

Abstract
Methylmalonic aciduria (MMA) cblB type is caused by mutations in the MMAB gene. This encodes the enzyme ATP:cob(I)alamin adenosyltransferase (ATR), which converts reduced cob(I)alamin to an active adenosylcobalamin cofactor. We recently reported the presence of destabilizing pathogenic mutations that retain some residual ATR activity. The aim of the present study was to seek pharmacological chaperones as a tailored therapy for stabilizing the ATR protein. High-throughput ligand screening of over 2000 compounds was performed; six were found to enhance the thermal stability of purified recombinant ATR. Further studies using a well-established bacterial system in which the recombinant ATR protein was expressed in the presence of these six compounds, showed them all to increase the stability of the wild-type ATR and the p.Ile96Thr mutant proteins. Compound V (N-{[(4-chlorophenyl)carbamothioyl]amino}-2-phenylacetamide) significantly increased this stability and did not act as an inhibitor of the purified protein. Importantly, compound V increased the activity of ATR in patient-derived fibroblasts harboring the destabilizing p.Ile96Thr mutation in a hemizygous state to within control range. When cobalamin was coadministrated with compound V, mutant ATR activity further improved. Oral administration of low doses of compound V to C57BL/6J mice for 12 days, led to increase in steady-state levels of ATR protein in liver and brain (disease-relevant organs). These results hold promise for the clinical use of pharmacological chaperones in MMA cblB type patients harboring chaperone-responsive mutations.
INTRODUCTION
Cobalamin, or vitamin B12, is an essential micronutrient that mammals must obtain from their diet. It is then processed via a complex pathway for the biosynthesis of methylcobalamin and adenosylcobalamin (AdoCbl), which, respectively, serve as cofactors of the cytosolic enzyme methionine synthase (MS, EC_2.1.1.13) and the mitochondrial enzyme methylmalonyl-CoA mutase (MUT, EC_5.4.99.2). The study of inborn defects of cobalamin metabolism has revealed nine complementation groups: cblA to cblJ and mut (1–4). In particular, defects in the mitochondrial enzyme methylmalonyl-CoA mutase (mut group) or in the synthesis of the AdoCbl cofactor (complementation groups cblA, cblB or cblD variant2) lead to isolated methylmalonic aciduria (MMA, MIM 251 000) (5).
The synthesis and transfer of AdoCbl to MUT involves three proteins, ATP:adenosylcobalamin transferase (ATR, EC_2.5.1.17), the cblA protein and the cblD protein (5–7). ATR is a homotrimer encoded by the MMAB gene (8,9). MUT is responsible for the isomerization of l-methylmalonyl-CoA into succinyl-CoA, which can enter the Krebs cycle, and MMA cblB type is caused by mutations in the gene encoding ATR (10).
MMA cblB patients suffer either a severe early-onset form of the disease, with neonatal ketoacidosis, lethargy, failure to thrive and encephalopathy, or a milder late-onset form usually diagnosed during infancy that has a less serious neurological outcome. While there is currently no cure for the disease, therapeutic options, including the dietary restriction of substrates, aim to reduce methylmalonic acid production and improve the clinical outcome. Pharmacological cobalamin supplementation is part of the usual treatment, although only 40% of patients show any biochemical response to it, and in many cases neurological manifestations persist. In those that do respond, significant improvements in clinical condition, growth and metabolic variables have been reported, although the levels of toxic metabolites never normalize (1).
Efforts are now being directed towards exploring alternative therapies based on identifying new targets in the context of personalized medicine. Understanding the molecular mechanisms of pathogenesis underlying genetic defects can help guide the design and development of new therapeutic approaches. Several studies have shown that many human diseases are caused by loss-of-function mutations that impair the correct folding of affected proteins (11–13). Over the past decade, pharmacological chaperones have emerged as novel therapeutic tools for rescuing misfolded proteins by stimulating and preserving their correct folding (14,15). Pharmacological chaperones are small, cell-permeable molecules that specifically bind and stabilize target proteins. Pharmacological chaperones have been investigated as a potential treatment for many genetic disorders that result from misfolded and/or unstable proteins, and proof-of-concept studies have been described for several human diseases such as lysosomal disorders, phenylketonuria and cystic fibrosis (12,16–19).
In a previous study, we reported the characterization of two mutations in the MMAB gene, p.Ile96Thr and p.Arg191Trp, both of which are associated with reduced ATR stability and residual activity (20). The aim of the present work was to identify compounds with pharmacological chaperone potential for ATR. We demonstrate that N-{[(4-chlorophenyl)carbamothioyl]amino}-2-phenylacetamide, here after referred to as compound V, enhanced the stability of wild-type and p.Ile96Thr mutant ATR. It also increased the residual activity of ATR in patient-derived fibroblasts harboring the latter mutation. The effect of compound V on the stabilization of recombinant wild-type and mutant ATR both as isolated recombinant protein and in patient-derived fibroblasts is further increased when tested simultaneously with cobalamin. Finally, oral loading of compound V in wild-type mice resulted in an increase in ATR stability in the liver and brain (disease-relevant organs).
RESULTS
High-throughput screening for pharmacological chaperones
A subset of 2000 compounds from the MyriaScreen Diversity Collection was screened by differential scanning fluorimetry (DSF). Data analysis revealed a group of six compounds that shifted the Tm of ATR between 6.0 and 17°C. These six compounds (I–VI) were selected for further studies (Figs 1 and 2).
Figure 1.

Differential scanning fluorimetry profiles for six candidate pharmacological chaperones. Thermal denaturation profiles of purified wild-type ATR (0.086 mg/ml) (fluorescence at 610 nm vs. temperature [°C]). In grey, curve for ATR incubated with DMSO (final concentration 2%); in black, purified ATR incubated with 0.04 mg/ml of compounds I–VI. The melting temperature (Tm) was taken as the temperature at which the maximum fluorescence was achieved; ΔTm was calculated as the difference between the Tm in the presence of a ligand and Tm in the presence of DMSO. The lower figure for each graph represents the data without scaling.
