

Abstract
Mortality from pancreatic ductal adenocarcinoma cancer (PDAC) is among the highest of any cancer and frontline therapy has changed little in years. Activation of endothelial nitric oxide synthase (eNOS or NOS III) has been implicated recently in the pathogenesis of PDAC. In this study, we used genetically engineered mouse and human xenograft models to evaluate the consequences of targeting eNOS in PDAC. Genetic deficiency in eNOS limited the development of pre-invasive pancreatic lesions and trended towards an extended lifespan in mice with advanced pancreatic cancer. These effects were also observed upon oral administration of the clinically evaluated NOS small molecule inhibitor L-NAME. Similarly, other transgenic models of oncogenic KRas-driven tumors responded to L-NAME treatment. Finally, these results were recapitulated in xenograft models of human pancreatic cancer, in which L-NAME was found to broadly inhibit tumorigenic growth. Taken together, our findings offer preclinical proof-of-principle to repurpose L-NAME for clinical investigations in treatment of PDAC and possibly other KRas-driven human cancers.
Keywords: pancreatic cancer, KRas, eNOS, xenograft, genetically engineered mice
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has five year survival rates <5% (1). Current frontline treatment for advanced PDAC, gemcitabine, was introduced thirteen years ago with only incremental progress made thereafter (2, 3). Thus, there is a need to develop new and less toxic therapeutic approaches that can be rapidly introduced into the clinic. In this regard, 90% of PDAC possess oncogenic mutations within the gene KRAS, which renders the encoded small GTPase constitutively GTP-bound and active (4). Oncogenic KRas can convert normal human ductal pancreatic epithelial cells to a tumorigenic state (5) and pancreatic expression of the activated oncogene leads to PanIN lesions that progress to PDAC in mice (6). Conversely, silencing oncogenic Ras in established tumors causes spontaneous regression (7). KRas is thus the initiating oncogene in PDAC, and like CML is addicted to BCR-Abl and hence sensitive to imatinib (8), pancreatic cancers required oncogenic KRas for continued growth.
Despite the pivotal role of oncogenic KRas in PDAC, it has proved challenging to inhibit (9). KRas exerts its tumorigenic functions by activating primarily three effector proteins, Raf kinases, phosphatidylinositol 3-kinases (PI3K), and Ral guanine nucleotide exchange factors. Pharmacological inhibitors of the first two pathways reduce tumor growth in some cancers, with numerous clinical trials underway (4). As such, targeting druggable components of oncogenic KRas signaling is one potential strategy to treat PDAC.
Of the three KRas effectors, only active PI3K, or its principal target AKT kinases, maintain xenograft tumor growth upon silencing oncogenic Ras (10), suggesting that pancreatic cancer cells become addicted to PI3K-AKT signaling. Consequently, components of this pathway represent attractive targets. While the families of PI3K and AKT proteins are druggable, they are comprised of highly related proteins involved in a large number of normal physiological processes, and general inhibitors of these kinases can be toxic (11). However, the AKT substrate endothelial Nitric Oxide Synthase (eNOS or NOS III) (12) has been detected in the active state in human PDAC tissues (13). eNOS is a member of the NOS family additionally comprised of neuronal NOS (nNOS or NOS I), and inducible NOS (iNOS or NOS II) that generate nitric oxide (12). Unlike AKT, eNOS plays a limited role in normal physiology, mainly in vasorelaxation (14), and eNOS-/- mice are viable (15). Moreover, mounting evidence suggests that inhibition of eNOS has anti-tumor effects. Specifically, eNOS-/- mice are resistant to DMBA/TPA chemical carcinogenesis (13) and PDGF-induced gliomagenesis (16), while peptide-mediated inhibition of eNOS decreases tumor vascular permeability and tumor growth in hepatocarcinoma and lung carcinoma xenograft models (17). In regards to PDAC, shRNA knockdown of eNOS reduces tumor growth of two PDAC cell lines with highly phosphorylated eNOS (13). Thus, inhibiting eNOS may be a way to indirectly exploit the reliance of pancreatic cancer cells on oncogenic KRas for tumorigenesis. In this regard, the NOS inhibitor NG-nitro-l-arginine methyl ester (L-NAME), which is moderately selective for eNOS and nNOS over iNOS (12), was developed and clinically evaluated in phase II trials for cardiogenic (18) and septic (19-21) shock, and in numerous other clinical trials, including those involving normal healthy subjects (22). This drug is relatively benign compared to conventional cytotoxic chemotherapy; the major side effect of chronic administration is hypertension (23). These findings support the possibility that eNOS could be targeted by simply repurposing the drug L-NAME to treat PDAC. However, nitric oxide can both inhibit and enhance tumorigenesis (24), and the effect of ablating the eNOS gene on PDAC was unknown. Even if genetic ablation of eNOS inhibited PDAC, it was unknown whether this could be translated into a clinically relevant approach using a small molecule NOS inhibitor. We thus evaluated the impact of genetically and pharmacologically inhibiting eNOS on PDAC.
Materials and Methods
Cell lines
AsPC-1, CAPAN-1, CFPac-1, HPAC, HPAF-II, MiaPaCa-2, PANC-1 and SW-1990 (purchased from ATCC) were not independently authenticated. Tumor tissue from KPC mice was minced in collagenase V (Sigma-Aldrich) for 30 minutes at 37°C, after which cells were cultured in DMEM+10%FBS for 4 passages.
Mouse pancreatic cancer models
Pdx-1-Cretg/+(6);eNOS+/-(15) mice were interbred with LSL-KrasG12D(26);eNOS+/- to generate eNOS+/+ and eNOS-/- KC (LSL-KrasG12D/+;Pdx-1-Cretg/+) littermates. KPC (LSL-KrasG12D/+;Pdx-1-Cretg/+;LSL-Trp53R172H/+(25)) mice were generated in a similar manner except the final step consisted of crossing littermates that were either both eNOS wild type or null. KC and KPC mice were randomly assigned to received water untreated or treated with 1g/L L-NAME (Sigma-Aldrich) (27) after weaning until endpoints (330±7 days of age or at moribundity endpoints), according to a Duke IACUC-approved protocol.
Xenograft assays
107 cells suspended in 100μl of Matrigel (BD Biosciences) were injected subcutaneously into flanks of SCID/beige mice (Charles River) and resultant tumors measured thrice weekly (30). Mice were treated with 1g/L L-NAME (see above) beginning on the day of xenograft injection, or once tumors reached a size of 0.75cm3, with 120mg/kg (i.p.) gemcitabine (Eli Lilly) twice weekly for two weeks once tumors reached a size of 0.75cm3, or with 10mg/kg (i.p.) amlodipine (Sigma) five times weekly as previously described (31), beginning on the day of xenograft injection. Equal volumes of PBS were injected in control mice.
