

Summary
The Bone Morphogenetic Proteins (BMP) are secreted ligands largely known for their functional roles in embryogenesis and tissue development. A number of structurally diverse extracellular antagonists inhibit BMP ligands to regulate signaling. The DAN family of antagonists represents the largest group of BMP inhibitors, however, little is known for how they mechanistically inhibit BMP ligands. Here, we present the structure of the DAN family member Protein Related to Dan and Cerberus (PRDC) solved by X-ray crystallography. The structure reveals an unexpected growth factor-like appearance with a novel dimerization mechanism that is formed through extensive β-strand contacts. Using site-directed mutagenesis coupled with in vitro and in vivo activity assays, we identified a BMP binding epitope on PRDC. We also determined that PRDC binds heparin with high affinity and that heparin binding to PRDC interferes with BMP antagonism. These results offer insight for how DAN family antagonists functionally inhibit BMP ligands.
Introduction
The Bone Morphogenetic Proteins (BMPs) define a specific subclass of signaling ligands belonging to the TGF-β superfamily, comprising nearly 40 structurally similar secreted proteins. During development, the BMPs play crucial roles in the maturation and differentiation of many tissue types, where they can function to activate or suppress other cellular signaling regimes (Nimmagadda et al., 2007). To date, many biological roles have been classified for this signaling family, including bone and cartilage development, oocyte and follicular development, as well as gut differentiation from mesoderm tissue (Bragdon et al., 2011). Furthermore, their roles in several disease states, including lung and kidney fibrosis, osteoporosis, and cardiovascular disease, have indicated their importance in adult homeostasis (Cai et al., 2012; Walsh et al., 2010).
At the molecular level, BMP ligands form stable disulfide-bonded dimers that transduce their signals by binding two Type I and two Type II receptors, leading to Type I receptor phosphorylation. Once activated, Type I receptors phosphorylate SMAD transcription factors, leading to gene regulation (Hinck, 2012). Although most BMP ligands directly activate the canonical SMAD 1/5/8 pathway, the overall signaling outcome is unique to each ligand and dependent on both the cellular state and signal strength. Because of this, extracellular control of these ligands is important for determining their role within particular cell types and stages of development. Therefore, specialized mechanisms have evolved to fine-tune and regulate signaling.
In vivo, over 20 antagonists have been identified that directly bind to and inhibit BMP/TGF-β signaling (Rider and Mulloy, 2010). Studies have shown that inhibition occurs through interference of ligand-receptor interactions at both the Type I and Type II receptor binding epitopes (Mueller and Nickel, 2012). Importantly, antagonists bind with high affinity to particular subsets of ligands, sometimes being limited to only a defined few (Monestier et al., 2012). But unlike the BMP/TGF-β growth factors, antagonists are structurally diverse and range from large-multi domain proteins (e.g. Follistatin- and Chordin-like proteins) to smaller, single-domain proteins, including the DAN family (Differential screening-selected gene Aberrative in Neuroblastoma) of proteins and Noggin (Bragdon et al., 2011). Because of this, widely different binding and inhibitory mechanisms are available for neutralizing ligand-receptor interactions, described by the ligand-bound structures of Noggin and Follistatin (Cash et al., 2009; Groppe et al., 2002; Thompson et al., 2005). Furthermore, analysis of these structures has led to an increased understanding for how ligand specificity and affinity are achieved (Cash et al., 2012). Despite this, only a handful of structures of these functional protein antagonists have been resolved, thus limiting our knowledge of BMP/TGF-β antagonism, especially in regards to the DAN family inhibitors.
The DAN family of proteins represents the largest collection of antagonists with 9 members canonically known for BMP inhibition (Avsian-Kretchmer and Hsueh, 2004). Furthermore, they represent the smallest BMP antagonists with typical sizes around 20 kDa. DAN family members have a central cysteine-rich domain, termed the DAN domain, which contains a cystine-knot motif with an eight-residue ring (Walsh et al., 2010). Interestingly, these antagonists show the greatest homology within their DAN domains but exhibit significant diversity and low conservation in their termini. Additionally, this group of proteins can be subdivided into two main groups based upon their ability to antagonize BMPs: 1) strong BMP antagonists, including PRDC, Gremlin, and Dan, and 2) weak BMP antagonists, including SOST and USAG-1, which also bind to the coreceptor LRP5/6 to antagonize Wnt signaling (Ellies et al., 2006; Hung et al., 2012; Sudo et al., 2004; Sun et al., 2006; van Bezooijen et al., 2004). However, antagonist features that account for this subdivision in BMP affinity have not been resolved, in part due to the limited information defining the BMP binding epitope. Moreover, only the NMR structures of SOST are available, which have provided minimal insight into DAN family mediated BMP antagonism (Veverka et al., 2009; Weidauer et al., 2009).
To clarify these differences in anti-BMP functionality, we present the crystal structure of the strong BMP antagonist, PRDC. This structure reveals a novel growth factor-like appearance where two monomers of PRDC are tightly interlaced via a significant stretch of backbone hydrogen bonds. Using this structure, we performed targeted mutagenesis studies to identify the BMP binding epitope. Our results indicate that BMP binding occurs within a partially exposed hydrophobic patch located at the dimer interface within the central portion of the DAN domain. Furthermore, structural comparison of PRDC and SOST has revealed several important features for differentiating their BMP ligand affinities.
Results
Determination and Overview of the X-ray Crystal Structure of PRDC
We recently determined that PRDC and Dan exist as stable non-disulfide bonded dimers, very different from the monomeric nature of SOST (Kattamuri et al., 2012b; Veverka et al., 2009; Weidauer et al., 2009). To understand the molecular differences between these family members and gain insight into DAN family mediated BMP-inhibition, we determined the crystal structure of PRDC to 2.25 Å using SeMet MAD phasing (Table 1). Initial observations show that four PRDC monomers are present in the asymmetric unit (ASU), forming two independent, head-to-tail protein dimers between Chains A and B and Chains C and D (Figure 1A). These dimers exist in a very rod-like conformation approximately 82 Å in length and roughly 30–35 Å in width and height. Additionally, bending of the dimer along the long axis gives PRDC a near perfect arch-like appearance, exposing large concave and convex surfaces. The majority of the structure is composed of very long and extended anti-parallel β-strands. Importantly, these dimers exist as a result of non-covalent interactions between neighboring monomer β-strands. Furthermore, α-helices are present on each monomer, flanking the long axes and bridging the dimer interface. The final refined model consists of residues Q46-V160 in Chain A, K50-V160 in Chain B, L60-V160 in Chain C, and K50-V160 in Chain D, where residues after the truncated signal sequence (R22-W45) and at the extreme C-terminus (N161-Q168) were unresolved.
Table 1.
