

Abstract
An intercellular “glymphatic” pathway clears cell waste from the brain and may reveal new targets for treating neurodegenerative diseases.
Essentially all neurodegenerative diseases are associated with misaccumulation of cellular waste products. Of these, misfolded or hyperphosphorylated proteins are among the most difficult for the brain to dispose. For example, tau and β-amyloid can accumulate as stable aggregates that are neurotoxic in conditions such as Alzheimer’s disease (1). Intracellular proteasomal degradation and autophagy are considered the principal means for removing proteins in the central nervous system, and the dysfunction of each has been causally associated with neurodegeneration (2). Yet many cytosolic proteins are released into the interstitial space in the brain, suggesting that extracellular disposal routes may also eliminate waste (3).
Throughout the body’s tissues, bulk flow of the fluid between cells, into the blood or lymph, plays an important role in the removal of potentially toxic metabolic by-products. Lymphatic vessels, which run in parallel with the blood vascular system, are the principal means by which tissues eliminate excess fluid and proteins. Although the density of lymph vessels generally correlates with tissue metabolic rate (4), the brain and spinal cord are curiously devoid of such a lymphatic tree. This is puzzling because the high metabolic activity of neurons predicts the need for rapid elimination of their metabolic byproducts. It was long thought that movement of the cerebrospinal fluid (CSF), which is produced in the choroid plexus of the brain and flows through its ventricles and basal cisterns, constitutes a “sink” for waste products to diffuse from the brain, for eventual clearance to the general circulation. However, the large tissue distances in most of the brain prevent diffusion and bulk flow from making this process efficient. Albumin, for instance, would require more than 100 hours to diffuse through 1 cm of brain tissue (5).
Two-photon imaging of live mice through a closed cranial window has since permitted the direct observation of CSF movement through the intact brain. This technique revealed that CSF is exchanged rapidly with interstitial fluid (ISF) in the brain by a highly organized, brain-wide pathway that consists of three serial elements: a paraarterial CSF influx route, a paravenous ISF clearance route, and an intracellular transastrocytic path that couples the two extra-cellular paravascular routes (6). Specifically, CSF passes through the para-arterial space that surrounds arteries; the space is bound by the abluminal surface of the blood vessel and the apical processes of astrocytes. Water channels called aquaporin 4 (AQP4) on the vascular endfeet of astrocytes (7) facilitate convective flow out of the para-arterial space and into the interstitial space (see the figure). As CSF exchanges with the ISF, vectorial convective fluxes drive waste products away from the arteries and toward the veins. ISF and its constituents then enter the paravenous space. As ISF exits the brain through the paravenous route, it reaches lymphatic vessels in the neck, and eventually returns its contents to the systemic circulation. Radio-label tracer studies indicate that 40 to 80% of large proteins and solutes are removed from the brain through this macroscopic clearance pathway (6). CSF can also exit through the arachnoid villi, which extend through the outer protective membrane layer of the brain and allow CSF to exit to the bloodstream, as well as at sites along the cavity and cranio-spinal nerve roots. Regardless of the route, its solutes and proteins ultimately reach the liver, where they are degraded. As such, the “glymphatic system”—so-named for its dependence on glial water channels and its adoption of a clearance function similar to that of the peripheral lymphatic system—avoids the need for local protein processing and degradation. Instead, it facilitates transport to the same central excretion and recycling sites used by other tissues.
Figure 1. Go with the flow.
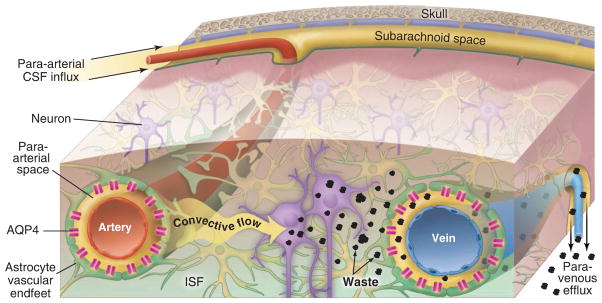
Convective glymphatic fluxes of CSF and ISF propel the waste products of neuron metabolism into the paravenous space, from which they are directed into lymphatic vessels and ultimately return to the general circulation for clearance by the kidney and liver.
Studies of mice genetically engineered to lack AQP4 showed that fluid flux through the glymphatic pathway relies on specific expression of this water channel along the apical membrane of vascular endfeet of astrocytes (6). When AQP4 is mislocated to the cell body of astrocytes or to astrocytic processes that do not abut the vasculature, as observed in traumatic brain injury or stroke (8, 9), clearance of soluble proteins through the glymphatic system declines substantially.