Figure 2.

Chemical structures and IUPAC names of the selected compounds. Six compounds, named I to VI, were found to stabilize ATR protein in a high-throughput screening of 2000 compounds.
ATR stability and activity in the presence of hit compounds
To determine whether compounds I–VI acted as pharmacological chaperones, their effect on the stability of wild-type ATR and the p.IleI96Thr mutant was studied. Table 1 summarizes the quantification of ATR by western blot analysis of the bacterial lysates grown in the presence of the potential chaperones. Compounds I–VI increased or tended to increase the steady-state levels of expressed wild-type and/or mutant ATR (Table 1) indicating an increase in ATR stability. Compound V (80 µm) had the largest effect on the mutant (Table 1). Compound VI was the most effective in stabilizing the wild-type type ATR (Table 1). Based on these results, compounds V and VI were selected for further studies.
Table 1.
Thermal stabilization in the presence of compounds I–VI from DSF screening, and relative stability of wild-type and p.Ile96Thr ATR in the presence of compounds as measured by quantitative immunoblottinga
| ΔTm (°C) | Wild-type |
p.Ile96Thr |
|||
|---|---|---|---|---|---|
| 40 µm | 80 µm | 40 µm | 80 µm | ||
| DMSOb | — | 1.0 | 1.0 | 1.0 | 1.0 |
| Compound I | 16.1 | 1.8 ± 0.4 (*) | 1.6 ± 1.2 | 0.8 ± 0.3 | 2.3 ± 1.3 |
| Compound II | 5.4 | 2 ± 1 | ND | 1.3 ± 0.1 | ND |
| Compound III | 8.5 | 0.7 | 0.9 ± 0.4 | 1.9 ± 0.5 | 2.1 ± 0.6(**) |
| Compound IV | 7.4 | 1.0 ± 0.2 | 3.4 ± 0.4(****) | 2.0 ± 0.6 | 2.2 ± 0.1(***) |
| Compound V | 13.6 | ND | 1.4 ± 1.0 | 1.6 ± 1.2 | 3 ± 1 (**) |
| Compound VI | 15.7 | 4 ± 2 (*) | 6.2 ± 0.5(****) | 2.1 ± 0.1 (****) | 1.7 ± 0.2(**) |
Bold figures highlight the most important effect of compounds V and VI.
ND, not determined.
aData obtained with bacterial extracts prepared from cultures grown in the presence of 40 or 80 µm of compounds I–VI, as described in Materials and Methods.
bData represent the mean ± SD of at least two independent determinations. In all cases the appropriate DMSO control was used and taken as the reference value (stability = 1) for the bacterial cultures grown in the presence of the compounds.
*P < 0.05.
**P < 0.01.
***P < 0.005.
****P < 0.001.
The effect of compounds V and VI was then analyzed by measuring the activity of the lysates of bacterial cultures expressing wild-type and mutants (p.Ile96Thr and p.Arg191Trp), grown in the presence of 100 µm compounds. While no effect could be observed for compound VI, in either wild-type or mutant extracts, the activity in the lysates of bacterial cultures grown with compound V increased 1.5-fold for wild-type and 8-fold for the destabilizing mutation p.Ile96Thr. For the p.Arg191Trp mutant, which is also unstable, no quantifiable enzyme activity could be measured in the lysates of cultures grown without compound V, but some activity could be detected in its presence (data not shown).
A clear stabilizing effect of the p.Ile96Thr by compound V was observed in the time-dependent quantification of mutant protein in the bacterial lysates (Fig. 3A and B). Similar time-course experiments were performed with lysates from bacterial cultures grown in the presence of 1 µg/ml (0.7 µm) OHCbl, which did not have any stabilizing effect on the expressed wild-type and only slight on mutant protein (Fig. 3A and B). Interestingly, the combined addition of compound V and OHCbl showed a slightly increased stabilizing effect of the mutant p.Ile96Thr compared with compound V alone (Fig. 3A and B).
Figure 3.

Effect of compounds V, VI and cobalamin on ATR stability and activity. (A) Effect of compound V on p.Ile96Thr ATR protein stability. Bacteria expressing wild-type or p.Ile96Thr mutant ATR were incubated with 80 µm compound V (final DMSO concentration 0.2%) or the equivalent amount of the vehicle (0.2% DMSO) or with 1 µg/ml (0.7 µm) of OHCbl or both. Crude cell extracts from cultures expressing wild-type or p.Ile96Thr ATR were incubated at 37°C, and aliquots were removed at different times. The aliquots were loaded onto SDS–PAGE gels and the protein was immunodetected by western blotting. Proteins were quantified by laser densitometry as the percentage density of each protein relative to its density at time 0. Compound-treated samples were compared with the DMSO-treated sample. (B) Quantification of the effect of compound V and/or OHCbl on stability of wild-type ATR and mutant p.Ile96Thr ATR. (C) Concentration-dependent stabilization of ATR by compound V and effect of OHCbl and ATP. The stabilization of ATR in the presence of compound V (0–200 µm) in the absence of OHcbl (black solid line, circles) and in the presence of OHCbl at 3 µm (dark grey solid line, triangles) and 12 µm (light grey solid line, squares) or 40 µm ATP (dotted line, squares). See main text for estimated Kd values for compound V binding. (D) Specific activity was measured using purified wild-type ATR from bacteria with 40 or 80 µm of either compound V or VI or the equivalent volume of DMSO (final DMSO concentration 0.4 or 0.8%). *P < 0.05, **P < 0.01.