X-ray diffraction data and structure refinement statistics.
| Nativea | SeMeta | |||
|---|---|---|---|---|
| Data collection | ||||
| Space group | P 21 | |||
| Unit cell dimensions | ||||
| a, b, c (Å) | 73.3, 65.6, 85.1 | 73.2, 65.8, 85.1 | ||
| α, β, γ (°) | 90, 105.5, 90 | 90, 105.2, 90 | ||
| Peak (E1) | Inflection (E2) | Remote (E3) | ||
|
|
||||
| Wavelength (Å) | 0.95740 | 0.9794 | 0.97961 | 0.95740 |
| Resolution (Å) | 2.25 (2.37-2.25) | 2.45 (2.90-2.45) | 2.90 (3.06-2.90) | 2.9 (3.06-2.90) |
| Rmerge | 0.058 (0.79) | 0.057 (.53) | 0.058 (0.40) | 0.063 (0.47) |
| Mn (I/sd)b | 15.6 (2.1) | 12.3 (2.6) | 13.3 (2.7) | 11.9 (2.2) |
| Completeness (%) | 99.8 (100.0) | 98.4 (99.4) | 98.3 (99.2) | 98.0 (99.1) |
| Redundancy | 6.1 (6.1) | 3.1 (3.2) | 3.1 (3.2) | 3.1 (3.2) |
| Refinement | ||||
| Resolution (Å) | 17.6-2.25 | |||
| No. reflections | 36,786 | |||
| Rwork (%)/Rfreec (%) | 20.2/22.2 | |||
| Number of atoms (molecules)/ | 3,748/84.2 | |||
| B-factors (Å2) | ||||
| Protein | 3,489/77.0 (main), 90.1 (side) | |||
| Water (113) | 113/69.9 | |||
| Citrate (3) | 36/128.6 | |||
| Ethylene glycol (12) | 168/116.8 | |||
| Glutathione (4) | 77/126 | |||
| RMSD from ideal geometry | ||||
| Bond lengths (Å) | 0.010 | |||
| Bond angles (°) | 1.20 | |||
| Ramachandran plot | 409 (95.1%) Favored | |||
| 18 (4.6%) Allowed | ||||
| 4 (0.9%) Outliers | ||||
Two crystals were used for data collection, one native and one SeMet.
Values in parentheses are for highest-resolution shell as defined in the resolution row.
Mn (I/sd) is defined as <merged<Ih>/sd(<Ih>) > ~= signal/noise.
Rfree was calculated from 5% of initial total number of reflections.
Figure 1. Crystal structure of PRDC.

(A) Ribbon representation of the X-ray crystal structure of PRDC with each monomer colored separately (green, blue). ‘Bottom’ view is shown in the center with corresponding ‘Side’ views rotated through the labeled axis. (B) Topology diagram of PRDC with the fingers colored (F1, green and F2, red). The β-strands in the wrist of PRDC are colored orange. The location of the dimer interface is annotated and colored light purple. (C) PRDC chains A–D were aligned using residues 72–160. The alignment results are depicted for the core DAN domain (left) and the N-terminal helix region (right). (D) PRDC monomer (chain B) colored by increasing crystallographic temperature factors from blue to red. See also Figure S1.
Interestingly, in comparison to the available structures of other cystine-knot containing proteins, our model shows that each PRDC monomer takes on an apparent growth factor-like fold, where the helical N-terminus leads into a concession of anti-parallel β-strands (β1-β4) that form a characteristic two-finger-wrist model (Figures 1A and 1B). Using this model and tracing along the length of the PRDC monomer, the protein can be broken apart into 4 major sections based upon cysteine spacing: 1) the N-terminus, amino acids H46-W72, containing the initial α-helix (α1), 2) Finger 1 (F1), amino acids C73-Q100, 3) the Wrist region (W), amino acids C101-F122, and 4) Finger 2 (F2), amino acids C123-V160 (Figure 1B). This two-finger-wrist arrangement is also found in the TGF-β/BMP ligands in addition to several antagonists, including the related DAN family protein, SOST (Hinck, 2012; Veverka et al., 2009; Weidauer et al., 2009). Furthermore, this arrangement is stabilized by a central cystine-knot motif (Figure 1B). For PRDC, the cystine-knot motif is formed by 6 conserved cysteines that form 3 disulfide bonds (C73-C123, C97-C155, and C101-C157). Additionally, a disulfide bond links F1 to F2 (C87-C137) towards the tips of the fingers (Figure 1B).
Structural Implications for Flexibility in the PRDC N-terminus
When comparing the different chains within the ASU, only minor deviations can be noted within the core DAN domains of the four PRDC monomers (Figure 1C). Despite this, differences are observed in the location and conformation of the N-terminal helix (Figures 1C and S1). In Chain A, the N-terminus forms an additional helix that extends across the dimer (Figures 1A and 1C), whereas for Chains B–D, the N-terminus points away from the opposing monomer into the solvent void (Figure S1). These differences can partially be explained by crystal packing interactions, where the N-terminus of Chain A interacts with other PRDC chains found in neighboring ASUs (Figure S1). Additionally, crystallographic temperature factors show the N-terminus within each chain to derive a high level of mobility, where the majority of the remaining structure appears significantly more static (Figure 1D). Furthermore, it can be clearly seen that the helical content within each of the four chains is significantly different (Figures 1C and S1). For instance, Chain B shows helical content from S56 to L52, where residues T63 through Y67 exist in the highly destabilized pi-helix form. For Chain D, helical content is found spanning residues Q57 to A54, where those residues mainly composing the pi-helix in Chain B lack any significant helical content. These structural differences, in addition to the significant abundance of helical content in Chain A and a lack thereof in Chain C, indicate that the N-terminus likely exhibits a significant amount of conformational sampling and local flexibility.
Interestingly, the helix found at the N-terminus of PRDC partially interacts with a large, underlying hydrophobic interface. This interface consists of several sizable hydrophobic amino acids, including F104, I106, and F117 from the wrist region (β2-β3) on one chain and W72, L77, F96, and Y98 from F1 (β1-β2) on the second chain (Figure 2C). These residues are partially buried by the N-terminal helix packing with the ‘top’ or convex surface of the dimer’s core domain, potentially stabilizing the protein dimer (Figure 2C). However, looking at the helical differences amongst the four different PRDC monomers and high temperature factors of the N-terminus, it is plausible that these hydrophobic residues experience significant and varying levels of solvent accessibility in solution. This is especially apparent when comparing the different chains of PRDC in the ASU, where Dimer CD shows a significant increase in solvent accessibility at this interface in comparison to Dimer AB. With this in mind, it is highly unlikely that the modest interactions taking place between PRDC’s N-terminus and core domain provide any stabilizing support to the central dimer interface.
Figure 2. Intermolecular contacts of PRDC dimers.

Top, PRDC dimer side view colored as in Fig 1A. Letters correspond to the general location of close-up views depicted in the panels below. (A) Close-up view of the anti-parallel interaction of β2 of each PRDC monomer. Backbone atoms are shown as sticks and mainchain H-bonds between strands are indicated. (B) Interactions of the wrist (blue) and F1 (green) regions on opposing monomers. View is down the long axis to highlight the 4-stranded β-sheet generated from merging the two monomers. (C) Interaction of the N-terminal helix (transparent) from Chain B (blue) with each monomer, representing the intra-chain (purple) and inter-chain (yellow) residues as sticks at the hydrophobic interface. (D) Zoomed in view showing the spatial relationship of C120 at the dimer interface, indicating the absence of a disulfide bond. See also Figure S2.