Human PDAC tissue samples
Resected archived primary PDAC specimens were provided for eNOS immunohistochemisty analysis devoid of all identifying information, in accordance with IRB protocols.
Grading of ductal lesions
H&E stained histologic sections were reviewed by two pathologists (DMC, MJS) blinded to the experimental groups. Examined slides consisted of a single longitudinal section of pancreas (head to tail) with adjacent small intestine and spleen from each mouse. Quantification of mPanIN lesions was accomplished by first determining the total number of anatomic pancreatic lobules per specimen. Lobules were counted and subsequently evaluated if they contained at least a single identifiable duct and surrounding circumscribed pancreatic acini and/or fibrosis. Within each lobule, the highest grade mPanIN lesion (normal, 1A, 1B, 2 or 3) was identified (28). Quantification of normal acinar area was determined in a blinded fashion from typically five randomly identified high-power fields from 15 or more pancreatic sections and expressed as a percentage of the total area.
Mouse squamous papilloma and carcinoma analysis
Vulvar papillomas were excised from KC mice at time of necropsy and weighed. Total number of new facial papillomas arising until endpoints were determined for each mouse.
eNOS immunohistochemistry
Heat induced epitope retrieval was performed on H&E stained sections followed by staining with an α-eNOS (1:70, Assay Designs 905-386) antibody (Vectastain Elite ABC kits, Vector Labs, were used for peroxidase-based detection). Photographs taken from the areas of strongest immunoreactivity were qualitatively assessed within the vessels (internal control), duct epithelium, stroma, pancreatic acini and adenocarcinom using a four tier scale (0-3+) in a blinded fashion by a pathologist (D.M.C.).
CD31 immunohistochemistry
Heat induced epitope retrieval was performed on H&E stained sections followed by staining with an αCD31 (1:100 Abcam ab2864 or 1:50 BD Pharmingen 550274) antibody (Vectastain Elite ABC kits, Vector Labs, were used for peroxidase-based detection). Usually five random high-power field images of pancreatic sections from six eNOS+/+ and five eNOS-/- KPC mice each at moribundity endpoints were analyzed using ImageJ software to quantitate pixels of CD31 reactivity per field, and an average calculated.
Ki67 immunohistochemistry
Heat induced epitope retrieval was performed on H&E stained sections followed by staining with an αKi67 (1:50 Dako M7249) antibody (Vectastain Elite ABC kits, Vector Labs, were used for peroxidase-based detection). Ki67 immunoreactive cells were counted from four to five random high-power field images of CFPac-1 xenograft tumor sections from seven tumors each from mice treated or untreated with 1g/L L-NAME, and an average calculated.
PCR of KRas alleles
DNA, purified from pancreatic, facial papilloma, or vulvar tumor tissue, was PCR amplified to detect wild type and recombined KRas alleles, as described previously (29).
RT-PCR of eNOS
RNA was purified from pancreatic tissue or tumor cell lines established from pancreatic tumors of KC or KPC mice in the absence or presence of the eNOS gene using the RNABee reagent (TelTest), then reverse transcribed using the Omniscript RT kit (Qiagen) and PCR amplified with the primers 5’-TCTTCCATCAAGAGATGGTCAA-3’ and 5’-TCATACTCATCCATGCACAGG-3’ to detect eNOS and 5’-GCACAGTCAAGGCCGAGAAT-3’ and 5’-GCCTTCTCCATGGTGGTGAA-3’ to detect GAPDH.
Blood pressure measurements
The average daily cohort blood pressure was determined by averaging the average of the last 10 of 15 blood pressure measurements from each conscious mouse in the cohort using a computerized tail cuff monitor (Hatteras Industries) as previously described (32). Average daily blood pressure was calculated from measurements taken on 26 days over an eight week span.
eNOS and HRas activation status
CFPac-1 cells transfected with 6μg pCMV-neo-HA-eNOS using Fugene 6 (Roche) were treated for 2 hours with DMSO or 20μM LY294002 (Cell Signaling Technologies). These cells, KPC cell lines grown overnight in 0.5% FCS, or subcutaneous tumors derived from CFPac-1 cells in cohorts of mice untreated or treated with L-NAME were lysed in RIPA buffer, and resolved by SDS-PAGE and probed using αAKT, αSer1177 Phospho-eNOS or αSer473 Phospho-AKT (Cell Signaling Technology) or αHRas (Santa Cruz) antibodies. GTP-bound HRas was detected as previously described (13). Quantitation of phospho-eNOS and HRas-GTP levels was done using ImageJ software. Results were normalized to total AKT and total HRas levels, respectively.
L-NAME dose titration
24 hours after HPAF-II, PANC-1, or CAPAN-2 cells were plated (4,000 cells per well), cells were treated with 2μl L-NAME in an 11-point dilution standard curve with concentrations ranging from 10μM diluted 1:3 down to 0.2nM then 24 hours later assayed wit the Cell Titer-Glo Luminescent Cell Viability Assay (Promega). Luminescence was detected with an Analyst HT Luminometer and graphed in a dose-response curve to calculate pIC50 values.
Statistical Analyses
For two cohort xenograft studies, tumor volumes of untreated and treated mice on the final day of the experiment were compared using a two-sided unpaired t test. For xenograft studies with multiple arms, one-way ANOVA was used with Tukey’s Multiple Comparison Test. For comparison of individual arms in blood pressure studies, for comparison of individual arms in studies analyzing the area of normal acinar tissue remaining in KC pancreata, for studies comparing relative CD31 and KI-67 immunoreactivity in tumors, and for comparison of facial papilloma appearances in the mice, a two sided unpaired t test was again used. For comparison of vulvar papillomas the Mann-Whitney U test was performed. For analysis of normal ducts and PanIN-1A lesions, chi-squared analysis was performed to compare each arm to the control untreated mice. Ducts were grouped into two groups (e.g. normal versus abnormal, or PanIN-1A versus all other) and chi-squared analysis was performed. Kaplan-Meier survival curves were generated for each of the KPC mice cohorts and P values were calculated using the log-rank (Mantel-Cox) test. All statistical analyses were performed using Graphpad Prism v5 (Graphpad Software).
