthesis, a solution with 1 molar equivalent of aminoethyl acrylamide (Enamine, Kiev, Ukraine) in dimethyl sulfoxide was added dropwise to the reaction solution while stirring under nitrogen. After 2 hours, the final product was precipitated in cold diethyl ether, filtered, and dried under vacuum. Successful synthesis of PEGDI and Aam-PEG-I was confirmed with FTIR spectroscopy in a method like that described above. Aam-PEG-I structure was confirmed with 1H-NMR spectroscopy as described above. Aam-PEG-I: 1H-NMR (CDCl3): 3.6 ppm (m, -OCH2CH2-), 3.3 ppm (m, -NHCH2CH2NH-), 1.4 and 3.1 ppm (m, -NH[CH2]6N-), 5.1–5.4 and 7.3 ppm (s, -NH-), 6.1 ppm (dd, -CH=CH2), 5.8 and 6.2 ppm (dd, -CH=CH2).
Protein Functionalization with Acrylamide-PEG-Isocyanate
Scl2-2 and rat tail collagen type I were functionalized with Aam-PEG-I in a method that was modified from that described above. Briefly, Aam-PEG-I was dissolved in anyhydrous dimethylformamide and added dropwise to protein solutions in phosphate buffered saline (PBS) at room temperature. The molar ratio of Aam-PEG-I:NH2 was either 0.1:1 or 1:1 to provide proteins with low and high functionalization densities, respectively. After 2 hours of stirring, dialysis against RO water for 24 hours (MWCO = 20,000 Da) was used to purify functionalized proteins. FTIR spectroscopy confirmed functionalization of proteins with low and high Aam-PEG-I densities.
Preparation and Characterization of Bioactive PEG Hydrogels
PEG, PEG-Scl2-2, and PEG-collagen hydrogels were prepared by dissolving PEG (3.4 kDa) DA or PEG (3.4 kDa) DAA (10 wt%) and functionalized Scl2-2 or collagen (4 mg ml−1) in 20 mM acetic acid. A photoinitiator solution (1 mg Irgacure 2959 per 0.01 ml 70% ethanol) was added at 1 vol% of precursor solution. Solutions were pipetted between 0.5–1.5 mm spaced plates or into microcentrifuge tubes and crosslinked by 6 min exposure to long wave UV light (Intelli Ray Shuttered UV Flood Light, Integrated Dispensing Solutions, Inc., 365 nm, 4 mW/cm2).
To measure compressive modulus, six 8-mm diameter discs were punched from hydrogel sheets (1.5 mm thick) and swollen in RO water overnight. Samples were subjected to mechanical testing using a dynamic mechanical analyzer (RSAIII, TA Instruments) equipped with a parallel-plate compression clamp. Testing was performed under unconstrained compression at room temperature. Dynamic strain sweeps were used to determine the linear viscoelastic range for each hydrogel formulation. A strain within the upper region of the linear viscoelastic range was used in a constant strain frequency sweep. Tests were conducted between 0.79 and 79 Hz, and the compressive storage modulus was taken at 1.25 Hz.
For swelling assessments, six 8-mm diameter discs were punched from hydrogel sheets (1.5 mm thick) directly after polymerization. The hydrogel discs were swollen in RO water overnight and weighed to determine the equilibrium swelling mass (Ws). Then, samples were dried under vacuum overnight and weighed to assess dry (polymer) mass (Wd). The equilibrium volumetric swelling ratio, Q, was calculated from the equilibrium mass swelling ratio:
| [1] |
Protein Retention in Bioactive PEGDA Hydrogels
PEGDA-collagen and PEGDA-Scl2-2 gels were crosslinked directly into microcentrifuge tubes and swelled in PBS at 37°C with weekly solution changes. After a swelling time period of 1 day to 6 weeks, gels were dried under vacuum then incubated in a specified volume of 0.5M sodium hydroxide (NaOH) at 37°C for approximately 3 hours, or until complete hydrolysis occurred. Protein concentration in the hydrolyzed gel solutions was determined with a 3-(4-carboxy-benzoyl)-2-quinoline-carboxaldehyde (CBQCA) protein quantification kit (Molecular Probes, Life Technologies, Grand Island, NY), as specified by the manufacturer. Briefly, 10 μl aliquots of each sample were diluted in 125 μl of 0.1M sodium borate buffer. Then, 5 μl of 5 mM potassium cyanide (KCN) were added to each sample, followed by an addition of 10 μl of 5 mM CBQCA (dissolved to 40 mM in DMSO, then diluted to 5 mM in 0.1M sodium borate buffer). Each sample was tested in triplicate and run against a standard of the same protein type and functionalization density to prevent any errors that could occur due to different numbers of free amines on the varied functionalization densities and/or on the two protein types. After 1 to 3 hours of incubation in a black 96 well plate at room temperature with shaking, fluorescence was read using an Infinite M200 Pro plate reader (Tecan Group, Inc.; emission = 550 nm, excitation = 465 nm). Standard curves of fluorescence versus protein concentration were generated for each protein formulation, and the protein concentration (C, mg/ml) in the hydrolyzed gels was determined by inserting the measured fluorescence into a best-fit linear equation for corresponding standard curves. Protein retention in PEGDA gels at each time point (PRDA, mg/ml) was then calculated to be:
| [2] |
where VNaOH is the volume of the hydrolysis solution and V0 is the original gel volume in milliliters. Initial protein incorporation (I) was defined as the percentage of protein retained relative to the initial concentration (C0) after 1 day of swelling in PBS:
| [3] |
Percent protein retention over time (% PR) was measured as the percent difference between the protein retained at each time point (PRn) relative to the initial protein incorporation concentration taken after 1 day of swelling in PBS (PR1):
| [4] |
Protein Retention in Bioactive PEGDAA Hydrogels
PEGDAA-collagen and PEGDAA-Scl2-2 gels were prepared similarly to PEGDA gels and swelled in PBS at 37°C for up to 6 weeks. Due to the hydrolytic stability of PEGDAA gels, protein loss was measured to determine protein retention. It was confirmed that the two methods provided similar initial protein incorporation measurements (less than 10% difference) in PEGDA gels. Briefly, swelling solutions were retained throughout the specified time period, frozen at −80°C, and lyophilized to obtain the gel leachables. The lyophilized powders were dissolved in a known volume of phosphate buffered saline (PBS), and protein concentration of the resulting solution was determined with a CBQCA protein quantitation kit as described above. All samples were tested in triplicate against a standard of the same protein type and functionalization density. The concentration of lost protein (CL, mg/ml) in the hydrolyzed gels was determined by inserting the measured fluorescence into a best-fit linear equation for corresponding standard curves. Protein retention (PRDAA, mg/ml) was then calculated as:
| [5] |
where C0 and V0 are the original protein concentration (mg/ml) and gel volume (ml), respectively, and VPBS is the volume of PBS used to dissolved the lyophilized leachables (ml). Initial protein incorporation (I) was measured from PRDAA according to Equation 2, and percent protein retention was measured relative to the initial protein incorporation concentration according to Equation 4.
Cell Adhesion and Spreading
Bovine aortic endothelial cells (BAOECs) were used for all cell studies. Cells were cultured in vitro at 37°C/5% CO2 with Dulbecco’s Modified Eagle Medium (DMEM, high glucose GlutaMAX™, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen) and 1% Penicillin-Strepotomycin solution (Gibco). Cells were used between passages 7 and 10 after 10–14 days of culture for all studies.
Bioactive hydrogels were crosslinked between 0.5 mm spaced plates as described above and swelled for 3 hours in 70% ethanol. Subsequently, gels were swelled in sterile PBS for approximately 6 hours with solution changes every 2 hours to remove residual ethanol. Then, 6 mm diameter punches were taken from each hydrogel formulation and discs were swelled in PBS at 37°C for a specified time period (1 day to 6 weeks) with regular solution changes. Only bioactive hydrogel formulations with greater than 10% protein retention at 6 weeks were used in 6 week bioactivity retention studies. Following the desired time period, BAOECs were seeded onto specimens at 10,000 cells cm−1 for 3 hours at 37°C/5% CO2. Cells were then fixed with 3.7% gluteraldehyde and stained with rhodamine phalloidin (F-actin/cytoplasm, Invitrogen) and SYBRGreen (DNA/nucleus, Invitrogen). Representative images were obtained with a Nikon Eclipse TE2000-S with 3 field views per specimen and 4 specimens per hydrogel formulation. The number of cell nuclei per image was used as a quantitative assessment of cell adhesion on each test surface and was counted using the SYBRGreen stained images. Average cell spreading, or cell area, was quantified by applying the Photoshop “magic wand” tool to the image background of the rhodamine phalloidin stained images and adjusting the tool tolerance so that all extracellular regions were selected. The histogram function was utilized to evaluate the extracellular pixels (PEx). The average pixels per cell (Acell) for that image was quantified as:
| [6] |
where PT represents total image pixels and N is total cell nuclei. Pixels were then converted to microns using known objective scaling.
Statistical Analysis
All modulus, swelling, and protein retention data were expressed as the mean ± standard derivation of the mean. All cell adhesion and spreading data were expressed as the mean ± standard error of the mean. Statistical analysis was performed by an unpaired two-tailed student’s t-test. Statistical significance was accepted at p<0.05.