Analysis of the Structural Mechanisms for PRDC Dimerization
In agreement with our previous findings, PRDC exists as a seemingly stable non-disulfide linked dimer that buries over 1800Å2 per monomer with the opposing chain, similar to the large surface area buried by Follistatin in complex with Myostatin (~1800Å2) and greater than the monomer contacts of the BMP7 dimer (~1200Å2) (Cash et al., 2009; Groppe et al., 2002; Kattamuri et al., 2012b). Central to this interface is a long anti-parallel β-strand interaction between β2 of Chain A and β2 of Chain B. This interaction involves amino acids C97-Y105 from each monomer, providing 10 hydrogen bonds between opposing chains that compose greater than 1300Å2 of the dimer interface (Figure 2A). Furthermore, amino acids N102-Y105 are located in the wrist region, providing intimate contact between the wrist of one monomer and the fingertips of the adjacent monomer (Figure 2B). The loop of the wrist mainly contacts the adjacent monomer through van der Waals interactions. However, H109 and E114 of the wrist form hydrogen bonds with the backbone of F1 that further stabilize the intermolecular surfaces (Figure 2B). In addition, the end of the wrist loops back into an anti-parallel interaction with itself, altogether providing a 4-stranded β-sheet interaction between opposing fingers (Figure 2B). Overall, the extensive dimer interface of PRDC likely explains the extreme stability of the protein under both reducing and denaturing conditions, as previously described (Kattamuri et al., 2012b).
Prior to our recent study, it was predicted that PRDC dimerization (among other DAN family members) was driven by covalent interactions between the odd, unpaired cysteines of opposing monomers (Alvarez et al., 2009). This hypothesis was based upon the protein’s significant similarity to many TGF-β ligands, where disulfide bonds are important for functionally dimerizing these cytokines (Hinck, 2012). Contrary to this, we showed that PRDC forms noncovalent dimers independent of this unpaired cysteine (Kattamuri et al., 2012b). Examination of the PRDC structure provides clarification for this observation. Here, the unpaired cysteine (C120) is located in the center of β3 and the curvature of the core domain brings the two opposing β3 strands into proximity. However, despite being relatively close to one another, the unpaired cysteines on each opposing PRDC monomer are largely outside the range of acceptable disulfide bonding distances (8.2 Å between opposing Cα’s, Figure 2D). Additionally, F122 on each monomer forces an interaction that repels the opposing C120, making this interaction highly unlikely (Figure 2D). In sight of this, it should be noted that in our structure C120 is modified by glutathione, as observed in the electron density maps (Figure S2). Despite this, the PRDCC120S mutation in our previous studies (and in this study) was proven to be dimeric and maintain its functional specificity, alleviating concerns of a potentially disruptive protein modification.
SAXS Validation of the Dimeric PRDC Structure
In order to validate the overall structure, we analyzed PRDC in solution using small angle X-ray scattering (SAXS). Guinier analysis of the samples shows a significantly linear representation over the low q range, where serial dilution of the protein samples resulted in near perfect scaling of the calculated I(0) values (Figure 3A and 3B; Table S1). Additionally, computed Rg values obtained from both real and reciprocal space are in close agreement (Table S1).
Figure 3. SAXS analysis of WT and C120S PRDC.

(A) Guinier plot of PRDC WT and PRDCC120S. The low-resolution scattering angles depict well-ordered scattering with negligible aggregation or repulsion. (B) Intensity distribution of the SAXS scattering function with the simulated data plotted using FoXS and the PRDC crystal structure. (C) Pair-wise distance distribution function of PRDC and PRDCC120S. (D) Crystal structure combined surface and ribbon representations. (E–F) Superposition of the crystal structure (ribbon) with SAXS ab initio reconstructions of WT PRDC (D) and PRDCC120S (E). Two views are rotated through the long axis. Larger surface represents DAMAVER solution while smaller internal mesh represents the filtered Damfilt models. See also Figures S3 and S4 and Table S1.
Ab initio reconstructions of the SAXS data were used to generate a low-resolution envelope of PRDC, which matched well with the crystal structure (Figures 3C, 3D and 3E) as shown by low NSD scores (Table S1). Furthermore, PRDC in the solution state is in close agreement with the computed intensity profiles from the crystal structure, with a χ score of 0.69 calculated using FoXS against Dimer AB as compared to a significantly higher χ score of 2.01 when only using a monomer (Figures 3B and S3). Additionally, scattering data was collected for PRDCC120S, which lacks the ninth cysteine but exhibits native BMP antagonism, providing almost identical scattering and density profiles to those obtained for native PRDC (Figure 3). From these results, it becomes evident that the overall shape and curvature of PRDC in solution matches well with that seen in the crystal structure and that modification of the unpaired cystine does not perturb the structure.
It should be noted, however, that flexibility is inherent to the PRDC structure. This is indicated by the increasing scattering intensity of the 1-D data within the high q range of the Kratky plot (Figure S3). Additionally, Porod representation of the data recapitulates that a well defined plateau is formed in the I(q)•q3 versus q3 scattering profile but not in the I(q)•q4 versus q4 profile within the low q range (Figure S3). These results indicate inherit flexibility within PRDC where, presumably, this motion is localized to the N-terminus. Furthermore, while it is possible that the C-terminus contributes to this observation, N-terminal flexibility is likely much more significant, outlined by crystallographic temperature factors, structural inconsistencies, and a larger number of amino acids lacking solid diffraction density as compared to the C-terminus.
Mutational Analysis and Characterization of the PRDC BMP-binding Epitope
For general TGF-β signaling, strong hydrophobic interactions are utilized to promote affinity between both ligand:antagonist and ligand:receptor functional pairs (Groppe et al., 2002; Hinck, 2012; Mueller and Nickel, 2012; Thompson et al., 2005). However, it was suggested that the DAN family member Gremlin binding to BMP4 may be mediated through charge type interactions (Sun et al., 2006). To resolve this discrepancy, we measured the binding of PRDC to BMP ligands in the presence of increasing ionic strength. Our results show that the interaction of PRDC with BMP ligands was mostly unaffected even in the presence of 1M NaCl (Table 2). This indicates that the interactions of PRDC with BMP ligands are largely driven by hydrophobic interactions, similar to Noggin, Follistatin and their receptors.
Table 2.
Analysis of PRDC mutants by SPR and luciferase reporter assay.