The combined effect of compound V and OHCbl was further analyzed using isolated recombinant wild-type ATR protein. We first measured the concentration-dependent thermal stabilization of ATR by OHcbl using DSF. Concentrations up to 25 µm OHCbl did not affect the thermal stability of the protein, and similar unfolding curves as for the controls (shown in Fig. 1) were obtained. But higher concentrations of OHCbl destabilized ATR slightly (1.5°C) and gave poor curves due to quenching of fluorescence. Titration with compound V (0–200 µm) in the absence of OHCbl showed that ATR is thermally stabilized by the compound at concentrations approximately >2 µm (Fig. 3C), and by analyzing the effect of the ligand on the Tm of ATR as described by Cooper and McAuley-Hecht (21), we estimated a Kd = 7.4 ± 0.4 µm for compound V. In the presence of 0.75 µm OHCbl, this Kd was not affected, but higher concentrations of the substrate increased the affinity of compound V (Kd = 5.4 ± 0.5 µm with 3 µm OHCbl and Kd = 5.5 ± 0.4 with 12 µm), resulting in increased stabilization at similar concentrations of the compound (Fig. 3C). Comparatively, ATP, which had a negligible effect on the Tm for ATR denaturation up to 200 µm, did not have any effect on the concentration-dependent stabilization by compound V (Fig. 3C). The affinity of ATR for compound V appears comparable to the apparent Km values for ATP (6.4 ± 0.4 µm) and 3.7 ± 0.5 µm for cobalamin (22).
Given that pharmacological chaperones can act as enzyme inhibitors if they resemble the natural substrates (23), we tested the effect of compound V on the activity of recombinant ATR. The specific activity of purified recombinant human ATR was 0.23 ± 0.04 U/mg at the standard assay conditions. In the presence of 40 µm of compound V, the activity remained comparable to that of control samples (0.27 ± 0.02 U/mg) (Fig. 3D). Comparatively, with 40 µm of compound VI, another of the initial hit compounds, the specific activity decreased to 0.09 ± 0.05 U/mg, suggesting that compound VI, but not V might be inhibitory. At concentrations of 80 µm, no inhibitory effect was again obtained for compound V (specific activity of 0.30 ± 0.14 U/mg). For compound VI at 80 µm, the inhibition observed at lower concentration appears reversed, suggesting a complex pattern of stability and activity effects for this inhibitory potential pharmacological chaperone.
Given that compound V showed a clear stabilizing effect on wild-type and mutant ATR and did not seem to be inhibitory, it was selected for further testing on cellular and animal models.
Recovery of residual activity in a cellular disease model
The therapeutic potential of compound V was further evaluated by its ability to increase total cellular ATR activity using patient-derived transformed fibroblasts. Its effect at 40 and 80 µm on the propionate oxidation metabolic pathway was assessed using immortalized dermal fibroblasts from a patient bearing the mutations p.Ile96Thr and p.Ser174fs (20) incubated for 48 and 72 h with the compound. [14C]-propionate uptake was quantified as an indirect measurement of ATR activity. Incubation with compound V at 80 µm for 48 h induced a slight increase in the residual activity (data not shown), while 72 h of treatment significantly increased ATR activity (5.4-fold). Indeed, it reached control range levels. An effect due to DMSO was ruled out since residual activity in the presence or absence of DMSO was comparable. In the same way as described earlier, the effect of compound V and OHCbl on the activity and stability of ATR was assessed. According to the results obtained with the patient-derived fibroblasts, OHCbl increased the incorporation of 14C propionate (3.5-fold). 14C propionate incorporation was further stimulated (∼17-fold) when compound V and OHCbl (1 µg/ml) were simultaneously present (Fig. 4A). The levels of mitochondrial ATR protein were also slightly increased when both compound V and OHcbl were added (Fig. 4B).
Figure 4.

Effect of compound V and cobalamin in a cellular disease model. Immortalized patient (harboring the mutations p.Ile96Thr/p.Ser174fs) and healthy-derived fibroblasts were incubated in the presence of 80 µm of compound V or the equivalent volume of DMSO (0.1 or 0.2%), and in the presence of 1 µg/ml of cobalamin. After 72 h, ATR activity was indirectly measured via [14C]-propionate incorporation into acid-precipitable material in intact cells grown in basal medium. (B) Western blot analysis for ATR protein using mitochondria isolated from healthy and patient-derived fibroblasts. The cells were treated for 72 h with 80 µm of compound V or 1 µg/ml of OHCbl or both substances. Mitochondrial Hsp60 was used as a protein control loading.
Supplementation with compound V to mice: effect on liver and brain ATR
In addition to in vitro and ex vivo studies, experiments with whole organisms are pivotal for determining a drug's pharmacokinetics to assess whether it is adequately absorbed, distributed and excreted. To this end, the effect of compound V on the stability of endogenous ATR was examined in a wild-type mouse strain since a cblB mouse model is currently unavailable. Two doses (0.25 and 5 mg/kg/day) of compound V were administered orally every day for 12 days. No secondary effects of compound exposure (such as weight loss, automutilation, tumors, abnormal movements, etc.) were observed. The results showed an increase in the amount of immunoquantified ATR at the 5 mg/kg/day dose in liver and brain, two disease-relevant organs (Fig. 5), supporting the stabilizing effect of compound V in vivo.
Figure 5.

Effect of compound V in wild-type mice. Two groups of six wild-type C57BL/6 mice were orally treated for 12 days with 0.25 mg/kg/day or 5 mg/kg/day of compound V. The control animals (five mice) were treated with 10% DMSO in a 9% glucose solution, i.e. the vehicle for compound V administration. After treatment, whole liver and whole brain extracts were prepared and ATR protein levels detected by immunoblotting (represented as the mean ± SD for each group). *P < 0.05.