RESULTS AND DISCUSSION
Protein Functionalization with Acr-PEG-NHS
Fourier transform infrared (FTIR) spectroscopy was utilized to confirm functionalization of collagen and Scl2-2 with a range of Acr-PEG-NHS densities, Figure 3. FTIR absorbance peaks assigned to the amide of the protein (C=O) at 1650 cm−1 and the ether backbone of the Acr-PEG-NHS (C-O-C) at 1110 cm−1 were present in the purified products. As the reaction ratios of Acr-PEG-NHS-to-lysine were increased from 0.1:1 to 1:1, a corollary increase in the relative peak height ratios of ether-to-amide was also observed. This confirmed that proteins with a range of functionalization levels were synthesized. Additionally, relative peak ratios of different batches of functionalized proteins remained constant, indicating that similar functionalization levels were achieved between batches. For these studies, collagen and Scl2-2 were functionalized with 0.1:1, 0.5:1, and 1:1 molar ratios of Acr-PEG-NHS-to-lysine to yield 0.1×, 0.5×, and 1× functionalization densities, respectively.
Figure 3.

Transmission FTIR spectra of functionalized (A) collagen and (B) Scl2-2. Ratio of 1110 cm−1 (ether of PEG) to 1650 cm−1 (amide of protein) peaks decreases with decreasing functionalization density.
Protein Functionalization with Acrylamide-PEG-Isocyanate Linker
Acrylamide-PEG-isocyanate (Aam-PEG-I) was synthesized in which the hydrolyzable acrylate group of Acr-PEG-NHS was replaced with a biostable acrylamide group, Figure 2. FTIR spectroscopy was used to confirm key structural features during the two-step reaction of Aam-PEG-I, Figure 4A. First, isocyanate endcapping of PEG diamine was indicated by the introduction of an isocyanate peak at 2250 cm−1 and urea peaks at 1560 and 1650 cm−1. In the second step, relative decreases in the isocyanate peak and increases in the urea peaks confirmed successful formation of the Aam-PEG-I linker. The integration ratio of acryloyl protons to backbone methyl protons and presence of HDI methyl protons in the 1H NMR spectra confirmed the structure and final average molecular weight of Aam-PEG-I, Figure 4B. Endgroup conversion ratios of greater than 80% and final molecular weights between 3800 and 4200 Da were observed for all candidate linkers, indicating that minimal coupling occurred during the synthesis.
Figure 4.

(A) Transmission FTIR spectra of PEG (3.4 kDa) diisocyanate (PEGDI) synthesized from PEG (3.4 kDa) diamine and acrylamide-PEG (3.4 kDa)-isocyanate (Aam-PEG-I) synthesized from the resulting PEGDI. (B) 1H NMR spectra of Aam-PEG-I. (C) Transmission FTIR spectra of Aam-PEG-I-functionalized collagen and Scl2-2. Ratio of 1110 cm−1 (ether) to 1650 cm−1 (amide) peaks decreases with decreasing functionalization density.
The final product was utilized to functionalize collagen and Scl2-2 with low (0.1×) and high (1×) functionalization densities. FTIR absorbance peak ratios of the ether of the Aam-PEG-I (1110 cm−1) were decreased relative to the amide of the proteins (1650 cm−1) as functionalization density was decreased, Figure 4C. Relative peak ratios were constant between different batches of Aam-PEG-I functionalized proteins, indicating that consistent levels of functionalization were achieved. This data confirmed that Aam-PEG-I can be used to functionalize proteins with varied functionalization densities similarly to the traditional Acr-PEG-NHS linker.
Bioactive Hydrogel Modulus and Swelling Ratio
To confirm that protein functionalization density and linker type do not alter bulk network properties, 10% PEG (3.4 kDa) DAA gels were prepared with Acr-PEG-NHS- and Aam-PEG-I-functionalized collagen at high (1×) and low (0.1×) functionalization densities, and compressive modulus and swelling ratio were measured relative to 10% PEG (3.4 kDa) DAA gel controls, Figure 5. No significant differences in modulus or swelling were observed for any of the tested formulations. This was expected, as the protein component comprises less than 1% of the network, and properties should be dominated by the synthetic PEG matrix. Our previous studies have demonstrated similarly negligible effects from adding functionalized Scl2-2 into PEG-based hydrogel scaffolds.6
Figure 5.