| Mutanta,b | kon (104 s−1) | koff (10−3 s−1) | KD (nM) | Fold SPRc (WT) | IC50 (nM) | Fold Lucc (WT) |
|---|---|---|---|---|---|---|
| PRDC-WT | 9.1 | 0.8 | 9.2 | 1.0 | 0.5 | 1.0 |
| 7.3 | 0.8 | 12 | 1.0 | |||
| PRDC-WT (1M NaCl) | 12.5 | 2.2 | 18 | 2.0 | ||
| 13.2 | 2.0 | 16 | 1.3 | |||
| L68A | 10.9 | 2.0 | 18 | 2.0 | 1.2 | 2.5 |
| 9.5 | 2.3 | 25 | 2.1 | |||
| W72A | 2.2 | 1.2 | 49 | 5.3 | 3.6 | 7.6 |
| 2.8 | 1.6 | 55 | 4.7 | |||
| F96A | 43.0 | 8.1 | 19 | 2.1 | 3.0 | 6.3 |
| 6.5 | 2.2 | 35 | 3.0 | |||
| Y98A | 4.3 | 1.8 | 42 | 4.5 | 8.4 | 18 |
| 3.3 | 2.1 | 63 | 5.4 | |||
| F104A | 36.4 | 7.7 | 21 | 2.3 | 23 | 49 |
| 11.6 | 2.6 | 22 | 1.9 | |||
| Y105A | 3.0 | 1.9 | 64 | 7.0 | 9.3 | 20 |
| 2.2 | 2.7 | 122 | 10 | |||
| F117A | 5.6 | 2.0 | 36 | 3.9 | 30 | 63 |
| 4.5 | 2.6 | 57 | 4.9 | |||
| F122A | 21.0 | 0.9 | 4.1 | 0.5 | 0.3 | 0.7 |
| 10.6 | 1.1 | 10 | 0.9 | |||
| L140A | 21.2 | 0.9 | 4.0 | 0.4 | 0.4 | 0.7 |
| 13.6 | 1.2 | 8.7 | 0.7 | |||
| F144A | 5.6 | 0.8 | 14 | 1.5 | 0.5 | 1.1 |
| 2.9 | 0.9 | 30 | 2.6 | |||
| I146A | 13.8 | 0.6 | 4.5 | 0.5 | 0.4 | 0.7 |
| 15.2 | 0.6 | 3.8 | 0.3 |
SPR analysis was performed with BMP2 & BMP4 and the luciferase reporter assay was performed with BMP2.
BMP2 and BMP4 results are shown in the first and second rows respectively.
SPR binding was performed in 0.5 M NaCl unless otherwise noted.
Fold represents the ratio of the (WT/mutant) activity for each ligand.
See also Figure S5.
With this in mind, we analyzed the surface of PRDC using the SPIDDER (buried surface area analysis) and ClusPro2.0 (molecular docking prediction) servers to identify surface exposed hydrophobic residues that potentially function to bind BMP ligands (Kozakov et al., 2010; Porollo and Meller, 2007). From this analysis, we identified surface exposed amino acids that clustered in mainly two locations: (1) the fingertip region and (2) the central, convex (top) surface of the DAN domain, including the previously mentioned interface with the N-terminal helix (Figures 3A and 4B). We then performed selective site-directed mutagenesis and individually converted each of these residues to an alanine. We have previously shown that active (dimer) and inactive (monomer) species of PRDC could be isolated after refolding. Therefore, during purification, we pooled mutant PRDC protein that exhibited similar elution profiles (to WT) from the heparin and subsequent S75 SEC columns (Figure S4). Taken together with the fact that all mutants exhibited a characteristic shift when analyzed by SDS-PAGE following oxidative refolding, we believe our PRDC mutants to be correctly folded and dimeric (Figure S4).
Figure 4. Identification of a BMP binding epitope for PRDC.
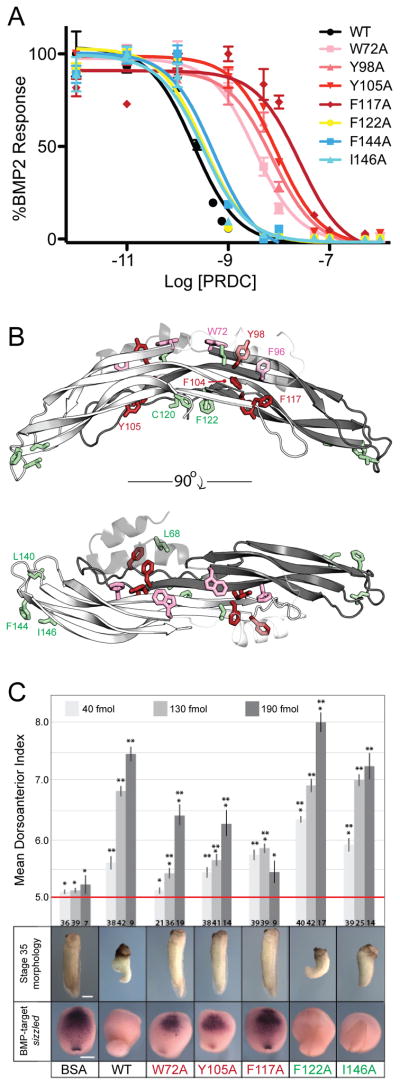
(A) Effect of mutations of PRDC on the inhibition of BMP2 signaling using BRE-C2C12 cells. To determine IC50 values, BMP2 (1nM) was incubated with increasing concentrations of PRDC proteins. Each concentration was tested in triplicate and the curves represent the average of three individual experiments (error bars represent +/−SEM). (B) Side and top view of the PRDC dimer highlighting the luciferase reporter assay results in Table 2. Residues that were mutated are shown in stick and colored; green=minimal effect; pink > 5–10 fold decrease; red > 10-fold decrease. (C) In vivo effects of PRDC mutants in Xenopus development. PRDC mutant proteins (40 fmols, 130 fmols or 190 fmols) were injected into the blastocoel cavity of Xenopus embryos at stage 9. (top) Embryos were scored for dorsal morphology at stage 35 using the dorsoanterior index (5= normal development and 10= extreme dorsalization). The histogram indicates the mean dorsoanterior index +/−SEM *p<0.01 compared to WT PRCD and **p<0.01 compared to BSA injected in pairwise t-tests. (middle) Representative examples of the stage 35 morphology from the 190 fmol injection. (bottom) Sibling embryos were assayed by in situ hybridization for expression of the BMP-target gene sizzled at stage 18 and representative embryos shown for the 140 fmol injection. Each protein was tested in at least two separate injection experiments and the total number of embryos assayed is indicated at the bottom of each column. See also Figure S6.
Initially, we analyzed the direct binding of PRDC proteins to BMP2 and BMP4 by surface plasmon resonance (SPR) (Table 2; Figure S5). These experiments revealed that residues within the fingertip epitope, in addition to F122A on the concave surface of PRDC, did not drastically alter BMP binding. However, we found that the W72A mutation, which exhibits significant surface exposure within the central portion of the convex dimer surface, moderately reduced the affinity of PRDC for BMP ligands. Extending from this finding, we then mutated nearby residues lying within the vicinity of W72. From this analysis, we found that disruption of the hydrophobic residues composing the interface between the convex surface and the N-terminal helix significantly reduced PRDC’s affinity for BMP, with Y98A, F104A, Y105A, and F117A showing the greatest effects (Table 2; Figure 4B).
Extending from this, we further recapitulated our results using a BMP-responsive cell-based reporter assay (Kattamuri et al., 2012b). As previously determined, WT PRDC has an IC50 value around 1nM, where similar values were observed for the PRDC mutants lying within the fingertip and the concave portion of the DAN domain, including F122A, L140A, F144A, and I146A (Table 2 and Figure 4A). Furthermore, mutations in the central, convex surface of the DAN domain (W72A, Y98A, F104A, Y105A, and F117A) had a reduced ability to inhibit BMP signaling, indicating the importance of these residues in deriving BMP antagonism (Table 2; Figures 4A and 4B).