Results
eNOS expression during PDAC development
LSL-KrasG12D/+;Pdx-1-Cretg/+ (KC) mice conditionally express endogenous mutant KrasG12D in the pancreas develop pancreatic intraepithelial neoplastic lesions (PanINs), presumed precursors to invasive disease that at low frequency progress to PDAC (6). LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cretg/+ (KPC) mice conditionally express endogenous mutant KrasG12D and p53R172H in the pancreas, which leads to lethal PDAC similar to human PDAC (33, 34). Capitalizing on the ability to model different stages of pancreatic cancer in mice we measured eNOS levels by immunohistochemistry in the pancreas of two wild type, KC, and KPC animals. As previously reported (35), eNOS was confined to endothelial cells in the normal pancreas (+2 to +3 staining in a scale of 0 to +3), with no visible staining in ducts or acini. Variable eNOS reactivity (0 to +2) was observed in PanIN lesions in KC mice. Patchy (+2) to a widespread blush (+1) with focal eNOS staining was detected in adenocarcinomas of KPC mice, which was lost in eNOS-/- KPC mice (Fig. 1A). eNOS was detected at variable levels by RT-PCR in three normal pancreatic tissue, presumably from the endothelial cells, in all four pancreatic samples from KC mice, and 11 of 13 pancreatic samples from KPC mice at the time these tissues developed PanIN or adenocarcinomas. Eight of the 13 tumor cell lines devoid of stromal tissue that were derived from pancreata of KPC mice also expressed eNOS. As a negative control, eNOS was not detected in the pancreata analyzed from one normal, four KC, or one KPC eNOS-/- mice (Fig. 1B). In humans, eNOS was previously shown to be elevated, particularly in the vasculature, in pancreatic cancer cell lines and specimens (36), and a number of human PDAC cell lines exhibit activated eNOS (13). In agreement, four of nine human pancreatic cancer specimens had regions of eNOS positivity (e.g. Supplementary Fig. S1).
Figure 1. eNOS levels during murine pancreatic tumorigenesis.
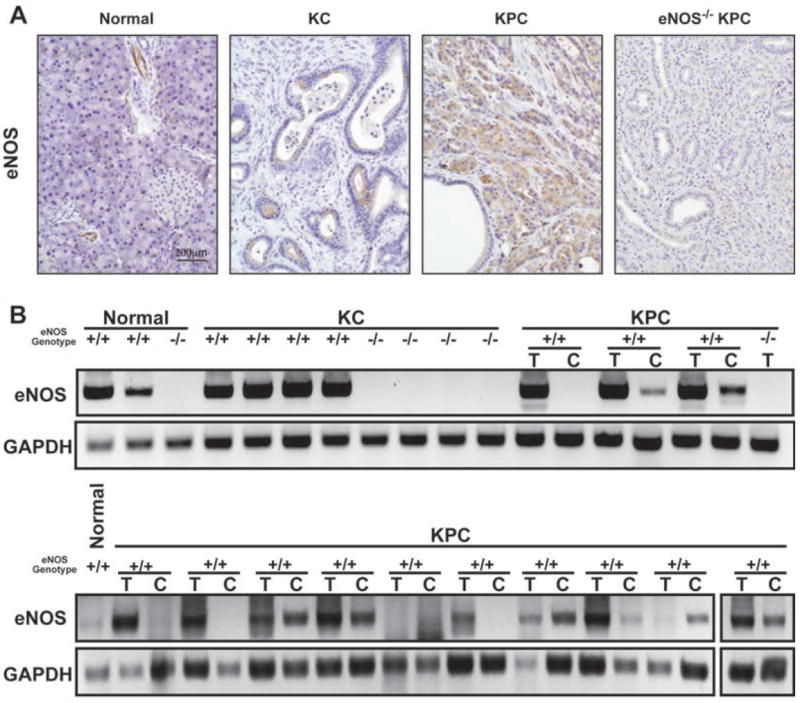
(A) eNOS protein and (B) mRNA, as detected by immunohistochemical staining and semi-quantitative RT-PCR, in the pancreata of normal (e.g. LSL-KrasG12D/+ mice that lack Pdx-Cre), KC (330 days of age), KPC, or eNOS-/- KPC mouse (at the mortality endpoint). T, pancreatic tumor tissue. C, cancer cell line derived from the matched tumor.
Genetic ablation of eNOS decreases PanIN development
To test whether ablation of the eNOS gene disrupts the onset of pancreatic tumorigenesis, KC and eNOS-/- KC mice were aged to ~330 days to allow a spectrum of PanINs to develop (6). Pancreata were removed, stained, and the area of remaining normal acinar tissue quantified from usually five high-power fields per pancreas from cohorts of 12 to 16 mice (amounting to eighty random high-power fields analyzed) as well as the highest grade ductal PanIN lesion per lobule scored from >2600 lobules per group.
As previously reported (6), most of the pancreata in KC animals was replaced with abnormal tissue consisting of varying grades of PanIN lesions with abundant surrounding fibrotic stroma and associated chronic inflammatory cells (Fig. 2A). Quantification revealed that the average area of normal acinar tissue remaining was reduced to 17% (Fig. 2B), with 94.5% of lobules having abnormal ducts, mainly consisting of low grade PanIN-1A lesions (Fig. 2C). eNOS-/- KC mice exhibited over twice as much normal pancreatic tissue area (P<0.001, Fig. 2, A and B), a significant (P<0.05) drop in lobules with PanIN-1A lesions, and an accompanying significant (P<0.0001) doubling in the number of lobules with normal ducts (Fig. 2C).
Figure 2. Analysis of pancreatic lesions in eNOS-/- and L-NAME-treated KC mice.