Molecular docking
The molecular docking of compound V (24,25) resulted in 49 clusters being identified in eight different pockets of the protein. The top-score cluster had a significantly lower FullFitness (26) than the others. This binding site is formed by residues at the C-terminal end of each subunit in the trimeric ATR (residues 228–240), and the loop 165–175, close to residue Phe170, which has been reported as a potential candidate residue for binding the hydrophobic surfaces of cobalamin (9). The site is distant from the ATP binding site (Fig. 6A). In this conformation, compound V could be able to form three hydrogen bonds with the backbone amide of Ala169 and the carboxyl acid side chain of Glu235, making it a favorable binding site (Fig. 6B). The location of this binding pocket is consistent with the results obtained by DSF on the synergistic stabilizing effect of OHCbl and compound V and the absence of effect by ATP (Fig. 3C). Thus, binding of OHCbl and compound V to their respective adjacent sites might lead to a greater packing and conformational stability of this part of the protein, and notably the loop 165–175, than obtained by compound V binding alone.
Figure 6.
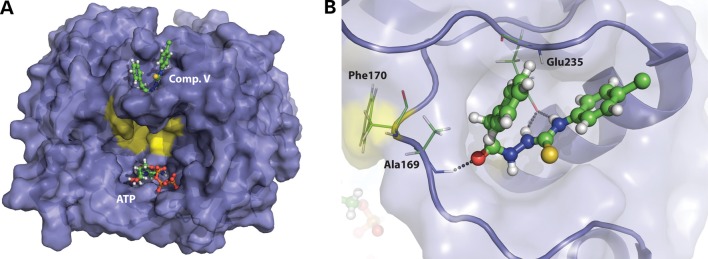
Proposed binding site of compound V. (A) Overview of ATR (PDB ID 2IDX) showing the location of the proposed binding site for compound V in one of the protein subunits (top score position from SwissDock with highest FullFitness) (26), the location of ATP, and the residues proposed to interact with cobalamin (yellow). (B) A detailed view of the proposed binding site of compound V (ball-and-stick). The residues Ala169 and Glu235 are shown with sticks and the potential hydrogen bonds to compound V with dotted lines. Phe170, which is implicated in cobalamin binding, is also shown.
DISCUSSION
Research into understanding how missense mutations trigger protein degradation is a growing field, the goal of which is to find new therapeutic options for protein misfolding diseases (27). Particular efforts have been directed towards the use of drugs that increase the amount of native-state protein (12,15) through increasing its stability and reducing the degradation of mutated but biologically active protein. Such research could lead to new orphan drugs for treating rare diseases within the context of personalized medicine (12,17,18,28–30). In the present study, we searched for compounds that enhance ATR enzyme stability, opening up the possibility of a new therapeutic strategy for the treatment of MMA cblB, which envisions a tailored therapy for patients harboring destabilizing mutations that could be used in combination with MMA conventional treatment.
Combinations of in vitro, ex vivo and in vivo approaches were used successfully in the search for compounds capable of stabilizing the ATR protein. DSF-based high-throughput screening (HTS) identified compounds that stabilized purified recombinant wild-type ATR and the p.Ile96Thr mutant to different degrees. One of these compounds, compound V, also stabilized the p.Ile96Thr protein ex vivo, and was associated with a dose-dependent increase in ATR activity in the patient-derived cell line bearing the mutations p.Ile96Thr/-p.Ser174fs (20). No inhibitory effects of the compounds on the purified ATR protein were observed. A broad concentration window for compound V administration could be used for increasing a patient ATR activity up to the therapeutic range and is therefore available for a future therapeutic trial in selected cblB patients (18,31).
The promising preliminary results obtained with compound V with the rare destabilizing mutations p.Ile96Thr and p.Arg191Trp (20) open up the possibility of analyzing the response of other misfolding mutations of ATR (9,31,32). An in vitro evaluation of compound V effects is needed to determine which patients it might be beneficial for.
The efficacies of the potential pharmacological chaperones found by HTS were varied (Table 1) and it is possible that their co-administration might lead to synergistic effects, increasing the residual activity of mutant ATR, and combinations of the hit compounds could be tested for each mutation background. While the threshold for clinical efficiency in MMA is unknown, small increases in residual activity (5–10%) might be sufficient to attenuate a severe clinical phenotype (17,18,32). The synergistic effect of OHCbl and compound V on the activity and stability of ATR is noteworthy. The results suggest that vitamin B12 administrated alone is not a natural chaperone; no increase in protein was detected in any experiment, and the vitamin did not cause any increase in the Tm of recombinant wild-type ATR, as seen by DSF. Nevertheless, wild-type ATR is indeed stabilized in the presence of both substances, as well as the mutant enzyme in the bacterial and cellular model. The modeled structure of compound V bound to ATR, adjacent to the proposed cobalamine site (Fig. 6), appears to support the synergistic effect of both substances.
The present study suggests a possible clinical application for pharmacological chaperones in the treatment of MMA cblB type. However, putative toxic effects of compound V need to be assessed in an appropriate cblB animal model (33,34). The proof-of-concept work performed in the present study made use of wild-type mice since no available ATR-deficient mouse model carrying missense alleles was available. The results obtained in these mice in vivo suggest that compound V is efficiently absorbed and transported to tissues that are affected in MMA and that it crosses the cellular and mitochondrial membranes and increases ATR stability at a relatively low dose (5 mg/kg/day).
The ultimate goal in MMA treatment research is to prevent, or slow, the development of symptoms of the disease, including metabolic instability, strokes and kidney failure. Currently there is some controversy regarding the source of neurological dysfunction: it might be caused by methylmalonic acid produced in the neurons and/or glia in brain, or by it being made elsewhere in the body and being transported across the blood–brain barrier (BBB) (35). If the brain is responsible for its production, a pharmacological chaperone would have to cross the BBB to be effective. The results obtained with mouse brain extracts are very encouraging since they suggest that compound V could cross the BBB.
In conclusion, the present work identifies a potential pharmacological chaperone (compound V) with in vitro efficacy in patient-derived fibroblasts carrying the p.Ile96Thr mutant protein and in wild-type mice. It is therefore a good candidate for therapeutic trials in selected cblB patients. Further, the excellent effects of cobalamin—reported over decades—in the treatment of cblB MMA patients might be improved by co-administration of compound V. Compound V might also be subjected to chemical modification to enhance its effectiveness; this warrants further investigation.