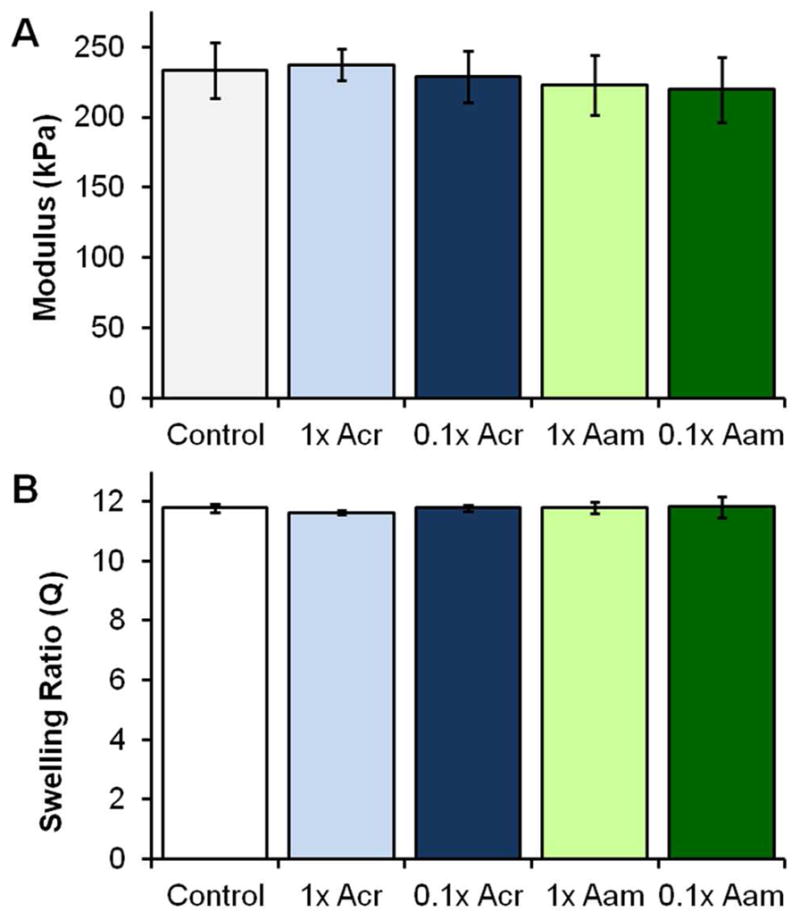
Compressive modulus and swelling ratio of 10% PEG (3.4 kDa) DAA gels with no protein (Control), Acr-PEG-NHS-functionalized collagen at high (1× Acr) and low (0.1× Acr) functionalization densites, and Aam-PEG-I-functionalized collagen at high (1× Aam) and low (0.1× Aam) functionalization densities. No significant differences in either modulus or swelling ratio were measured between any of the tested groups (p>0.05). n=6.
Initial Protein Incorporation and Bioactivity
PEGDA was crosslinked with each of the Acr-PEG-NHS-functionalized collagen and Scl2-2 proteins at an initial concentration of 4 mg protein ml−1. After swelling the gels in PBS for 24 hours to remove uncrosslinked proteins, they were hydrolyzed in 0.5M NaOH. CBQCA was run on the resulting solutions relative to non-swollen controls to quantify the concentration of proteins remaining in the gels and determine the initial protein incorporation during crosslinking for each functionalization density, Figure 6A. As functionalization density is decreased, the initial incorporation of both collagen and Scl2-2 was reduced from ~80% to ~60%. This was expected given that the lower functionalization density proteins have fewer available crosslinking sites. Despite having a reduced protein concentration after 1 day of swelling, gels with lower functionalization density proteins demonstrated significantly increased levels of BAOEC adhesion after 3 hours of incubation, Figure 6B and C. While spreading on PEG-collagen gels was not significantly affected by functionalization density, spreading on PEG-Scl2-2 gels was significantly increased (p<0.05) by reducing functionalization from 1× to 0.1× (from 643 ± 95 to 1088 ± 126 mm2/cell). Given that cell interactions with the bioactive PEG hydrogel are limited to the functionalized proteins, these results suggest that the PEG linkers could block integrin binding to functionalized proteins and reduce bioactivity. While similar effects have been observed with PEGylated drugs, this has not been documented with bioactive hydrogels.18 Monfardini et al. reported that branched PEG linkers reduce bioactivity relative to linear linkers.29 While linear PEG was used here, subsequent crosslinking into a PEG network may provide a similar effect. It can be concluded from these findings that reducing protein functionalization density is a facile method for improving cell-material interactions with bioactive hydrogels by reducing the steric hindrance around integrin binding sites.
Figure 6.