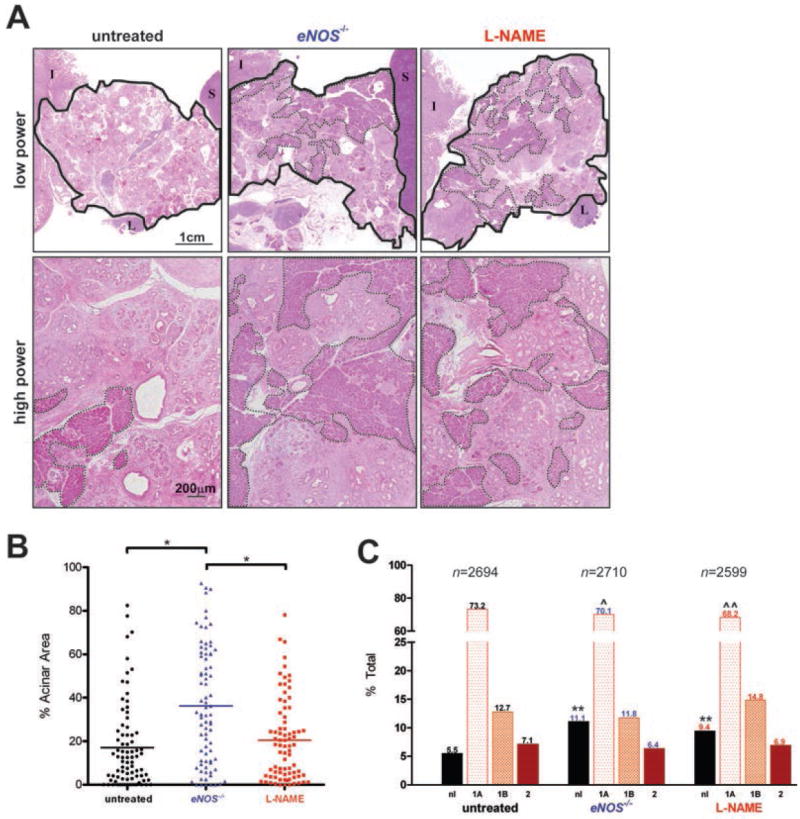
(A) Whole mount (low-power) and magnified (high-power) H&E stained pancreata (solid line: outline of pancreas, dashed line: normal acinar tissue, I: intestine, L: lymph node, S: spleen). (B) Average area of normal acini from typically five random high-power fields per mouse (Bar, mean % normal acini. *P<0.001) and (C) % of lobules with the highest grade lesion being normal duct (nl), PanIN-1A (1A), PanIN-1B (1B), or PanIN-2 (2). n, graded lobules. **P<0.0001, normal versus abnormal ducts compared to untreated, and ˆP<0.05 or ˆˆP<0.0001, PanIN-1A versus all other ducts compared to untreated, in the indicated cohorts of 12 to 16 KC mice untreated, L-NAME-treated or eNOS-/-.
Genetic ablation of eNOS decreases development of other oncogenic KRas-driven tumors
KC mice develop vulvar and facial papillomas (Fig. 3A, B) due to a recombined oncogenic KrasG12D allele (Fig. 3C) likely due to Pdx-1 restricted Cre expression (6, 37, 38). eNOS-/- KC mice exhibited a significant (P<0.05) 50% decrease in the number of facial papillomas and a significant (P<0.05) 85% decrease in the weight of vulvar papillomas per mouse at the time of sacrifice (~330 days) compared to KC mice (Fig. 3D).
Figure 3. Analysis of facial and vulvar tumors in eNOS-/- and L-NAME-treated KC mice.
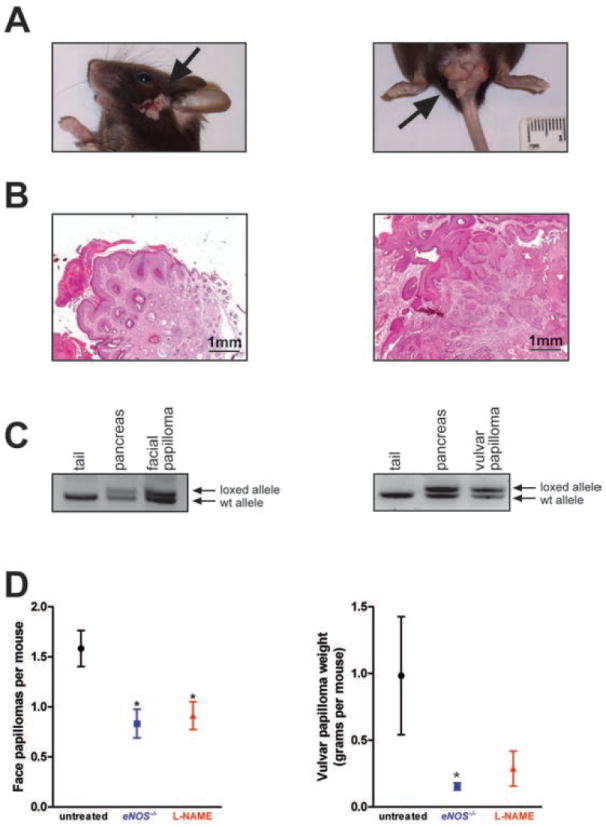
(A) Gross photograph and (B) H&E stained section of a representative facial squamous papilloma (left) and a vulvar papilloma (right) from KC mice. (C) PCR analysis with primers specific for either the wild type Kras allele (wt allele) or the LSL-KrasG12D allele after Cre excision (loxed allele). Samples include DNA isolated from the tail (negative control sample, no Cre excision), pancreas (positive control sample, Cre excision) and a facial (left) and a vulvar (right) tumor. (D) Mean ± SEM of the total number of new facial tumors (left) developing over the course of 330 days in cohorts (n=24) of KC mice either untreated or treated with L-NAME, or eNOS-/- KC mice. Mean weight ± SEM of vulvar tumors (right) arising at 330 days of age in females in cohorts of KC mice either untreated (n=9) or treated with L-NAME (n=7) and in a cohort of eNOS-/- KC mice (n=11). *P<0.05 compared to untreated.
Genetic ablation of eNOS trends towards an increase in the lifespan of mice with lethal PDAC
To test whether genetic ablation of eNOS provides a survival benefit, the most clinically relevant endpoint, eNOS null alleles were crossed into the KPC background, and littermates generated over the course of a year were used to populate cohorts of 35 KPC and 32 eNOS-/- KPC mice. Mice were euthanized at moribundity endpoints known to immediately precede death (33). Consistent with previous studies, KPC mice had a median survival of 142 days (33) while eNOS-/- KPC mice exhibited a median survival of 176 days, a trend that although did not reach significance (P=0.093, HR=0.709, CI95=0.431-1.167), corresponded to an increase of 34 days or nearly 25% of the control lifespan (Fig. 4A).
Figure 4. Lifespan of eNOS-/- and L-NAME-treated KPC mice.
