

Abstract
Genetic mutations in NLGN4X (neuroligin 4), including point mutations and copy number variants (CNVs), have been associated with susceptibility to autism spectrum disorders (ASDs). However, it is unclear how mutations in NLGN4X result in neurodevelopmental defects. Here, we used neural stem cells (NSCs) as in vitro models to explore the impacts of NLGN4X knockdown on neurodevelopment. Using two shRNAmir-based vectors targeting NLGN4X and one control shRNAmir vector, we modulated NLGN4X expression and differentiated these NSCs into mature neurons. We monitored the neurodevelopmental process at Weeks 0, 0.5, 1, 2, 4 and 6, based on morphological analysis and whole-genome gene expression profiling. At the cellular level, in NSCs with NLGN4X knockdown, we observed increasingly delayed neuronal development and compromised neurite formation, starting from Week 2 through Week 6 post differentiation. At the molecular level, we identified multiple pathways, such as neurogenesis, neuron differentiation and muscle development, which are increasingly disturbed in cells with NLGN4X knockdown. Notably, several postsynaptic genes, including DLG4, NLGN1 and NLGN3, also have decreased expression. Based on in vitro models, NLGN4X knockdown directly impacts neurodevelopmental process during the formation of neurons and their connections. Our functional genomics study highlights the utility of NSCs models in understanding the functional roles of CNVs in affecting neurodevelopment and conferring susceptibility to neurodevelopmental diseases.
INTRODUCTION
In the past few years, genetic studies have implicated a strong connection between the synaptic complex with a range of neurodevelopmental or neuropsychiatric disorders, such as autism and schizophrenia (1–4). The key component of this complex include presynaptic neurexins, postsynaptic neuroligins (NLGNs), SHANKs, postsynaptic density protein 95 (PSD-95) and disks large-associated proteins (DLGAPs). Among them, neuroligins represent some of the most well-known postsynaptic molecules that play key roles in establishing cell adhesions with presynaptic neurexins at the synaptic junction (5–8). All neuroligins contain an N-terminal hydrophobic sequence with the characteristics of a cleaved signal peptide followed by a large esterase homology domain, a highly conserved single transmembrane region and a short cytoplasmic domain. Among the four members of the neuroligin family, NLGN4X is located in the X chromosome; it has 63–73% amino acid identity with the other neuroligins (9). The NLGN4X gene contains six exons and codes for a protein of 816 amino acids, and has a widespread gene expression pattern (9).
Multiple genetic studies have now implicated a significant role for neuroligins, especially NLGN4X, in the susceptibility to several neurodevelopmental disorders. Xp22.3 deletions that include the NLGN4X gene have long been associated with autism (10). In 2003, a study screened mutations in NLGN3 and NLGN4X in a cohort with autism spectrum disorders (ASDs), and identified de novo frameshift mutations in NLGN4X in one affected individual's mother (11). Another frameshift mutation in NLGN4X was observed in all affected individuals in a large French family, in which 10 males had X-linked mental retardation, 2 had autism and 1 had pervasive developmental disorder (12). A few follow-up studies suggested that missense changes in NLGN4X may also contribute to autism susceptibility (13,14), although a study on Quebec population failed to identify NLGN4X mutations in 96 subjects affected with autism (15). Another family-based association analysis in 100 families with ASDs yielded only modest associations at NLGN4X and other neuroligins, suggesting that NLGN4X mutations probably represent only rare causes of autism (16). Several recent studies have also linked various NLGN4X deletions with autism susceptibility (17). Additionally, novel de novo splice variants in NLGN4X have also been observed in autistic individuals (18). In summary, mounting evidence suggested that very rare and loss-of-function mutations in NLGN4X, including deletions, are associated with susceptibility to ASDs.
Despite these prior genetic studies, it is still unclear how mutations in NLGN4X confer susceptibility to multiple related diseases, and what biological processes are compromised due to NLGN4X haploinsufficiency. These types of questions can be partially addressed in animal models by behavioral and molecular studies. In 2008, Jamain et al established a mouse model with loss-of-function mutation in Nlgn4, and found that these mice have highly selective deficits in reciprocal social interactions and communication that are reminiscent of ASDs in humans (19). Recently, the same group demonstrated that Nlgn4 null mouse may serve as a construct-valid model of heritable monogenic autism (20). However, analysis on models derived from human tissues may provide complementary insights into mouse models. Recently, in vitro cellular models, such as neurons derived from humans (21) or induced pluropotent stem cells (hiPSCs) (22–25), have been increasingly recognized as important tools to gain insights into the functional roles of genetic mutations during neurodevelopment. Several models for Rett syndrome (26), schizophrenia (27) and fragile X mental retardation syndrome (28), have already been reported, implicating the potential utility of using in vitro human-based models to study functional impacts of genetic mutations on neurodevelopment. We have also recently established neural stem cell (NSC) models derived from both hiPSCs and human embryonic stem cells, and demonstrated that knockdown of NRXN1 expression in NSCs compromise neurodevelopmental pathways, based on gene expression profiling at Week 4 post differentiation (29). Other groups have also examined the utility of hiPSCs-derived neurons to investigate synapse formation and function, and found that HEK-293T cells expressing NLGN3 and NLGN4X, but not those containing autism-associated mutations, are able to induce presynaptic differentiation in hiPSCs-derived neurons (30).
In the current study, we addressed a central hypothesis that if deletions of NLGN4X influence susceptibility to autism, then knockdown of gene expression levels for NLGN4X may influence the course of neurodevelopment in an in vitro model. Similar to a previous study (29), we used NSCs as models, and used shRNAmir to re-create NLGN4X haploinsufficiency; however, we are particularly interested in the time course relationships between gene knockdown and genome function, so we measured gene expression levels across six time points, after differentiating the NSCs into neurons. To address the concern on nonspecific knockdown, we evaluated two different knockdown vectors in the study. With this in vitro model, we attempted to observe whether there are cellular and molecular differences between NSCs with or without NLGN4X knockdown, and related these observations to prior knowledge on the potential functionality of neuroligins in synaptic development and function.
RESULTS
Normal developmental time course in neural stem cells
We differentiated NSCs into neurons during a 6-week period, and collected RNA samples at multiple time points (Weeks 0, 0.5, 1, 2, 4, 6) for whole-genome gene expression profiling. For each time point, two to three replicates were assayed. We constructed a hierarchical clustering plot on their expression levels, and found that these cells fall into two major groups (Fig. 1). The three replicates in Week 4 and two replicates in Week 6 cluster together, and are separated from those samples collected at Weeks 0, 0.5, 1 and 2. This observation is also consistent with Kanopka et al (21), where they demonstrated that Week 4 is a critical time point during differentiation of primary normal human neuronal progenitors, when a significant number of genes associated with ASDs are either induced or repressed. Additionally, Kanopka et al identified 4427 genes that have expression alterations during developmental course at Week 4 (21). We found that 1696 of 4427 genes from the Kanopka et al study are also identified as differentially expressed between Weeks 0 and 4 in our study. Notably, several prominent synaptic genes, such as NRXN1, NRXN2, NLGN1, NLGN2, HOMER1, HOMER2, GRM3 and GRIA2, are found in our results, suggesting that these genes have altered gene expression during the normal developmental time course.
Figure 1.