(A) Protein quantitation (CBQCA) data of hydrolyzed PEGDA-Scl2-2 and PEGDA-collagen hydrogels after 1 day of swelling in PBS at 37°C compared to non-hydrated controls. Initial protein incorporation decreases with decreasing functionalization density. n=3; mean ± standard deviation; p<0.05 for each decrease in functionalization density. (B) General increases in BAOEC adhesion were seen with decreases in protein functionalization density. n=4 samples, 3 images per sample for a total of 12 images; mean ± standard error; *p<0.05 relative to all samples, †p<0.05 relative to Scl2-2 0.5× and Scl2-2 1×, ‡p<0.05 relative to Scl2-2 1×. (C) Rhodamine phalloidin and SYBR Green stained BAOECs adhered to PEGDA-collagen and PEGDA-Scl2-2 hydrogels after a 3 hour incubation period. Scale bar applies to all images.
It should be noted that EC interactions with PEG-Scl2-2 hydrogels were consistently lower than those on PEG-collagen hydrogels. This is to be expected, as collagen contains three α1β1 and α2β1 integrin binding sites whereas Scl2-2 only contains one.26 However, reducing Scl2-2 functionalization density to 0.1× provided comparable EC adhesion and spreading to PEG-collagen 1× and 0.5× gels. While similar effects could likely be achieved by increasing Scl2-2 concentration, the use of larger amounts of protein increases scaffold cost.9,30–31 Additionally, concentration increases can be limited by solubility of large molecules such as growth factors and proteins.32 Thus, reducing functionalization density provides an alternate route for increasing cell interactions that is facile, adaptable, and cost effective.
Protein and Bioactivity Retention: Acr-PEG-NHS
Although reduced functionalization density enhanced cell interactions, it was hypothesized that it may also reduce protein retention within the hydrogel over time. Indeed, following 6 weeks of swelling in PBS at 37°C, both collagen and Scl2-2 levels in PEGDA hydrogels were reduced by 91 to 100%, Figure 7A. These large protein losses were attributed to hydrolysis of the PEGDA network. While PEGDA hydrogels are typically considered biostable, the acrylate esters are susceptible to hydrolytic degradation that results in alterations of both scaffold properties and protein levels over time.33–36 PEGDA is suitable for relatively long-term bioactive factor delivery applications on the order of months, but it cannot be used in applications that require several years of bioactivity. To address this, PEG diacrylamide (PEGDAA), a PEG-based hydrogel system that is comparable to traditional PEGDA hydrogels but with improved hydrolytic stability was utilized, Figure 2.27 PEGDAA was previously employed by Elbert et al. to improve conjugate addition reaction bond stability between bioactive peptides and PEG hydrogels.37 In the current study, increasing network stability with PEGDAA hydrogels resulted in significantly increased protein retention compared to PEGDA hydrogels (from 0–9% to 14–67%) after 6 weeks of swelling in PBS at 37°C, Figure 7B. Thus, PEGDAA enhanced sustained network protein levels and may provide a tool for better controlling long-term delivery of drugs and presentation of bioactive agents. It should be noted that the ether linkages in PEG hydrogels are also susceptible to oxidative degradation, which has been proposed as an alternate in vivo degradation mechanism.36 It is likely that in vivo degradation of PEGDA hydrogels is due to some combination of both hydrolytic and oxidative degradation; therefore, PEGDAA should enhance overall in vivo stability compared to PEGDA even if oxidation does occur.27,35,38
Figure 7.

Protein retention in (A) PEGDA and (B) PEGDAA hydrogels over 6 weeks of swelling in PBS at 37°C. n=3; mean ± standard deviation; *p<0.05 relative to previous time point. (C) BAOEC adhesion and spreading on bioactive PEGDAA hydrogels after 6 weeks of swelling in PBS at 37°C. Measurements normalized to 1× samples at 1 day of swelling. n=4 samples, 3 images per sample for a total of 12 images; mean ± standard error; *p<0.05 relative to 1 day sample.