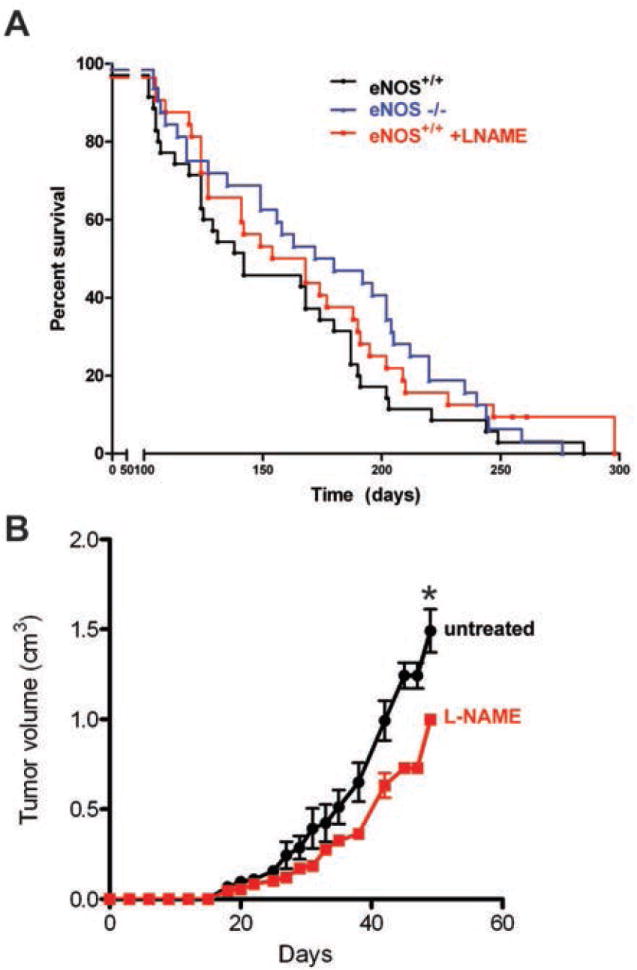
(A) Kaplan-Meier survival curve of cohorts of KPC mice untreated (n=35) or treated with L-NAME (n=32) and eNOS-/- KPC mice (n=32). (B) mean size ± SEM of subcutaneous xenograft tumors generated by a cell line derived from a KPC adenocarcinoma injected in isogenic immunocompetent cohorts of mice randomly assigned to either be untreated (n=4) or treated with L-NAME (n=4). *P<0.05. One L-NAME treated mouse was euthanized at day 29 for reasons unrelated to tumor size.
L-NAME treatment decreases development of preinvasive pancreatic lesions
To test whether pharmacologic inhibition of eNOS impacts pancreatic cancer, KC mice were treated with L-NAME. This drug was chosen because it has a ten-fold preference for eNOS and nNOS over iNOS (12), can be orally dosed, a quality that would facilitate administration in the clinic, is one of only two NOS inhibitors to have been brought to a phase II clinical trial for the treatment of shock (18-21), and is a relatively benign drug, with the main side effect of chronic administration being hypertension and resultant end organ damage, such as left ventricular hypertrophy and glomerulosclerosis (23), all of which can be prevented by co-administration of antihypertensives (39). A cohort of 16 KC mice was provided with L-NAME-treated water at a dose established to increase blood pressure, indicating effective eNOS inhibition (see below and Ref. 15) at weaning until the termination of the experiment (~330 days of age). The L-NAME-treated group trended, although did not reach significance, towards retaining more normal tissue architecture than untreated controls, with the mean percent of remaining normal acinar tissue increasing to 20.5% in 16 mice analyzed (Fig. 2A,B), exhibited a significant (P<0.0001) decrease in lobules with PanIN-1A lesions and a nearly two-fold significant (P<0.0001) increase in lobules with normal ducts in 12 mice analyzed (Fig. 2C). Anecdotally, mice progressing to invasive PDAC was also reduced, with three untreated, two eNOS-/- and one L-NAME treated KC mice developing PDAC.
L-NAME treatment can inhibit other oncogenic KRas-driven tumors
L-NAME treatment significantly (P<0.05) halved the number of facial papillomas and trended, but did not reach significance, towards an almost three quarters reduction in the weight of vulvar papillomas in KC mice (Fig. 3D).
L-NAME treatment trends towards an increase the lifespan of mice with lethal PDAC
To address whether L-NAME increases survival in a PDAC setting, 32 KPC mice treated as above with L-NAME were monitored for moribundity endpoints. All-cause mortality was assessed to include any deaths that may be due to adverse effects from the drug itself. Median survival for the L-NAME-treated mice was 161 days, a trend that although did not reach significance (P=0.208, HR=0.725, CI95=0.439-1.196), reflected a 19-day survival advantage over untreated KPC mice (Fig. 4A). While this effect could be ascribed to an initial decrease in PanIN lesions, as observed in KC mice treated with L-NAME (Fig. 2), L-NAME could similarly inhibit the adenocarcinomas directly. Indeed, the final tumor volume of a PDAC cell line established from a KPC mouse was significantly (P<0.05) reduced in mice treated with L-NAME (Fig. 4B).
L-NAME treatment decreases tumorigenic growth of human PDAC cell lines
To evaluate the potential of targeting NOS in a human cell setting, an important consideration given that oncogenic Ras signaling can exhibit species differences (30, 40), eight KRAS mutation-positive human PDAC cell lines were each injected into eight or more immunocompromised mice, of which half were untreated and half were treated with L-NAME. Five cell lines demonstrated a significant response (P<0.05), two lines trended towards a response, and one did not respond to L-NAME, typically halving the tumor size by the termination of the experiment (Fig. 5A and Supplementary Fig. S2A-G). Anecdotally, the one cell line not affected by L-NAME, PANC-1, was the only line with undetectable levels of activated eNOS (13). In agreement, N-nitro-l-arginine (L-NNA), an insoluble active metabolite of L-NAME, halved both subcutaneous and orthotopic xenograft tumor growth of the human PDAC cell line L3.6pl (41) and aminoguanidine, a broad NOS inhibitor with some specificity for iNOS, decreased tumor growth when given at high doses (42).
Figure 5. Tumorigenic growth of human pancreatic cancer cells treated with L-NAME.
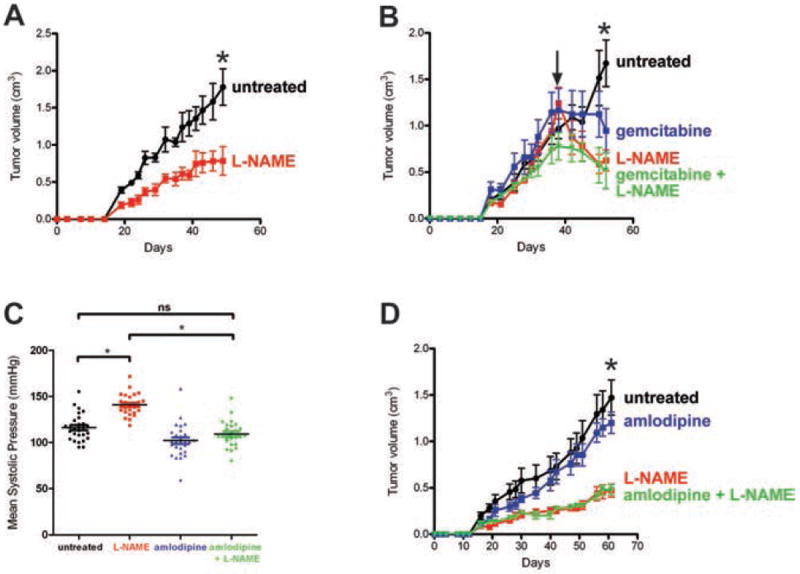
(A) Mean size ± SEM of subcutaneous tumors derived from CFPac-1 cells in cohorts (n=5) of mice untreated or L-NAME-treated. *P<0.05. (B) Mean size ± SEM of subcutaneous CFPac-1 tumors in cohorts of mice (n=4 to 5) that, upon reaching an established size of 0.75cm3 (arrow), were untreated or treated with L-NAME, gemcitabine, or L-NAME + gemcitabine. P<0.05, *each treatment compared to untreated. (C) The daily mean systolic pressure taken on 26 days over 8 weeks for the cohorts of mice (n=4 to 5) with subcutaneous CFPac-1 tumors that were either untreated or treated with L-NAME, amlodipine, or L-NAME + amlodipine. *P<0.05. ns, non-significant. (D) Mean size ± SEM of CFPac-1 xenografts in animals corresponding to treatment groups described in (C). *P<0.05, L-NAME ± amlodipine compared to untreated.