MATERIALS AND METHODS
Compounds tested for pharmacological chaperone potential
The library of small compounds tested in this study was the commercially available MyriaScreen Diversity Collection (Sigma-Aldrich, St Louis, MO, USA), where the compounds are dissolved to a final concentration of 2 mg/ml in 100% DMSO. The hit compounds from the screening were consecutively ordered from Sigma-Aldrich, prepared at concentrations of 10 mm in 100% DMSO and stored at −20°C, and their chemical structure confirmed by HPLC-MS/MS and NMR spectroscopy.
All experiments presented in this study were performed by the addition of compound dissolved in DMSO to the different assays and parallel control experiments were carried out maintaining the same concentration of DMSO (but without compound).
ATR expression, preparation of bacterial crude extracts and protein purification
Human ATR was expressed in Escherichia coli (BL21Star™ DE3 One Shot Cells) transformed with the NL173 plasmid. This plasmid, which is based on the pET41a vector, encoded either wild-type or mutant (p.Ile96Thr or p.Arg191Trp) ATR lacking the mitochondrial targeting sequence (20). Protein expression was induced with 1 mm IPTG; 5 h after induction the cells were harvested by centrifugation (4°C, 6000 rpm for 10 min) and frozen at −70°C until needed. Cell pellets were resuspended in 50 mm potassium phosphate buffer pH 8, 100 mm NaCl, 1 mm DTT, 1× Complete Mini, EDTA-free Protease Inhibitor Cocktail (Roche Applied Sciences, Indianapolis, IN, USA), 1 mm EDTA, 0.05 mg/ml DNAse and 0.3 mg/ml lysozyme. After 1 h, the cell suspension was sonicated and centrifuged (4°C, 13 000 rpm for 15 min) to obtain the crude soluble cell extract, which was diluted to 1 mg/ml. This crude extract was used in protein stabilization studies and in crude bacterial extract activity assays. For further purification of ATR, the extract was subjected to ammonium sulfate precipitation and subsequent anion-exchange chromatography using a HiTrap Sepharose Q column (GE Healthcare, Buckinghamshire, UK) as previously described (36). In all purification steps, the protein concentration was determined by the Bradford assay (Bio-Rad Laboratories, Munchen, Germany). Fractions eluted from the Sepharose Q column were subjected to SDS–PAGE to identify those enriched in ATR. Purified ATR was centrifuged at 10 000g for 10 min at 4°C to precipitate aggregates, and then loaded onto a Superdex 200 HiLoad 16/60 size-exclusion chromatography column (GE Healthcare). The elution peak corresponding to the trimeric ATR form was recovered and used for further experiments. Pure protein concentration was estimated by measuring the absorbance at 280 nm in a Nanodrop spectrophotometer (Thermo Scientific) using the theoretical molar extinction coefficient (16 305 M−1 cm−1) estimated from the amino acid composition for ATR.
High-throughput screening for pharmacological chaperones
Ligand-induced ATR stabilization was assessed by an HTS protocol using DSF (37), monitoring the thermal denaturation of purified ATR in the presence of the extrinsic fluorescent probe SYPRO Orange (Sigma-Aldrich). Protein solutions containing 0.086 mg/ml of ATR (3.2 μm ATR subunit) in 20 mm Na-Hepes, pH 7.0, 200 mm NaCl and 5× SYPRO Orange, were dispensed (49 μl) into 96-well PCR plates (LightCycler480 Multiwell Plate 96, Roche). One microliter of each potential chaperone compound dissolved in DMSO was added to each well to produce final concentrations of 0.04 mg/ml compound and 2% DMSO. Plates were incubated at room temperature for 30 min before loading into a Light Cycler 480 (Roche Applied Science, Indianapolis, IN, USA) for thermal denaturation. Control experiments with 2% DMSO were routinely performed. Unfolding curves were registered from 20 to 99°C, at a scan rate of 2°C/min. The increase in SYPRO Orange fluorescence intensity associated with protein unfolding (λexcitation = 465 nm, λemission = 610 nm) was monitored as a measure of thermal denaturation. Unfolding curves were recorded from 20 to 95°C, at a scan rate of 2°C/min. The experimental unfolding curves were smoothed, normalized and analyzed using in-house software. The midpoint melting temperature (Tm) was calculated as the temperature at which half the protein is in the unfolded state. A compound qualified as a ‘hit’ if it increased the Tm of the protein by ≥6°C.
The concentration-dependent stabilization of ATR 0.15 mg/ml (6.35 µm ATR subunit) by ATP and hydroxocobalamin (OHCbl) (0–200 µm) was carried out using DSF at otherwise similar sample preparation, pre-incubation and scanning conditions as in the HTS protocol. Following this experiment, thermal stabilization titrations with compound V (0–200 µm) were carried out in the presence or absence of either 40 µm ATP or 0.75, 3 and 12 µm of OHCbl.
Effect of the compounds on ATR stability in a prokaryotic expression system
ATR stability was assessed as previously described (20). Basically, ATR expression was induced with 1 mm IPTG in 10 ml E. coli LB cultures for 5 h in the presence of 40 or 80 µm compound dissolved in DMSO (final concentration of DMSO 0.1 or 0.2%, respectively) or the corresponding concentration of DMSO. To determine a possible synergistic effect of compound V and OHCbl on ATR stability, the same experiment was performed in the presence of 1 µg/ml (0.7 µm), of OHcbl a concentration commonly used to stimulate cells to incorporate 14C propionate. Additionally, E. coli were cultured for 5 h in the presence of 80 µm of compound V dissolved in DMSO added together with OHCbl dissolved in water. The crude extracts, prepared as described earlier, were diluted to 1 mg protein/ml. Aliquots of equal amounts of total protein were incubated at 37°C, and equal sample volumes were removed at different times for immediate analysis by SDS–PAGE or frozen until gel loading.