The effect of the observed protein loss over time on cell adhesion and spreading was investigated on bioactive hydrogel formulations that retained at least 10% of their protein at 6 weeks. Thus, none of the PEGDA-Acr-PEG-NHS systems were tested in bioactivity retention studies. Analysis of 3 hour BAOEC adhesion and spreading on PEGDAA hydrogels with Acr-PEG-NHS-functionalized collagen and Scl2-2 after swelling in PBS at 37°C for up to 6 weeks revealed that comparable adhesion and spreading levels were maintained for high functionalization density proteins (1×), Figure 7C. However, gels with low functionalization density proteins (collagen 0.1×; Scl2-2 0.5×, and 0.1×) had significantly reduced BAOEC adhesion and spreading relative to controls (swollen in PBS at 37°C for 1 day). This corresponded to the large decreases in protein concentrations in low functionalization density gels, and could be expected due to decreased cell adhesion that typically occurs with decreased bioactive factor concentration.30–31,39
Protein and Bioactivity Retention: Aam-PEG-I
Although the PEGDAA network provided increased stability that enhanced protein retention, there was still a significant reduction in protein concentration over time that corresponded with loss of bioactivity, especially at low functionalization densities. It was hypothesized that hydrolysis of the acrylate esters of the Acr-PEG-NHS linkers could also contribute to the observed protein losses. To test this hypothesis, Aam-PEG-I, a PEG linker with improved hydrolytic stability, was utilized to investigate the effects of linker stability on long-term protein and bioactivity retention in bioactive PEG hydrogels, Figure 2. It was found that increasing linker stability with Aam-PEG-I resulted in significantly improved protein retention in both PEGDA and PEGDAA gels over 6 weeks of swelling compared with Acr-PEG-NHS functionalized proteins, Figure 8A. Thus, increasing functionalization linker stability with Aam-PEG-I appears to have an even greater effect on long-term protein retention than increasing network stability with PEGDAA when compared with the traditional Acr-PEG-NHS/PEGDA system. The functionalization linker developed here demonstrates strong potential for use as a protein, drug, or peptide functionalization linker in a wide range of systems to provide a sustained presence of bioactive factors.
Figure 8.
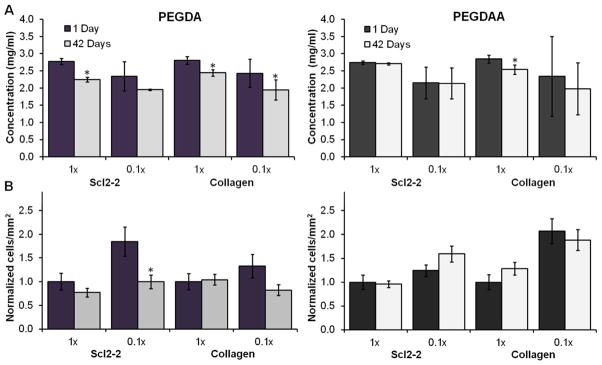
(A) Aam-PEG-I-functionalized protein retention in PEGDA and PEGDAA hydrogels after 6 weeks of swelling in PBS at 37°C. n=6; mean ± standard deviation; *p<0.05 relative to 1 day time point. (B) 3 hour BAOEC adhesion on bioactive PEG hydrogels with Aam-PEG-I-functionalized proteins after swelling gels in PBS for 6 weeks at 37°C. Measurements normalized to 1× samples at 1 day of swelling. n=4 samples, 3 images per sample for a total of 12 images; mean ± standard error; *p<0.05 relative to 1 day time point.
BAOEC adhesion and spreading on PEGDAA hydrogels with collagen- and Scl2-2-Aam at 3 hours revealed that reducing the functionalization density with Aam-PEG-I produces similar increases in initial cell-material interactions as the traditional Acr-PEG-NHS system for both collagen and Scl2-2. Both protein systems had significant increases in BAOEC adhesion and spreading (40–70% and 10–40%, respectively) with reduction of functionalization density from 1× to 0.1×, and all analogous gel systems had statistically similar BAOEC interactions for the two linker types. Upon confirmation of similar initial bioactivity with the two linker systems, PEGDA and PEGDAA gels with collagen- and Scl2-2-Aam were swollen in PBS for 6 weeks at 37°C to evaluate bioactivity retention in the new system.
In all PEGDAA gels with Aam-PEG-I, no significant alterations in BAOEC adhesion were measured after swelling gels for 6 weeks at 37°C, and gels with low functionalization density proteins maintained their enhanced BAOEC adhesion compared to high functionalization density protein gels, Figure 8B. Thus, the use of Aam-PEG-I allows for reduced functionalization density to increase bioactivity without losses over time. Similarly, bioactivity of PEGDA gels with Aam-PEG-I-functionalized proteins at low functionalization densities was similarly or better retained than analogous PEGDAA gels with Acr-PEG-NHS-functionalized proteins. No significant changes in BAOEC spreading were measured for any of the Aam-PEG-I systems after 6 weeks of swelling. These results indicate that the use of a biostable linker and network significantly increases protein and bioactivity retention compared to the traditional PEGDA/Acr-PEG-NHS system. Additionally, the significant improvements in both protein and bioactivity retention with Aam-PEG-I-functionalized proteins in PEGDA hydrogels indicate that this linker could be utilized in degradable matrices to provide control over bioactive factor delivery independently of linker hydrolysis.