PRDC Mutant Activity Monitored In Vivo
To test the ability of WT and mutant PRDC proteins to inhibit BMP signaling in vivo, we used a dorsal-ventral patterning assay in Xenopus embryos. Axial pattering of Xenopus embryos is regulated by the balance between endogenous BMP4 and BMP7, which promote ventral-posterior “tail” fate and BMP-antagonists which promote dorsal-anterior “head” structure (Marom et al., 1999). Experimental inhibition of BMP signaling in the early gastrula results in enhanced dorsal-anterior development and reduced tail structure. A dose range of purified WT or mutant PRDC proteins (40 fmols, 130 fmols and 190 fmols) were injected into the blastoceol cavity of stage 9 embryos. These were assayed for expression of the BMP-target gene sizzled in the ventral mesoderm at stage 20 and scored for morphology at stage 35 using the well-established dorsoanterior index, where a score or 5 is normal development and a score of 10 represents complete dorsalization, indicative of strong BMP-repression (Kattamuri et al., 2012b).
As previous published, WT PRDC exhibited potent BMP-inhibition, causing severe dorsalization and reduction of sizzled expression (Kattamuri et al., 2012b). Recapitulating the above-mentioned cell-culture and BMP-binding assays, the F122A and I146A mutants retained BMP-antagonist activity in vivo causing a dorsalized phenotype comparable to WT. In contrast, all of the other PRDC mutations tested, which lie within the convex epitope of PRDC, resulted in a dramatic reduction (but not complete absence) of BMP-inhibiting activity (Figure 4C).
With these results, we conclude that a significant portion of the PRDC BMP-binding epitope resides within the DAN domain, where those hydrophobic residues involved in the vicinity of the helix interface derive the highest affinity for this interaction (Figure 4B). Furthermore, large hydrophobic amino acids within the fingertips and on the concave surface of the protein dimer do not provide any functionality to this interaction, suggesting their lack of necessity for BMP binding in both the in vitro and in vivo context.
Characterization of PRDC Electrostatics and Implications for Heparin Interactions
It has been suggested that PRDC and other DAN family antagonists potentially bind to heparin/heparan oligosaccharides (Rider and Mulloy, 2010). However, only the interaction of SOST with heparin has been characterized, where this interaction does not appear to interfere with its function (Veverka et al., 2009). For PRDC, heparin affinity is strongly suggested, where the protein has an apparent predicted pI of 9.3 that is driven by 26 arginines and lysines, many of which occur in tandem and match up to those found in the SOST protein (Figure 6A). Furthermore, looking at the electrostatic surface of the PRDC structure, a large electropositive potential spans the entirety of the convex surface of the dimer (Figure 5B). Interestingly, this positive potential spans over both N-terminal helices and central core domains from each monomer, mainly through residues in β4 of F2. In contrast, the bottom of PRDC contains a linear stretch of negative surface charges (Figure 5B). Therefore, it can be expected that heparin would have a strong potential to associate with the convex and N-terminal epitopes of PRDC.
Figure 6. Comparison of PRDC to other DAN family members.

(A) Sequence alignment of mouse PRDC and other human DAN family members. Human and mouse PRDC amino acid sequences are identical in 160/165 positions. Numbers above alignment pertain to mouse PRDC. Conserved cysteines are highlighted yellow with a solid line connecting cystine-knot residues and a dashed line indicating the disulfide-bond linking the fingers. The unpaired cysteine is colored orange. Secondary structure elements are shown for PRDC (top, black) and SOST (bottom, gray) with red brackets indicating the extent of the DAN domain. Residues of PRDC analyzed by site-directed mutagenesis are colored consistent with Figure 4B. Putative heparin binding residues are colored blue for PRDC (open box) and SOST (solid box). SOST residues that interact with LRP5/6 are shown as bold. Columns following the alignment indicate the number of residues not included in the alignment at both the N- and C-terminus. (B) Ribbon representation of a monomer of PRDC (left) and the NMR structure of SOST (right). (C) Superposition of PRDC and SOST with inset depicting differences in the Wrist region. See also Figure S7.
Figure 5. PRDC heparin interactions.
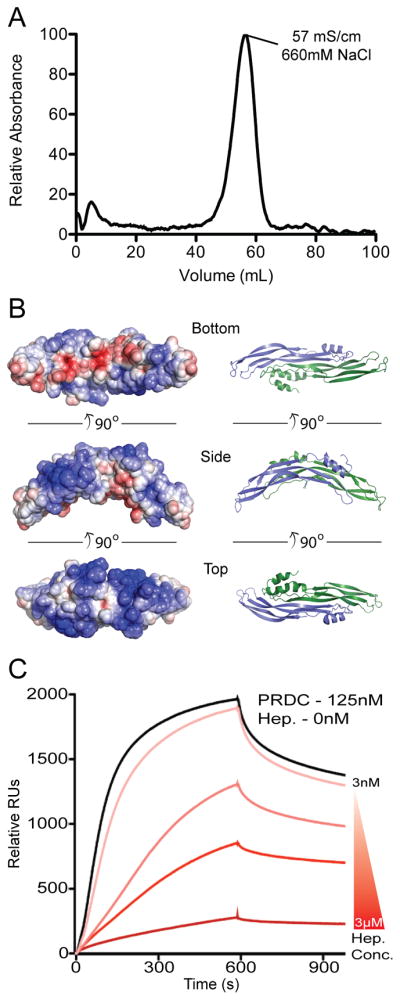
(A) PRDC (100 μg) was loaded onto a 1 ml heparin FF column at pH 7.5. The trace represents the elution profile generated from applying a gradient from 100mM to 1M NaCl. PRDC bound to the column and eluted at high salt concentrations. (B) Three views of PRDC shown from the bottom, side and top depicting the electrostatic surface potential on the left colored on a scale of −5 to 5 kbT/ec (red to blue) with the corresponding ribbon representation on the right. (C) SPR sensorgrams of PRDC-BMP2 interactions in the presence of heparin. Curves represent PRDC at 125nM preincubated with heparin at concentrations (from top to bottom) of 3, 30, 300, 3000nM.
In order to gain insight into the interactions between PRDC and heparin oligosaccharides, we determined the relative affinity of PRDC for heparin using affinity chromatography. As expected, PRDC was capable of strongly binding to a heparin column at physiological pH (Figure 5A). Furthermore, using a biosensor chip containing immobilized heparin, we performed kinetic analyses using purified PRDC as the analyte (Figure S6). From this, we determined that PRDC has a relatively strong affinity (KD 54nM) for heparin oligosaccharides.
From here, we wanted to determine what implication heparin binding has for PRDC mediated antagonism. Therefore, we pre-incubated PRDC with varying amounts of heparin oligosaccharides and measured the effect on BMP binding using an immobilized BMP2 biosensor chip. Our results show a significant decrease in the overall response units of PRDC binding in the presence of heparin (Figure 5C). In fact, this effect was magnified with increasing concentrations of heparin, where 3μM heparin in the presence of 125nM PRDC was nearly capable of completely abrogating the binding interaction, suggesting that heparin competes with BMP binding to PRDC.