An interesting question is whether the glymphatic system plays a role in spreading fibrillary tau aggregates through the interstitial space in neurodegenerative disease. The injection of brain extracts from mice containing an aggregation-prone form of human tau protein, into the brains of mice expressing wild-type human tau, induces self-assembly of the wild-type human tau into filaments. This results in the pathological spread of tau aggregates from the injection site to distant brain regions (2, 3, 10).
Perhaps the most persuasive example of CSF recycling as the cause of dispersing the initial seeds of tau tangles is after traumatic brain injury. As a result of axon damage, the tau concentration in CSF increases by as much as a factor of 40,000 (11). Consequently, as the heavily tau-laden CSF enters the brain tissue through the paraarterial space, it is taken up by cells closest to the paravascular boundary, thereby generating the typical paravascular predominance of tau-immunoreactive neurofibrillary tangles (12). Similarly, glymphatic CSF influx may also act as a constant source for delivering β-amyloid, which could contribute to the growth of para-arterial deposits in cerebral amyloid angiopathy. In turn, the same para-arterial space that normally functions as a low-resistance influx path for CSF will narrow as the amyloid plaques enlarge, slowing glymphatic clearance and thus accelerating amyloid deposition (13).
As such, studies of the multiple pathways involved in glymphatic clearance may identify new targets for treating neurodegenerative diseases. For example, mislocation of AQP4 water channels may contribute to neurodegenerative disease progression. Thus, potentiating the insertion and activity of AQP4 channels in astrocytic vascular endfeet might mitigate or even reverse the course of protein-associated neurodegenerative disorders.
Can the efficiency of glymphatic clearance be assessed? Preclinical analysis in rats shows that magnetic resonance imaging can provide a brain-wide map of both glymphatic influx and efflux, by which clearance kinetics can be derived and compared across subjects (14, 15). By extending this approach to humans, it may be possible to identify patients at risk for developing Alzheimer’s disease who would benefit from therapeutic intervention before symptomatic neurodegeneration ensues. Similarly, this type of analysis might allow the monitoring of treatment responses, as well as the identification of genetic markers that predict enhanced susceptibility to glymphatic decline. Such an approach also may be suitable for victims of brain injury who develop chronic traumatic encephalopathy, which is characterized by paravascular tau tangles and premature neuronal degeneration (12). There are currently no definitive diagnostics that identify susceptible individuals, and thus no means by which to achieve early clinical intervention. Recognition that the brain, like all other organs, uses both local and organ-wide mechanisms for clearing interstitial protein waste may offer new insights into the pathophysiology and prophylaxis of neurodegeneration, as well as injuries and proteinopathies of the human central nervous system.
References
- 1.Mucke L, Selkoe DJ. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frost B, Diamond MI. Nat Rev Neurosci. 2010;11:155. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker LC, Diamond MI, Duff KE, Hyman BT. J Am Med Assoc Neurol. 2013;70:304. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loukas M, et al. Clin Anat. 2011;24:807. doi: 10.1002/ca.21194. [DOI] [PubMed] [Google Scholar]
- 5.Cserr HF. Physiol Rev. 1971;51:273. doi: 10.1152/physrev.1971.51.2.273. [DOI] [PubMed] [Google Scholar]
- 6.Iliff JJ, et al. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagelhus EA, Mathiisen TM, Ottersen OP. Neuroscience. 2004;129:905. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 8.Iliff JJ, Nedergaard M. Stroke. 2013;44(Suppl 1):S93. doi: 10.1161/STROKEAHA.112.678698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren Z, et al. J Cereb Blood Flow Metab. 2013;33:834. doi: 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clavaguera F, et al. Nat Cell Biol. 2009;11:909. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zemlan FP, et al. Brain Res. 2002;947:131. doi: 10.1016/s0006-8993(02)02920-7. [DOI] [PubMed] [Google Scholar]
- 12.Goldstein LE, et al. Sci Transl Med. 2012;4:134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weller RO, Preston SD, Subash M, Carare RO. Alzheimers Res Ther. 2009;1:6. doi: 10.1186/alzrt6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iliff JJ, et al. J Clin Invest. 2013;123:1299. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, et al. J Transl Med. 2013;11:107. doi: 10.1186/1479-5876-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]