Enzyme activity assays
ATR activity was measured as described by Johnson et al. (38) with minor modifications. Briefly, 1 ml of reaction mixture (200 mm Tris–HCl pH 8.0, 1.6 mm KH2PO4, 2.8 mm MgCl2 and 100 mm KCl) was dispensed into cuvettes and kept at 37°C. Specific activity was measured with 40 µm ATP and 60 μm OHCbl (Sigma-Aldrich). In indicated experiments, 0–100 µm of compound V or VI were added to the reaction mixture. After titanium (III) citrate (30 µL) was added, the reaction was started by the addition of 10 µg of purified wild-type ATR. Alternatively, 50–100 µg of crude extracts from bacterial cultures expressing wild-type, p.Ile96Thr or p.Arg191Trp, grown in the presence of 0–100 µm of compound V or VI, were used to start the reaction. Anaerobic procedures were followed in the preparation of the reaction mix, the protein solution, compound solutions and the titanium (III) citrate solution. The reduction in absorbance at 388 nm was monitored to calculate the concentration of AdoCbl, with ΔA388 taken as 24.9 cm−1 mm−1, and specific activity was expressed as µmol of product generated per minute and milligram of protein (U/mg). Experiments were performed in triplicate.
Biochemical data on the p.Ile96Thr-bearing patient and biochemical chaperone analysis in patient fibroblasts
The patient with the genotype c.287T>C/c.584G>A (p.Ile96Thr/p.Ser174fs) has been described earlier (20). This patient was placed under protein restriction, carnitine supplementation and oral OHCbl administration (5 ml/day). During therapy, the patient showed >1000 mmol/mol creatinine, plasma C3 levels of ∼10 µm and normal plasma odd-chain fatty-acid levels (<0.4%). Urine methylmalonic acid varied (<200–800 mmol/mol creatinine). A dermal fibroblast cell line from this patient and healthy control fibroblasts were stably transformed with pBabe retroviral vector (kindly provided by Dr J.A. Enriquez, Centro Nacional de Enfermedades Cardiovasculares, Madrid). 5 × 105 transformed cells were plated and grown at 37°C for 24 h in MEM medium supplemented with 2 mm glutamine, 10% fetal bovine serum and antibiotics. After treatment, the culture medium was discarded and replaced with fresh medium containing 40 or 80 µm of compound V for 48 or 72 h, or the equivalent volume of DMSO for controls. After this time, cell lines were grown for 18 h in the presence of [14C]-propionate, and were harvested by trypsinization, followed by centrifugation, and were used to determine ATR activity, which was indirectly measured by [14C]-propionate incorporation into acid-precipitable material in intact cells grown in basal medium (39). Assays were performed in triplicate and results were examined statistically using an F-test analysis of variance followed by a one-tailed, paired t-test. Significance was set at P < 0.05.
Alternatively, mitochondrial isolation from cell pellets was performed using anti-TOM22 MicroBeads (Mitochondrial Isolation Kit, Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, 8.0 × 105 cells from a healthy control and patient immortalized fibroblasts were seeded in T75 flasks and incubated overnight at 37°C, and then for 72 h with 80 µm of compound V, 1 µg/ml of OHCbl or both. The cells were harvested by trypsinization and the mitochondria isolated following the manufacturer’s protocol. Mitochondrial pellets were resuspended in 50 µL of storage buffer.
Animal studies
Seventeen female 12-week-old wild-type mice (C57BL/6J) were purchased from Harlan Laboratories (Harlan Laboratories Inc., Indianapolis, IN, USA). These were housed under standard conditions in compliance with European Community Guidelines (Directive 86/609/EEC) at the Centro de Biología Molecular Severo Ochoa, and maintained at a temperature of 22 ± 1°C under a 12 h/12 h light/dark cycle. All had ad libitum access to food and water and were handled in strict accordance with national and institutional animal welfare guidelines. Two groups of six animals (23–25 g) were orally loaded with a 25 µl suspension containing either 0.25 or 5 mg/ml/day of compound V in 10% DMSO plus 9% glucose for 12 days. The control group was treated only with the 10% DMSO and 9% glucose. Approximately, 30 min after the last dose, the animals were sacrificed using CO2. The livers and brains were rapidly removed and divided into two equal parts, one of which was frozen in liquid nitrogen for further studies. Fresh liver extracts were prepared in phosphate buffered saline with protease inhibitors; fresh brain extracts were prepared in RIPPA buffer [50 mm Tris–HCl, pH 7.4, 1% Triton, 150 mm NaCl, 1 mm EDTA, 1 mm NaF, 1 mm Na3VO4, 1× Complete Mini EDTA-free Protease Inhibitor Cocktail (Roche Applied Sciences) and 0.25% Na-deoxycholate]. These samples were homogenized using a Tissue Lyser II (QIAGEN, Hilden, Germany) and the extracts clarified by centrifugation at 13 000 rpm for 30 min at 4°C. The supernatants were stored at −70°C prior to SDS–PAGE and immunoblotting analyses.
SDS–PAGE and immunoblotting
Samples were loaded onto pre-cast polyacrylamide NuPAGE SDS–PAGE Bis–Tris 10% gels in the case of bacterial crude extract or 4–12% gels for the murine tissue extracts (75 µg of liver or 150 µg of total brain extract) and separated using NuPAGE SDS MES running buffer (Life Technologies, Grand Island, NY, USA). Proteins were transferred to a nitrocellulose transfer membrane using the iBLOT gel Device System (Life Technologies). Poinceau staining was used to monitor equal protein loading. Immunodetection was performed using commercially available anti-ATR antibodies (ProteinTech Group, Inc., Chicago, IL, USA) as primary antibodies (diluted 1:1000 for the bacterial crude extracts, and 1:500 for the mouse tissue extracts). The secondary antibody used was conjugated goat-anti-mouse IgG-horseradish peroxidase (1:10 000 for bacterial extracts and 1:5000 for mouse tissue extracts) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The mouse tissue extract membranes were also immune stained with a polyclonal antibody against mitochondrial Hsp60 (1:10 000). Signals were visualized using the Enhanced Chemiluminescence System (GE Healthcare, Munich, Germany) and the relative amount of protein determined using a GS-800 calibrated densitometer running Quantity One software (BioRad, Hercules, CA, USA). The prokaryotic stability experiments were performed in triplicate and were statistically examined using the F-test analysis of variance followed by a one-tailed paired t-test. Significance was set at P < 0.05.