To evaluate the effect of treating established human tumors, once tumors from CFPac-1 cells reached a size of 0.75 cm3, animals were left untreated or dosed with L-NAME. Within days of beginning L-NAME treatments, tumor size was reduced and at the termination of the experiment tumor size was significantly (P<0.05) halved (Fig. 5B). To compare L-NAME with the standard of care therapeutic gemcitabine (3), mice with established tumors were also treated with gemcitabine or a combination of gemcitabine and L-NAME. L-NAME treatments were as effective as gemcitabine at reducing tumor size, but when combined did not provide any further reduction (Fig. 5B).
L-NAME treatment decreases PDAC tumor growth in mice treated with an antihypertensive
The primary effect of L-NAME is elevated blood pressure (Fig. 5C and Ref. 23). Acute L-NAME dosing variably causes transient hypertension in some, but not all healthy human subjects (22). Chronic daily dosing of L-NAME over the course of six weeks caused hypertension and resultant end-organ consequences in rats (23, 39), although this was prevented by a wide spectrum of antihypertensives (39). Therefore, cohorts of four to five mice injected with CFPac-1 cells were untreated or treated with the antihypertensive amlodipine, L-NAME, or both drugs after which ten blood pressure measurements were taken a total of 26 days over the course of 8 weeks, and tumor volume was measured. Amlodipine significantly (P<0.05) reduced systolic blood pressure of L-NAME-treated animals, as previously reported (39), from 141±2.2 mmHg to the normal level of 109±2.7 mmHg (Fig. 5C), but had no effect on the anti-tumor activity of L-NAME, as the size of tumors in the L-NAME and L-NAME+amlodipine cohorts were identical in size, but nevertheless were significantly (P<0.05) smaller compared to the untreated or amlodipine treated mice at the termination of the experiment (Fig. 5D).
eNOS promotes tumorigenesis through effects in the tumor and stroma
KRas signaling can stimulate AKT (4), and AKT can phosphorylate S1177 and activate eNOS, which in turn can elevate the level of activated GTP-bound wild-type HRas and NRas to promote tumor growth (13, 14). In agreement, the PI3K inhibitor LY294002 reduced the level of activated αS1177 phosphorylated eNOS in CFPac-1 cells (Fig. 6A). Similarly, the level of GTP-bound HRas, as detected by affinity capture with the Raf1 RBD followed by immunoblot with an α-HRas antibody, was diminished in tumor cell lines devoid of stromal tissue derived from the pancreas of three eNOS-/- KPC mice compared to tumor cell lines from three control KPC mice (Fig. 6B) and in two CFPac-1 derived tumors from mice treated with L-NAME relative to tumors from two untreated mice (Fig 6C). Interestingly, L-NAME did not inhibit the in vitro proliferation of three different human PDAC cell lines (Supplementary Fig. S3), suggesting a role for eNOS in the tumor stroma as well. In this regard, oncogenic Ras activates eNOS to maintain tumor growth (13), and the first effect observed upon silencing Ras oncogene expression was an increase in apoptotic CD31+ cells (43). Activation of Ras also leads to secretion of pro-angiogenic cytokines (44, 45). Similarly, eNOS is known to play a role in angiogenesis (46, 47). In agreement, CD31 staining from typically five randomly chosen fields of PDAC tumors from five eNOS-/- KPC mice compared to tumors from six control KPC mice was significantly (P<0.05) reduced by one third (Fig. 6D), and Ki67 immunoreactivity from four to five randomly chosen fields from tumors arising in seven mice injected with the CFPac-1 human pancreatic cancer cell line and treated with L-NAME compared to tumors from seven control untreated mice was significantly (P<0.05) reduced by one half (Fig. 6E).
Figure 6. Analysis of phosphorylated eNOS, HRas-GTP, CD31 and Ki67 immunoreactivity in tumorigenic cells upon pharmacologic inhibition or loss of eNOS.