Aam-PEG-I allows for use of low functionalization densities for enhanced initial cell interactions while maintaining long term protein levels within the gel to sustain desired cell interactions. This linker in combination with a PEGDAA network would be valuable in long-term implantable devices in which prolonged cell-material interactions are desired, such as non-thrombogenic vascular devices that promote endothelialization. As in vivo endothelialization that occurs via migration from the anastomoses can be a slow process (on the order of 0.2 mm per day), retention of bioactive factors over longer time frames is required, and alterations in swelling or modulus and/or physical breakage that occur with gel degradation could disrupt this process.8,40 Additionally, the combination of linkers, networks, and functionalization schemes presented here provides the means to tune protein and drug delivery with enhanced precision. For example, a drug delivery device could be designed to address the different stages of wound healing with temporally-controlled release of multiple bioactive agents by functionalization with linkers of varied biostability.41 A similarly designed scaffold could be used to regulate stem cell differentiation in vitro or in vivo with controlled release of multiple cues.42
CONCLUSIONS
The ability to tune the mechanical, chemical, and biological properties of PEG-based hydrogels is highly valuable in the development of improved biomaterials for regenerative medicine and drug delivery. Functionalization of bioactive factors and subsequent incorporation into PEG hydrogels provides controllable bioactivity; however, PEG linkers used to functionalize proteins can also sterically hinder integrin binding sites and impair cell-material interactions. Reducing the density of PEG linkers during functionalization provides a facile method to significantly improve initial cell adhesion and spreading with many advantages over altering bioactive factor concentration, but this also reduces protein retention over time as a result of network and/or PEG linker hydrolysis. These studies demonstrate that this protein loss is substantial with almost total loss of protein observed after 6 weeks in low functionalization density systems. The novel Aam-PEG-I linker synthesized here presents a PEG linker option with significantly enhanced hydrolytic stability relative to the traditional Acr-PEG-NHS. When used at a low functionalization density in combination with PEGDAA, this system provides a hydrogel with significantly improved initial bioactivity and sustained protein retention. This system will allow for a controlled investigation of longer-term cell interactions in future studies without concerns over effects of protein loss over time. Although these findings were focused on the characterization of bioactive hydrogels for regenerative medicine, the common use of PEGylation to enhance circulation time of bioactive factors makes these findings highly relevant in drug delivery as well.
Acknowledgments
This work was supported by NIH R01 EB013297 and by the National Science Foundation Graduate Research Fellowship Program.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Hoffman AS. Adv Drug Delivery Rev. 2002;43:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 2.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1880. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 4.Peppas NA, Keys KB, Torres-Lugo M, Lowman AM. J Controlled Release. 1999;62:81–87. doi: 10.1016/s0168-3659(99)00027-9. [DOI] [PubMed] [Google Scholar]
- 5.Gombotz WR, Guanghui W, Horbett TA, Hoffman A. J Biomed Mater Res. 1991;25:1547–1562. doi: 10.1002/jbm.820251211. [DOI] [PubMed] [Google Scholar]
- 6.Cosgriff-Hernandez E, Hahn M, Wilems T, Munoz-Pinto DJ, Browning MB, Rivera J, Russell B, Höök M. Acta Biomater. 2010;6:3969–3977. doi: 10.1016/j.actbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Cruise GM, Hegre OD, Scharp DS, Hubbell JA. Biotechnol Bioeng. 1998;57:655–665. doi: 10.1002/(sici)1097-0290(19980320)57:6<655::aid-bit3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 8.Deible CR, Petrosko P, Johnson PC, Beckman EJ, Russell AJ, Wagner WR. Biomaterials. 1998;19:1885–1893. doi: 10.1016/s0142-9612(98)00098-2. [DOI] [PubMed] [Google Scholar]