Discussion
To gain a better understanding of DAN family-mediated BMP inhibition, we have resolved the structure of the potent BMP antagonist, PRDC. Through our structural and functional analysis, we show that PRDC exists as a noncovalent dimer, stabilized by a long stretch of backbone hydrogen bonds within β2 of the Wrist region. Analyzing the structure, we identified several surface exposed residues across the surface of the PRDC dimer. Through mutational analysis of these residues, we identified a large hydrophobic interface on the convex surface of PRDC, beneath the α1 helix, important for mediating PRDC’s anti-BMP functionality both in vitro and in vivo. These findings are in agreement with previous studies that show the DAN domain, and not the N-terminus, to be essential in mediating BMP antagonism (Sun et al., 2006). Lastly, we are the first to characterize PRDC as a strong heparin binding protein, where heparin surprisingly functions to hinder BMP binding.
Model for PRDC Mediated BMP Inhibition
Upon resolution of the PRDC structure, it was not obvious which hydrophobic residues were involved in BMP ligand recognition. Given that the mutations of C120 and F122 had little affect on binding, it does not appear that the concave surface is utilized for BMP interactions. This notion is further supported when considering that our bacterially produced WT PRDC, which is modified by glutathione at C120, exhibits nanomolar affinity for BMP ligands. Although hydrophobic residues in the fingertips appear poised to interact with BMP, they are not conserved across the DAN family (Figure 6A and S7). Furthermore, through our functional studies, we show that mutation of these amino acids had no effect on BMP binding or inhibition (Figure 4B; Table 2).
On the other hand, residues in the central part of the DAN domain, specifically those on the convex surface, are the most critical for BMP inhibition, including W72, Y98, F104, and F117. These residues form a hydrophobic patch that is weakly and partially buried through interactions with the N-terminal α1 helix. Furthermore, several of these large hydrophobic residues are conserved across the other strongly antagonizing DAN family members, including Gremlin, Dan, and Coco (Figure 6A and S7). However, our structural and solution-state data indicates that there is a high likelihood for N-terminal flexibility and conformational sampling. Therefore, we suggest an N-terminal latch model of BMP inhibition, where the N-terminus of PRDC can readily dissociate from the protein core to expose a larger BMP binding epitope, potentially decreasing the buried hydrophobic surface area by 200 Å2 per monomer (Figure 7). This hydrophobic epitope, once exposed, would then be capable of providing affinity for the PRDC:BMP interaction. Thermodynamically speaking, flexibility within the N-terminus could provide PRDC with a mechanism to shield the hydrophobic BMP-binding epitope from water, while eliminating susceptibility for unwanted non-specific interactions but allowing the necessary amino acids to be accessible in the presence of BMP ligands.
Figure 7. Proposed model of BMP ligand inhibition by PRDC.
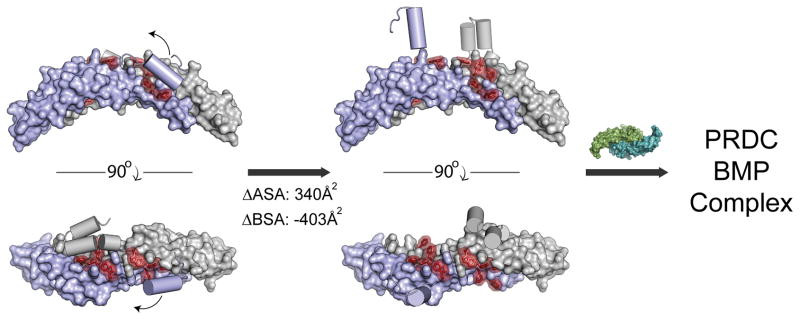
PRDC dimer AB (surface) with the N-terminal helices shown as cylinders and the residues implicated for BMP binding colored red. Prior to ligand binding, the N-terminal helix interacts with the DAN domain to partially cover hydrophobic residues in the BMP binding epitope. The N-terminal helix is loosely associated with the DAN domain and can be displaced by stochastic events or ligand binding to expose a larger hydrophobic interface. Numbers indicate the change in available surface area (ASA) and buried surface area (BSA) upon the dissociation of the helix from the core of the PRDC dimer.
In a different sense, Noggin forms a significant and stable interaction with BMP7 based upon a strong interaction developing from its N-terminus, where amino acids within its core, cysteine-rich domain are not critical for defining this interaction (Groppe et al., 2002). While the suggested N-terminal latch model for PRDC is very different and opposite in nature as compared to Noggin, it is plausible that the N-terminus of PRDC could provide an additional method for stabilizing the BMP binding interaction, deriving ligand specificity, and/or occupying the receptor binding sites to inhibit receptor activation. However, based upon our results and the previously reported finding that Gremlin maintains its antagonist functionality towards BMP4 even upon complete alteration of it N-terminus, we believe that PRDC derives the majority of its activity from its DAN-domain, independent of its N-terminus (Sun et al., 2006). Certainly, further crystallographic studies of these proteins and their complexes are needed to provide functional insight into the expanse of the BMP-binding epitope.
Implications for Heparin:PRDC Interaction
Through our heparin studies, we showed that PRDC tightly associates with heparin. In addition, this interaction abrogates the anti-BMP functionality of PRDC towards BMP2, drastically reducing its affinity for the ligand (Figure 5C). Since the majority of the electropositive potential of PRDC is located on the convex surface of the protein we postulate that heparin binds to this region of the protein dimer (Figure 5B). It is possible that heparin binding indirectly (local allosteric regulation) blocks the BMP binding epitope by binding the N-terminus and extending across the convex surface of the DAN domain, thus limiting the flexibility within α1 and forcing PRDC into a static or “closed” conformation. When this occurs, it would be expected that the important BMP binding residues on the convex surface would remain buried and hinder BMP binding. Alternatively, heparin may bind to PRDC (such as at the fingertips) and induce a global allosteric structural change by significantly contorting the structure of the protein or by directly competing for the BMP binding site.
Structural Comparison of PRDC, SOST, and Cystine-knot Containing Growth Factors
Direct comparison of PRDC and SOST monomers reveals an RMSD of 3.3 Å when aligning their cystine knot motifs, including F1 and F2 of their DAN domains. However, PRDC shows a number of significant structural differences in the N-terminus and Wrist region, accounting for an overall RMSD of 8.7 Å (Figure 6B). Interestingly, these same regions form important contacts at the dimer interface of PRDC, providing an explanation of the assembly differences of PRDC and SOST (dimer vs. monomer). In terms of BMP inhibition, there is limited conservation between PRDC and SOST for the hydrophobic residues defining the BMP-binding epitope (Figure 6A). Without these residues, it would be expected that SOST would be limited in terms of BMP-binding affinity or bind BMP utilizing a different epitope than PRDC. Certainly a key structural difference between the two DAN family members is dimerization. Therefore, a dimer with two BMP binding epitopes would offer an advantage to affinity when binding to BMP ligands, much like the Noggin dimer. Furthermore, lack of dimer formation might allow SOST to function as a more effective Wnt inhibitor. Recent studies have pinpointed the interaction of SOST with LRP5/6 to a linear stretch of residues within the wrist region of SOST (Holdsworth et al., 2012). Besides a lack of conservation with PRDC, these residues are packed at the dimer interface and would not be solvent accessible as in SOST. Therefore, differences in specific residues and overall oligimerization could differentiate BMP or Wnt antagonism. In the future, it will be interesting to see if those DAN family antagonists that exist as dimers solely exhibit anti-BMP functionality. In support of this notion, we have previously characterized Dan, which is very different than PRDC and SOST, to exist as a non-covalent dimer capable of inhibiting BMP signaling (Kattamuri et al., 2012b). In sight of this, it can be expected that Gremlin, a potent inhibitor of BMP signaling, also exists as a dimer.