Molecular docking
To identify potential binding sites of compound V, we performed an automated molecular-docking procedure using the web-based SwissDock program (24,25). The docking was performed using the ‘Accurate’ parameter at otherwise default parameters, with no region of interest defined (blind docking).
WEB RESOURCES
FUNDING
This work was funded by grants from the Fondo de Investigaciones Sanitarias (PI10/00455 to B.P.), the Fundación Ramón Areces to B.P., MITOLAB (S2010/BMD-2402 to B.P.), the Research Council of Norway (nr. 185181 to A.M.), the KG Jebsen Foundation, the Western Norway Health Authority (nr. 911618 to A.M.), Novo Seeds (Novo Nordisk Foundation to A.M.) and the National Institutes of Health (DK45776 to R.B.). A.J.F. was supported by a grant from the Instituto de Salud Carlos III. S.B. was supported by a grant from Fundação para a Ciência e Tecnologia of Portugal (SFRH/BD/45753/2008). An institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa is gratefully acknowledged.
ACKNOWLEDGEMENTS
We thank Carmen Gherasim (University of Michigan Medical School) for assistance with the measurement of ATR activity.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Fowler B., Leonard J.V., Baumgartner M.R. Causes of and diagnostic approach to methylmalonic acidurias. J. Inherit. Metab. Dis. 2008;31:350–360. doi: 10.1007/s10545-008-0839-4. [DOI] [PubMed] [Google Scholar]
- 2.Coelho D., Kim J.C., Miousse I.R., Fung S., du Moulin M., Buers I., Suormala T., Burda P., Frapolli M., Stucki M., et al. Mutations in ABCD4 cause a new inborn error of vitamin B(12) metabolism. Nat. Genet. 2012;44:1152–1155. doi: 10.1038/ng.2386. [DOI] [PubMed] [Google Scholar]
- 3.Coelho D., Suormala T., Stucki M., Lerner-Ellis J.P., Rosenblatt D.S., Newbold R.F., Baumgartner M.R., Fowler B. Gene identification for the cblD defect of vitamin B12 metabolism. N. Engl. J. Med. 2008;358:1454–1464. doi: 10.1056/NEJMoa072200. [DOI] [PubMed] [Google Scholar]
- 4.Rutsch F., Gailus S., Miousse I.R., Suormala T., Sagne C., Toliat M.R., Nurnberg G., Wittkampf T., Buers I., Sharifi A., et al. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 2009;41:234–239. doi: 10.1038/ng.294. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R., Gherasim C., Padovani D. The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 2009;13:477–484. doi: 10.1016/j.cbpa.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Froese D.S., Healy S., McDonald M., Kochan G., Oppermann U., Niesen F.H., Gravel R.A. Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol. Genet. Metab. 2010;100:29–36. doi: 10.1016/j.ymgme.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padovani D., Labunska T., Palfey B.A., Ballou D.P., Banerjee R. Adenosyltransferase tailors and delivers coenzyme B12. Nat. Chem. Biol. 2008;4:194–196. doi: 10.1038/nchembio.67. [DOI] [PubMed] [Google Scholar]
- 8.Dobson C.M., Wai T., Leclerc D., Wilson A., Wu X., Dore C., Hudson T., Rosenblatt D.S., Gravel R.A. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc. Natl Acad. Sci. USA. 2002;99:15554–15559. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schubert H.L., Hill C.P. Structure of ATP-Bound Human ATP:Cobalamin Adenosyltransferase. Biochemistry. 2006;45:15188–15196. doi: 10.1021/bi061396f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fenton W.A., Gravel R.A., Rosenberg L.E. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C.R., Beaudet A.L., Sly W., Valle D., editors. New York: McGraw-Hill; 2001. pp. 2165–2190. [Google Scholar]
- 11.Gregersen N., Bross P., Vang S., Christensen J.H. Protein misfolding and human disease. Annu. Rev. Genomics. Hum. Genet. 2006;7:103–124. doi: 10.1146/annurev.genom.7.080505.115737. [DOI] [PubMed] [Google Scholar]
- 12.Martinez A., Calvo A.C., Teigen K., Pey A.L. Rescuing proteins of low kinetic stability by chaperones and natural ligands phenylketonuria, a case study. Prog. Mol. Biol. Transl. Sci. 2008;83:89–134. doi: 10.1016/S0079-6603(08)00603-X. [DOI] [PubMed] [Google Scholar]
- 13.Cheung J.C., Deber C.M. Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry. 2008;47:1465–1473. doi: 10.1021/bi702209s. [DOI] [PubMed] [Google Scholar]
- 14.Loo T.W., Clarke D.M. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev. Mol. Med. 2007;9:1–18. doi: 10.1017/S1462399407000361. [DOI] [PubMed] [Google Scholar]
- 15.Conn P.M., Janovick J.A. Drug development and the cellular quality control system. Trends Pharmacol. Sci. 2009;30:228–233. doi: 10.1016/j.tips.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Bonnefont J.P., Bastin J., Laforet P., Aubey F., Mogenet A., Romano S., Ricquier D., Gobin-Limballe S., Vassault A., Behin A., et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin. Pharmacol. Therap. 2010;88:101–108. doi: 10.1038/clpt.2010.55. [DOI] [PubMed] [Google Scholar]
- 17.Parenti G., Zuppaldi A., Gabriela Pittis M., Rosaria Tuzzi M., Annunziata I., Meroni G., Porto C., Donaudy F., Rossi B., Rossi M., et al. Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Mol. Ther. 2007;15:508–514. doi: 10.1038/sj.mt.6300074. [DOI] [PubMed] [Google Scholar]