- 9.Mann B, Schmedlen R, West JL. Biomaterials. 2001;22:439–444. doi: 10.1016/s0142-9612(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 10.Zustiak SP, Wei Y, Leach JB. Tissue Eng. 2013;19:160–171. doi: 10.1089/ten.teb.2012.0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hermanson GT. Bioconjugate Techniques. 2. Academic Press; 2008. [Google Scholar]
- 12.Sebra RP, Masters KS, Bowman CN, Anseth KS. Langmuir. 2005;21:10907–10911. doi: 10.1021/la052101m. [DOI] [PubMed] [Google Scholar]
- 13.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 14.Greenwald RB, Choe YH, McGuire J, Conover CD. Adv Drug Delivery Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 15.Gobin AS, West JL. Biotechnol Progr. 2003;19:1781–1785. doi: 10.1021/bp0341390. [DOI] [PubMed] [Google Scholar]
- 16.DeLong SA, Moon JJ, West JL. Biomaterials. 2005;26:3227–3234. doi: 10.1016/j.biomaterials.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 17.Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF. J Biol Chem. 1977;252:3582–3586. [PubMed] [Google Scholar]
- 18.Pasut G, Veronese FM. Prog Polym Sci. 2007;32:933–961. [Google Scholar]
- 19.Esposito P, Barbero L, Caccia P, Caliceti P, D’Antonio M, Piquet G, Veronese FM. Adv Drug Delivery Rev. 2003;55:1279–1291. doi: 10.1016/s0169-409x(03)00109-1. [DOI] [PubMed] [Google Scholar]
- 20.Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. Biomacromolecules. 2000;1:31–38. doi: 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- 21.Brown FR, Hopfinger AJ, Blout ER. J Mol Biol. 1972;63:101–115. doi: 10.1016/0022-2836(72)90524-4. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Keene DR, Bujnicki JM, Hook M, Lukomski S. J Biol Chem. 2002;277:27312–27318. doi: 10.1074/jbc.M201163200. [DOI] [PubMed] [Google Scholar]
- 23.Han R, Zwiefka A, Caswell C, Xu Y, Keene D, Lukomska E, Zhao Z, Höök M, Lukomski S. Appl Microbiol Biotechnol. 2006;72:109–115. doi: 10.1007/s00253-006-0387-5. [DOI] [PubMed] [Google Scholar]
- 24.Browning MB, Dempsey D, Guiza V, Becerra S, Rivera J, Russell B, Höök M, Clubb F, Miller M, Fossum T, Dong JF, Bergeron AL, Hahn M, Cosgriff-Hernandez E. Acta Biomater. 2012;8:1010–1021. doi: 10.1016/j.actbio.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 25.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. J Biol Chem. 2000;275:35–40. doi: 10.1074/jbc.275.1.35. [DOI] [PubMed] [Google Scholar]
- 26.Sweeney SM, DiLullo G, Slater SJ, Martinez J, Iozzo RV, Lauer-Fields JL, Fields GB, Antonio JDS. J Biol Chem. 2003;278:30516–30524. doi: 10.1074/jbc.M304237200. [DOI] [PubMed] [Google Scholar]
- 27.Browning MB, Cosgriff-Hernandez EM. Biomacromolecules. 2012;13:779–786. doi: 10.1021/bm201707z. [DOI] [PubMed] [Google Scholar]
- 28.Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Biomaterials. 2006;27:2519–2524. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 29.Monfardini C, Schiavon O, Caliceti P, Morpurgo M, Harris JM, Veronese FM. Bioconjugate Chem. 1995;6:62–69. doi: 10.1021/bc00031a006. [DOI] [PubMed] [Google Scholar]
- 30.Civerchia-Perez L, Faris B, LaPointe G, Beldekas J, Leibowitz H, Franzblau C. Proc Natl Acad Sci U S A. 1980;77:2064–2068. doi: 10.1073/pnas.77.4.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hern DL, Hubbell JA. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 32.Trevino SR, Scholtz JM, Pace CN. J Pharm Sci. 2008;97:4155–4166. doi: 10.1002/jps.21327. [DOI] [PubMed] [Google Scholar]
- 33.Benoit DSW, Durney AR, Anseth KS. Tissue Eng. 2006;12:1163–1673. doi: 10.1089/ten.2006.12.1663. [DOI] [PubMed] [Google Scholar]
- 34.Bryant SJ, Anseth KS. J Biomed Mater Res. 2002;59:63–72. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- 35.Bryant SJ, Anseth KS. J Biomed Mater Res, Part A. 2003;64A:70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 36.Lynn AD, Kyriakides TR, Bryant SJ. J Biomed Mater Res. 2009;93A:941–953. doi: 10.1002/jbm.a.32595. [DOI] [PubMed] [Google Scholar]
- 37.Elbert DL, Hubbell JA. Biomacromolecules. 2001;2:430–441. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- 38.Larsen IB, Munksgaard EC. Eur J Oral Sci. 1991;99:254–261. [Google Scholar]
- 39.Mann B, Gobin A, Tsai A, Schmedlen R, West J. Biomaterials. 2001;22:3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 40.Clowes AW, Gown AM, Hanson SR, Reidy MA. Am J Pathol. 1985;118:43–54. [PMC free article] [PubMed] [Google Scholar]
- 41.Chen RR, Mooney DJ. Pharm Res. 2003;20:1103–1112. doi: 10.1023/a:1025034925152. [DOI] [PubMed] [Google Scholar]
- 42.Bae SE, Choi DH, Han DK, Park K. J Controlled Release. 2010;143:23–30. doi: 10.1016/j.jconrel.2009.12.024. [DOI] [PubMed] [Google Scholar]