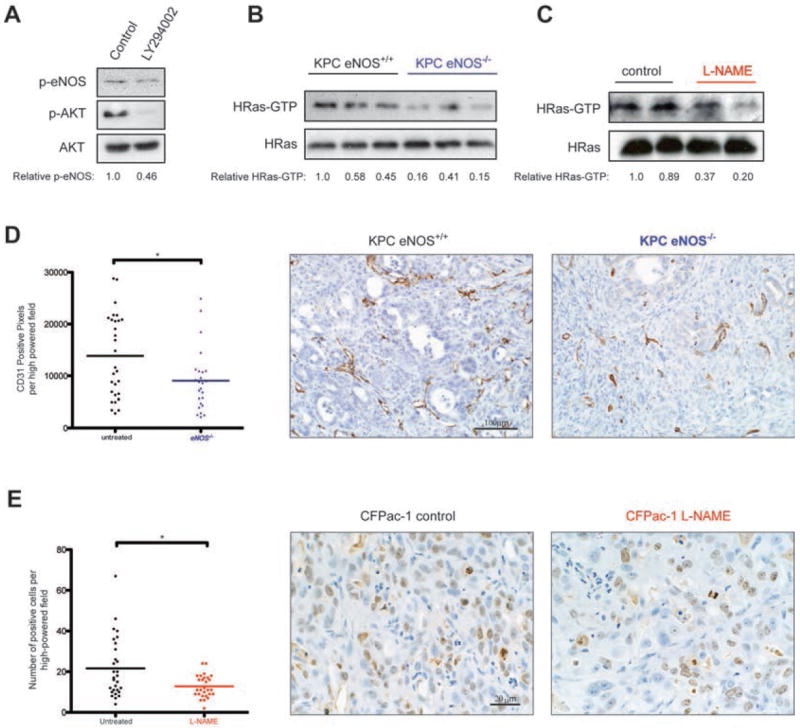
(A) Phospho-eNOS (normalized quantitation shown beneath), phospho-AKT, and AKT levels in CFPac-1 cells treated without (control) and with PI3K inhibitor LY294002. (B,C) HRas-GTP, as assessed by capture with GST-tagged RBD followed by immunoblot with an αHRas antibody, and total HRas levels (normalized quantitation shown beneath) in (B) cell lines established from eNOS+/+ and eNOS-/- KPC tumors and (C) tumors derived from CFPac-1 cells injected into mice treated or untreated with L-NAME for 7 weeks. (D) Representative example and CD31 immunoreactivity per field from typically five random high-power fields from pancreata of eNOS+/+ (n=6) and eNOS-/- (n=5) KPC mice. Bar, mean pixels per high-power field. *P<0.05. (E) Representative example and number of Ki67-positive cells per field from four to five random high-power fields from untreated control (n=7) and L-NAME-treated (n=7) CFPac-1 xenografts. Bar, mean number of cells per high-power field. *P<0.05
Discussion
Loss of eNOS or L-NAME reduced the development of pancreatic lesions, facial papillomas, and vulvar papillomas in the KC mouse model of preinvasive pancreatic cancer. eNOS ablation and L-NAME treatment also trended, but did not reach significance, towards a 34 and 19 day increase in survival in KPC mice, corresponding to a 25% and 13% increase in lifespan compared to that of control KPC mice, respectively. In terms of human cell settings, eNOS was detected in human PDAC samples, although admittedly this was a small sample set, and L-NAME broadly inhibited the tumorigenic growth of human PDAC cell lines. Given the variability in eNOS levels detected in PDAC samples, we cannot rule out that the more general inhibition of pancreatic tumorigenesis observed in multiple animal model systems may be more limited in a clinical setting. Similarly, given that L-NAME trended but did not reach significance towards a 19-day increase in survival in the metastatic KPC mouse model, we also cannot rule out the possibility that the drug may not be effective in the highly aggressive late stage of the disease in humans. With these caveats in mind, these data nevertheless suggest that eNOS is a potential therapeutic target that could be inhibited with the available drug L-NAME for the treatment of PDAC.
KC mice treated daily with L-NAME for roughly ten months had no overt adverse effects, and analysis of all-cause mortality in KPC mice, which would take into account deaths due to adverse drug events, was actually decreased in the L-NAME treatment group compared to untreated controls. In fact, the undesirable effects of L-NAME on blood pressure were ameliorated with a commonly used antihypertensive with no change to the antitumor activity of L-NAME. Indeed, L-NAME has been used in many clinical trials, including those involving normal human subjects (22), suggesting that this drug may have low toxicities in a cancer setting.
Since eNOS knockout and L-NAME treatments exhibited similar effects on tumorigenesis in KC and KPC mice, and since L-NAME inhibits eNOS in vivo, as measured by an increase in blood pressure, the antitumor effects of L-NAME can be attributed, at least in part, to eNOS inhibition. Nevertheless, we cannot rule out the possibility that this may also be a consequence of inhibiting other NOS isoforms. Similarly, the cell type sensitive to eNOS inhibition during the development of pancreatic cancer remains to be determined. NO can be produced both by tumor and stromal cells (24). In regards to the tumor, eNOS is detected in PDAC tumor cells (Fig. 1A), loss of the gene reduced the level of GTP-bound wild-type HRas (Fig. 6B), and knockdown of eNOS in two pancreatic cancer cell lines reduced their tumorigenic potential (13). In regards to the stroma, L-NAME did not decrease the viability of pancreatic cancer cell lines in vitro (Supplementary Fig. S3). Moreover, both Ras and eNOS promote angiogenesis (44-47), eNOS-/- animals are deficient in endothelial progenitor cell mobilization and neovascularization (48), and CD31 and Ki67 staining were reduced upon inhibiting eNOS (Figs. 6D,E). Thus, activation of stromal eNOS, perhaps by paracrine signaling from the tumor or other sources, may also promote tumorigenesis, for example, through effects on angiogenesis. An orthotopic model of pancreatic cancer would help resolve the issue of which tissue eNOS functions, and additionally, in the context of human cells. Regardless of the source of eNOS, the net result of genetic ablation and/or pharmacologic inhibition of eNOS in the tested pancreatic cancer models was a reduction in tumorigenesis.
The inhibition of eNOS may have therapeutic utility in malignancies beyond pancreatic cancer as oncogenic KRas-driven vulvar and facial papillomas responded to L-NAME treatment and exhibited reduced tumor growth in an eNOS-/- background. This effect may extend even beyond KRAS mutation-positive cancers. Specifically, eNOS-/- mice are also resistant to DMBA/TPA chemical carcinogenesis (13) and were recently reported to have prolonged survival in a PDGF-induced glioma mouse model (16). Similarly, peptide-mediated inhibition of eNOS decreases tumor vascular permeability and tumor growth in hepatocarcinoma and lung carcinoma xenograft models (17). Finally, eNOS-generated NO can regulate recruitment of pericytes and stabilization of angiogenic vessels in a xenograft model of murine melanoma (49). Thus, we suggest that eNOS is an attractive target that can be inhibited with the available drug L-NAME, which could potentially be exploited for the treatment of PDAC and perhaps other oncogenic Ras-driven cancers.
Supplementary Material
Acknowledgments
We thank David Kirsch for advice and reagents, Kiyomi Goto, Mariam Wahidi and Tom Ribar for technical assistance.
Grant Support
This work was supported by grants from the NCI (CA123031) and the Edward Spiegel Fund of the Lymphoma Foundation (CMC), the Alexander and Margaret Stewart Trust (SDK and CMC), and the American Foundation of Aging Research (BLL), and a K08 award from the NCI (SDK).