- 18.Benjamin E.R., Khanna R., Schilling A., Flanagan J.J., Pellegrino L.J., Brignol N., Lun Y., Guillen D., Ranes B.E., Frascella M., et al. Co-administration with the pharmacological chaperone AT1001 increases recombinant human alpha-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012;20:717–726. doi: 10.1038/mt.2011.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos-Sierra S., Kirchmair J., Perna A.M., Reiss D., Kemter K., Roschinger W., Glossmann H., Gersting S.W., Muntau A.C., Wolber G., et al. Novel pharmacological chaperones that correct phenylketonuria in mice. Hum. Mol. Genet. 2012;21:1877–1887. doi: 10.1093/hmg/dds001. [DOI] [PubMed] [Google Scholar]
- 20.Jorge-Finnigan A., Aguado C., Sanchez-Alcudia R., Abia D., Richard E., Merinero B., Gamez A., Banerjee R., Desviat L.R., Ugarte M., et al. Functional and structural analysis of five mutations identified in methylmalonic aciduria cblB type. Hum. Mutat. 2010;31:1033–1042. doi: 10.1002/humu.21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper A., McAuley-Hecht K.E. Microcalorimetry and the molecular recognition of peptides and proteins. Philos. Trans. R. Soc. A. 1993;345:23–35. [Google Scholar]
- 22.Zhang J., Dobson C.M., Wu X., Lerner-Ellis J., Rosenblatt D.S., Gravel R.A. Impact of cblB mutations on the function of ATP:cob(I)alamin adenosyltransferase in disorders of vitamin B12 metabolism. Mol. Genet. Metab. 2006;87:315–322. doi: 10.1016/j.ymgme.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Valenzano K.J., Khanna R., Powe A.C., Boyd R., Lee G., Flanagan J.J., Benjamin E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay. Drug Dev. Technol. 2011;9:213–235. doi: 10.1089/adt.2011.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grosdidier A., Zoete V., Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grosdidier A., Zoete V., Michielin O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011;32:2149–2159. doi: 10.1002/jcc.21797. [DOI] [PubMed] [Google Scholar]
- 26.Grosdidier A., Zoete V., Michielin O. EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins. 2007;67:1010–1025. doi: 10.1002/prot.21367. [DOI] [PubMed] [Google Scholar]
- 27.Wong E., Cuervo A.M. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harbor Perspect. Biol. 2:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calvo A.C., Scherer T., Pey A.L., Ying M., Winge I., McKinney J., Haavik J., Thony B., Martinez A. Effect of pharmacological chaperones on brain tyrosine hydroxylase and tryptophan hydroxylase 2. J. Neurochem. 2010;114:853–863. doi: 10.1111/j.1471-4159.2010.06821.x. [DOI] [PubMed] [Google Scholar]
- 29.Lee W.C., Kang D., Causevic E., Herdt A.R., Eckman E.A., Eckman C.B. Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J. Neurosci. 2010;30:5489–5497. doi: 10.1523/JNEUROSCI.6383-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khanna R., Benjamin E., Pellegrino L., Schilling A., Rigat B., Soska R., Nafar H., Ranes B., Feng J., Lun Y., et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. The FEBS J. 2010;277:1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamin E.R., Flanagan J.J., Schilling A., Chang H.H., Agarwal L., Katz E., Wu X., Pine C., Wustman B., Desnick R.J., et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009;32:424–440. doi: 10.1007/s10545-009-1077-0. [DOI] [PubMed] [Google Scholar]
- 32.Fan C., Bobik T.A. Functional characterization and mutation analysis of human ATP:Cob(I)alamin adenosyltransferase. Biochemistry. 2008;47:2806–2813. doi: 10.1021/bi800084a. [DOI] [PubMed] [Google Scholar]
- 33.Klotz U. The role of pharmacogenetics in the metabolism of antiepileptic drugs: pharmacokinetic and therapeutic implications. Clin. Pharmacokinet. 2007;46:271–279. doi: 10.2165/00003088-200746040-00001. [DOI] [PubMed] [Google Scholar]
- 34.Dawood S., Leyland-Jones B. Pharmacology and pharmacogenetics of chemotherapeutic agents. Cancer Invest. 2009;27:482–488. doi: 10.1080/07357900802574660. [DOI] [PubMed] [Google Scholar]
- 35.Ballhausen D., Mittaz L., Boulat O., Bonafe L., Braissant O. Evidence for catabolic pathway of propionate metabolism in CNS: expression pattern of methylmalonyl-CoA mutase and propionyl-CoA carboxylase alpha-subunit in developing and adult rat brain. Neuroscience. 2009;164:578–587. doi: 10.1016/j.neuroscience.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 36.Leal N.A., Olteanu H., Banerjee R., Bobik T.A. Human ATP:Cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J. Biol. Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 37.Niesen F.H., Berglund H., Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 38.Johnson C.L., Pechonick E., Park S.D., Havemann G.D., Leal N.A., Bobik T.A. Functional genomic, biochemical, and genetic characterization of the Salmonella pduO gene, an ATP:cob(I)alamin adenosyltransferase gene. J. Bacteriol. 2001;183:1577–1584. doi: 10.1128/JB.183.5.1577-1584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Cerda C., Merinero B., Sanz P., Jimenez A., Garcia M.J., Urbon A., Diaz Recasens J., Ramos C., Ayuso C., Ugarte M. Successful first trimester diagnosis in a pregnancy at risk for propionic acidaemia. J. Inherit. Metab. Dis. 1989;12(Suppl. 2):274–276. doi: 10.1007/BF03335396. [DOI] [PubMed] [Google Scholar]