When comparing the overall structure of PRDC and SOST to several other cystine-knot containing proteins, a genuine growth factor-like fold is observed. This is illustrated by the conservation of both the central cystine-ring as well as the tandem β-strands that allow for the formation of the finger-wrist fold (Figure 8A). Because of this conservation to several cytokine ligands (including VEGF-C, BMP7, FSH-B, and Artemin), it can be expected that either PRDC or SOST evolved from agonists and/or maintain agonist-like functions (Groppe et al., 2002; Jiang et al., 2012; Leppanen et al., 2010; Silvian et al., 2006). Intriguingly, PRDC adopts a head-to-tail dimerization mechanism, much different than Noggin (head-to-head), but similar to a number of other growth factors (Figure 8B). This type of dimerization is commonly mediated by important structural elements (typically helices) in their Wrist regions, which are absent in SOST. As this type of head-to-tail dimerization is often necessary to symmetrically bring into proximity and activate matching sets of receptors, it brings into question the role of PRDC and other DAN family members as signaling ligands. Interestingly, agonist activity has been supported for Gremlin, which functions to bind and activate the angiogenesis promoting receptor VEGFR2 (PRDC known as Gremlin 2 with 53.9% identity overall, 66.4% within the DAN domain alone, Figure 8A) (Chiodelli et al., 2011). Therefore, it is possible that DAN family antagonists simply evolved from their counterpart signaling ligands, similar to the Inhibin subfamily of protein inhibitors. Because of the high conservation between Gremlin and PRDC, it is plausible that PRDC could also function as a cytokine.
Figure 8. Structural comparison of cystine-knot monomers and dimers.

(A) Ribbon representations of a single chain for several cystine-knot proteins, colored as rainbow from the N- (blue) to C-terminus. Each structure was superimposed with the PRDC monomer (Chain B). PDB identifiers are indicated along with the Z-score from a pair-wise alignment using the Dali-lite server. (B) Comparison of the dimerization mechanisms of PRDC to other cystine-knot proteins. The PRDC dimer exhibits a growth factor-like architecture with a head-to-tail assembly, similar to BMP7 and VEGF-C, whereas Noggin assembles in a head-to-head fashion.
DAN family molecules are intriguing in that, as a whole, they can interact with multiple pathways, including TGF-β, Wnt and VEGF. To our knowledge, these characteristics have not been identified in other TGF-β ligand antagonists. Why multiple BMP antagonists have evolved has remained a perplexing question. Certainly, having redundancy during critical developmental stages provides a means towards higher fidelity. However, the unique folds and features of these different antagonists provide the cell with alternative mechanisms for signal inhibition, regulation, and, for DAN family members, the ability to cross-talk with other pathways. As such, DAN family proteins might provide a scaffold to develop therapeutics that can functionally target multiple pathways. The structure of PRDC and the identification of the BMP binding epitope provide an important first step towards this goal. Furthermore, the structure of PRDC provides an unprecedented glimpse into a BMP antagonist prior to ligand binding and will provide insight into the molecular transitions that occur upon binding once the structure of a DAN family member bound to a BMP ligand becomes available.
Experimental Procedures
Production of PRDC
Protein expression and purification was achieved using a modified version of our previously published protocol (Kattamuri et al., 2012a). Following refolding, protein was purified under denaturing conditions using heparin-affinity chromatography and size exclusion chromatography (SEC). Selenomethionine substituted PRDC was generated as previously described (Deng et al., 2012).
X-ray structure determination of PRDC
PRDC crystals (both native and SeMet) were grown by hanging-drop using 0.1 M Sodium Citrate at pH 5.5, and 9.25–10% PEG 3350 and 5 mg/ml of PRDC. The cryoprotectant included 20% PEG 3350 and 15% glycerol. Diffraction data were collected at the Advanced Photon Source (23ID-B GM/CA) at Argonne National Laboratory and processed as previously described (Deng et al., 2012).
Small-angle X-ray scattering
SAXS data were collected at beam line 12-ID-B and 18-ID-D at the Advanced Photon Source as previously described (Deng et al., 2013). Generated SAXS models were compared to the PRDC crystal structure using FoXS (Schneidman-Duhovny et al., 2010). For the final represented Ab initio reconstructions, a minimum of 10 Gasbor ensembles was used to generate averaged models using DAMAVER (Volkov and Svergun, 2003).
Surface plasmon resonance
All experiments were performed as previously described (Kattamuri et al., 2012b). Association (ka) and dissociation (kd) rate constants were determined by globally fitting the sensogram curves to a 1:1 Langmuir model available in the BIAevaluation 4.1.1 software (Biacore Inc). The dissociation constant (KD) was calculated using the formula KD = kd/ka.
Luciferase reporter assay
A C2C12 cell line stably transfected with the luciferase gene (BRE-C2C12) under the control of a BMP responsive promoter (kindly provided by Dr. Gareth Inman of the Beatson Institute for Cancer Research) was used to measure BMP activity and inhibition as previously described (Kattamuri et al., 2012b).
Xenopus embryo BMP target gene assay
Embryo manipulations and microinjections were performed as previously described and staged according to the normal table of development for Xenopus laevis (Kattamuri et al., 2012b). To assay PRDC activity in vivo, we injected the blastocoel cavities of stage 9 Xenopus embryos with 1 μM or 10 μM either WT or mutant PRDC. The total injection volume was adjusted to a constant 40 nl using phosphate-buffered saline with 0.1% BSA. After injection, embryos were cultured at room temperature until stage 20, fixed overnight at 4°C in MEMFA and analyzed for expression of the BMP target gene sizzled via whole-mount in situ hybridization as previously described (Kattamuri et al., 2012b). The antisense sizzled in situ probe was prepared using T7 RNA polymerase with SalI-linearized pCMV-Sport6-sizzled plasmid template (IMAGE clone 4057152 obtained from Open Biosystems).
Supplementary Material
Highlights.
2.2 Å crystal structure of a BMP antagonist from the DAN family
Structure adopts a dimer with growth-factor like features
Identification of the BMP binding epitope to the central DAN domain
Proposed structural mechanism for BMP binding involving the N-terminus
Acknowledgments
We wish to thank members of the Thompson Laboratory for critically reading the manuscript and Dr. Eek-hoon Jho, Univerisity of Seoul, Korea, for providing the Mus musculus PRDC cDNA. This work was supported by research grants from the National Institute of General Medical Sciences (R01 GM084186) to TT and (R01 GM38060) to RL. Xenopus experiments were supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK070858) to AZ.
Footnotes
Accession Numbers
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under the ID code 4JPH.