Footnotes
Disclosure of Portential Conflicts of Interest: CMC, BBA and DFK submitted a patent application to inhibit eNOS for the treatment of Ras mutation-positive cancers. CMC and KSB had a collaboration with GlaxoSmithKline to develop eNOS-specific inhibitors.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Ko AH, Tempero MA. Treatment of metastatic pancreatic cancer. J Natl Compr Canc Netw. 2005;3:627–36. doi: 10.6004/jnccn.2005.0036. [DOI] [PubMed] [Google Scholar]
- 4.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 5.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–53. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 6.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 7.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 8.Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–48. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 9.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 10.Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381–92. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 11.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–9. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–76. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 15.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–81. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–52. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gratton JP, Lin MI, Yu J, Weiss ED, Jiang ZL, Fairchild TA, et al. Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell. 2003;4:31–9. doi: 10.1016/s1535-6108(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 18.Cotter G, Kaluski E, Milo O, Blatt A, Salah A, Hendler A, et al. LINCS: L-NAME (a NO synthase inhibitor) in the treatment of refractory cardiogenic shock: a prospective randomized study. Eur Heart J. 2003;24:1287–95. doi: 10.1016/s0195-668x(03)00193-3. [DOI] [PubMed] [Google Scholar]
- 19.Avontuur JA, Buijk SL, Bruining HA. Distribution and metabolism of N(G)-nitro-L-arginine methyl ester in patients with septic shock. Eur J Clin Pharmacol. 1998;54:627–31. doi: 10.1007/s002280050525. [DOI] [PubMed] [Google Scholar]
- 20.Avontuur JA, Tutein Nolthenius RP, Buijk SL, Kanhai KJ, Bruining HA. Effect of L-NAME, an inhibitor of nitric oxide synthesis, on cardiopulmonary function in human septic shock. Chest. 1998;113:1640–6. doi: 10.1378/chest.113.6.1640. [DOI] [PubMed] [Google Scholar]
- 21.Kiehl MG, Ostermann H, Meyer J, Kienast J. Nitric oxide synthase inhibition by L-NAME in leukocytopenic patients with severe septic shock. Intensive Care Med. 1997;23:561–6. doi: 10.1007/s001340050373. [DOI] [PubMed] [Google Scholar]
- 22.Morgan DR, Silke B, Dixon LJ, Allen PB, Hanratty CG, McVeigh GE. Central and peripheral haemodynamic effects of L-NAME infusion in healthy volunteers. Eur J Clin Pharmacol. 2003;59:195–9. doi: 10.1007/s00228-003-0601-7. [DOI] [PubMed] [Google Scholar]
- 23.Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. 1992;90:278–81. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–34. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 25.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seligsohn EE, Bill A. Effects of NG-nitro-L-arginine methyl ester on the cardiovascular system of the anaesthetized rabbit and on the cardiovascular response to thyrotropin-releasing hormone. Br J Pharmacol. 1993;109:1219–25. doi: 10.1111/j.1476-5381.1993.tb13752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Mancera PA, Tuveson DA. Physiological analysis of oncogenic K-ras. Methods Enzymol. 2006;407:676–90. doi: 10.1016/S0076-6879(05)07053-9. [DOI] [PubMed] [Google Scholar]
- 30.Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu G, Liang X, Xie X, Su M, Zhao S. Diverse effects of chronic treatment with losartan, fosinopril, and amlodipine on apoptosis, angiotensin II in the left ventricle of hypertensive rats. Int J Cardiol. 2001;81:123–9. doi: 10.1016/s0167-5273(01)00539-3. discussion 9-30. [DOI] [PubMed] [Google Scholar]
- 32.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–5. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 33.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 34.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Worl J, Wiesand M, Mayer B, Greskotter KR, Neuhuber WL. Neuronal and endothelial nitric oxide synthase immunoreactivity and NADPH-diaphorase staining in rat and human pancreas: influence of fixation. Histochemistry. 1994;102:353–64. doi: 10.1007/BF00268906. [DOI] [PubMed] [Google Scholar]
- 36.Nussler AK, Gansauge S, Gansauge F, Fischer U, Butzer U, Kremsner PG, et al. Overexpression of endothelium-derived nitric oxide synthase isoform 3 in the vasculature of human pancreatic tumor biopsies. Langenbecks Arch Surg. 1998;383:474–80. doi: 10.1007/s004230050163. [DOI] [PubMed] [Google Scholar]
- 37.Gades NM, Ohash A, Mills LD, Rowley MA, Predmore KS, Marler RJ, et al. Spontaneous vulvar papillomas in a colony of mice used for pancreatic cancer research. Comp Med. 2008;58:271–5. [PMC free article] [PubMed] [Google Scholar]
- 38.Mazur PK, Gruner BM, Nakhai H, Sipos B, Zimber-Strobl U, Strobl LJ, et al. Identification of epidermal Pdx1 expression discloses different roles of Notch1 and Notch2 in murine Kras(G12D)-induced skin carcinogenesis in vivo. PLoS One. 2010;5:e13578. doi: 10.1371/journal.pone.0013578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akuzawa N, Nakamura T, Kurashina T, Saito Y, Hoshino J, Sakamoto H, et al. Antihypertensive agents prevent nephrosclerosis and left ventricular hypertrophy induced in rats by prolonged inhibition of nitric oxide synthesis. Am J Hypertens. 1998;11:697–707. doi: 10.1016/s0895-7061(98)00051-x. [DOI] [PubMed] [Google Scholar]
- 40.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–83. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Camp ER, Yang A, Liu W, Fan F, Somcio R, Hicklin DJ, et al. Roles of nitric oxide synthase inhibition and vascular endothelial growth factor receptor-2 inhibition on vascular morphology and function in an in vivo model of pancreatic cancer. Clin Cancer Res. 2006;12:2628–33. doi: 10.1158/1078-0432.CCR-05-2257. [DOI] [PubMed] [Google Scholar]
- 42.Mohamad NA, Cricco GP, Sambuco LA, Croci M, Medina VA, Gutierrez AS, et al. Aminoguanidine impedes human pancreatic tumor growth and metastasis development in nude mice. World J Gastroenterol. 2009;15:1065–71. doi: 10.3748/wjg.15.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang Y, Kim M, Carrasco D, Kung AL, Chin L, Weissleder R. In vivo assessment of RAS-dependent maintenance of tumor angiogenesis by real-time magnetic resonance imaging. Cancer Res. 2005;65:8324–30. doi: 10.1158/0008-5472.CAN-05-0027. [DOI] [PubMed] [Google Scholar]
- 44.Breier G, Blum S, Peli J, Groot M, Wild C, Risau W, et al. Transforming growth factor-beta and Ras regulate the VEGF/VEGF-receptor system during tumor angiogenesis. Int J Cancer. 2002;97:142–8. doi: 10.1002/ijc.1599. [DOI] [PubMed] [Google Scholar]
- 45.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–58. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 46.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–9. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee PC, Salyapongse AN, Bragdon GA, Shears LL, 2nd, Watkins SC, Edington HD, et al. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol. 1999;277:H1600–8. doi: 10.1152/ajpheart.1999.277.4.H1600. [DOI] [PubMed] [Google Scholar]
- 48.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–6. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 49.Kashiwagi S, Izumi Y, Gohongi T, Demou ZN, Xu L, Huang PL, et al. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest. 2005;115:1816–27. doi: 10.1172/JCI24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.