Supplemental Information includes seven figures, one table, and Supplemental Experimental Procedures and can be found with this article at doi:xxxxxxxxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez E, Cahoreau C, Combarnous Y. Comparative structure analyses of cystine knot-containing molecules with eight aminoacyl ring including glycoprotein hormones (GPH) alpha and beta subunits and GPH-related A2 (GPA2) and B5 (GPB5) molecules. Reprod Biol Endocrinol. 2009;7:90. doi: 10.1186/1477-7827-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsian-Kretchmer O, Hsueh AJW. Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol Endocrinol. 2004;18:1–12. doi: 10.1210/me.2003-0227. [DOI] [PubMed] [Google Scholar]
- Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23:609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Cai J, Pardali E, Sánchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. FEBS Lett. 2012;586:1993–2002. doi: 10.1016/j.febslet.2012.04.030. [DOI] [PubMed] [Google Scholar]
- Cash JN, Angerman EB, Kattamuri C, Nolan K, Zhao H, Sidis Y, Keutmann HT, Thompson TB. Structure of myostatin·follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J Biol Chem. 2012;287:1043–1053. doi: 10.1074/jbc.M111.270801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. Embo J. 2009;28:2662–2676. doi: 10.1038/emboj.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodelli P, Mitola S, Ravelli C, Oreste P, Rusnati M, Presta M. Heparan sulfate proteoglycans mediate the angiogenic activity of the vascular endothelial growth factor receptor-2 agonist gremlin. Arterioscler Thromb Vasc Biol. 2011;31:e116–e127. doi: 10.1161/ATVBAHA.111.235184. [DOI] [PubMed] [Google Scholar]
- Deng X, Morris J, Chaton C, Schroder GF, Davidson WS, Thompson TB. Small-angle X-ray Scattering of Apolipoprotein A-IV Reveals the Importance of Its Termini for Structural Stability. Journal of Biological Chemistry. 2013;288:4854–4866. doi: 10.1074/jbc.M112.436709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Morris J, Dressmen J, Tubb MR, Tso P, Jerome WG, Davidson WS, Thompson TB. The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure. 2012;20:767–779. doi: 10.1016/j.str.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies DL, Viviano B, McCarthy J, Rey J-P, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–1749. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, Affolter M, Vale WW, Belmonte JCI, Choe S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–642. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- Hinck AP. Structural studies of the TGF-βs and their receptors - insights into evolution of the TGF-β superfamily. FEBS Lett. 2012;586:1860–1870. doi: 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Holdsworth G, Slocombe P, Doyle C, Sweeney B, Veverka V, Le Riche K, Franklin RJ, Compson J, Brookings D, Turner J, et al. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of Wnt co-receptors. J Biol Chem. 2012;287:26464–26477. doi: 10.1074/jbc.M112.350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung W-T, Wu F-J, Wang C-J, Luo C-W. DAN (NBL1) Specifically Antagonizes BMP2 and BMP4 and Modulates the Actions of GDF9, BMP2, and BMP4 in the Rat Ovary 1. Biology of Reproduction. 2012;86:158, 1–9. doi: 10.1095/biolreprod.111.096172. [DOI] [PubMed] [Google Scholar]
- Jiang X, Liu H, Chen X, Chen PH, Fischer D, Sriraman V, Yu HN, Arkinstall S, He X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proceedings of the National Academy of Sciences. 2012;109:12491–12496. doi: 10.1073/pnas.1206643109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattamuri C, Luedeke DM, Thompson TB. Expression and purification of recombinant protein related to DAN and cerberus (PRDC) Protein Expr Purif. 2012a;82:389–395. doi: 10.1016/j.pep.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattamuri C, Luedeke DM, Nolan K, Rankin SA, Greis KD, Zorn AM, Thompson TB. Members of the DAN Family Are BMP Antagonists That Form Highly Stable Noncovalent Dimers. Journal of Molecular Biology. 2012b;424:313–327. doi: 10.1016/j.jmb.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Beglov D, Brenke R, Comeau SR, Shen Y, Li K, Zheng J, Vakili P, Paschalidis IC, et al. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins. 2010;78:3124–3130. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, Lankinen H, Goldman A, Ballmer-Hofer K, Alitalo K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proceedings of the National Academy of Sciences. 2010;107:2425–2430. doi: 10.1073/pnas.0914318107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom K, Fainsod A, Steinbeisser H. Patterning of the mesoderm involves several threshold responses to BMP-4 and Xwnt-8. Mech Dev. 1999;87:33–44. doi: 10.1016/s0925-4773(99)00137-9. [DOI] [PubMed] [Google Scholar]
- Monestier O, Brun C, Cocquempot O, Petit D, Blanquet V. GASP/WFIKKN Proteins: Evolutionary Aspects of Their Functions. PLoS ONE. 2012;7:e43710. doi: 10.1371/journal.pone.0043710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012;586:1846–1859. doi: 10.1016/j.febslet.2012.02.043. [DOI] [PubMed] [Google Scholar]
- Nimmagadda S, Geetha-Loganathan P, Scaal M, Christ B, Huang R. FGFs, Wnts and BMPs mediate induction of VEGFR-2 (Quek-1) expression during avian somite development. Dev Biol. 2007;305:421–429. doi: 10.1016/j.ydbio.2007.02.031. [DOI] [PubMed] [Google Scholar]
- Porollo A, Meller J. Prediction-based fingerprints of protein-protein interactions. Proteins. 2007;66:630–645. doi: 10.1002/prot.21248. [DOI] [PubMed] [Google Scholar]
- Rider CCC, Mulloy BB. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem J. 2010;429:1–12. doi: 10.1042/BJ20100305. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvian L, Jin P, Carmillo P, Boriack-Sjodin PA, Pelletier C, Rushe M, Gong B, Sah D, Pepinsky B, Rossomando A. Artemin crystal structure reveals insights into heparan sulfate binding. Biochemistry. 2006;45:6801–6812. doi: 10.1021/bi060035x. [DOI] [PubMed] [Google Scholar]
- Sudo S, Avsian-Kretchmer O, Wang LS, Hsueh AJW. Protein related to DAN and cerberus is a bone morphogenetic protein antagonist that participates in ovarian paracrine regulation. Journal of Biological Chemistry. 2004;279:23134–23141. doi: 10.1074/jbc.M402376200. [DOI] [PubMed] [Google Scholar]
- Sun J, Zhuang FF, Mullersman JE, Chen H, Robertson EJ, Warburton D, Liu YH, Shi W. BMP4 activation and secretion are negatively regulated by an intracellular gremlin-BMP4 interaction. Journal of Biological Chemistry. 2006;281:29349–29356. doi: 10.1074/jbc.M603833200. [DOI] [PubMed] [Google Scholar]
- Thompson TB, Lerch TF, Cook RW, Woodruff TK. The structure of the follistatin: activin complex reveals antagonism of both type I and type II receptor binding. Developmental Cell. 2005;9:535–543. doi: 10.1016/j.devcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- van Bezooijen RL, Roelen BA, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, Hamersma H, PAPAPOULOS SE, ten Dijke P, Löwik CW. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. The Journal of Experimental Medicine. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, Muzylak M, Greenslade K, Moore A, Zhang L, et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009;284:10890–10900. doi: 10.1074/jbc.M807994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DW, Godson C, Brazil DP, Martin F. Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends in Cell Biology. 2010;20:244–256. doi: 10.1016/j.tcb.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Weidauer SE, Schmieder P, Beerbaum M, Schmitz W, Oschkinat H, Mueller TD. NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun. 2009;380:160–165. doi: 10.1016/j.bbrc.2009.01.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.