Hierarchical clustering of whole-genome gene expression in NSCs post differentiation across six time points.
NLGN4X knockdown results in morphological differences in neural differentiation
To knockdown NLGN4X expression reliably in NSCs and differentiated neurons, we procured four commercially available shRNAmir vectors and tested them in HEK-293T cells. We confirmed that all of them can knockdown NLGN4X expression by a qPCR, but with varying efficiencies (Supplementary Material, Fig. S1). We selected two shRNAmir vectors, sh7 and sh8, in following studies, given that they generally resulted in high knockdown efficiency under the conditions of high cell confluence. We next tested these shRNAmir vectors in NSCs. After packaging control shRNAmir (shCon) or NLGN4X shRNAmir (sh7 and sh8) into lentivirus and infecting NSCs, we observed RFP signals within 1 week for >95% of all cells (Fig. 2A), suggesting efficient production of the shRNAmir. Seven days post infection, with doxycycline induction, we observed high knockdown efficiency (46–76%) in the NSCs for both sh7 and sh8 vectors, compared with NSCs with shCon, suggesting that the vectors can efficiently knockdown NLGN4X expression in a controlled manner (Fig. 2B).
Figure 2.

Evaluation of shRNAmir in knocking down NLGN4X gene expression in NSCs. (A) Expression of Red Fluorescence Protein (RFP) signals in NSCs at different time points post differentiation, suggesting successful infection of all three shRNAmir vectors in NSCs. (B) Knockdown efficiency of two shRNAmir vectors against NLGN4X in NSCs at different time points post differentiation, as measured by a qPCR and normalized against the NSCs treated with control shRNAmir.
We next examined whether there are developmental differences between the NSCs with or without NLGN4X knockdown, by examining cell morphology across a 6-week period after differentiating NSCs into the neuronal lineage. In the very early-stage post differentiation, the cells with NLGN4X knockdown and those without knockdown show typical NSC morphology (Fig. 3A). At Day 3, we observed signs that some neurons began to emerge in the culture dish with control shRNAmir, including neurites at the cell surface, but not in the two dishes with NLGN4X knockdown (Fig. 3B). At 7 days post differentiation, there were limited protrusion and branches of neurites in cells with NLGN4X knockdown, and we cannot identify obvious cell–cell connections; however, in dishes with control shRNAmir, we observed obvious neurites forming cell–cell interactions (Fig. 3B). After 2 weeks, when we examine the controls, we can see that a large number of neurons are clearly formed, and some axons are already visible in the cells treated with control shRNAmir (Fig. 3B). At the same time, when we examined the cells with NLGN4X knockdown, we observed less formation of neurons and inter-cell connections. Additionally, we examined zoomed-in photos of individual neuronal cells at different time points, and found similar patterns of defects in neurite formation (Supplementary Material, Fig. S2). These differences suggested that NLGN4X knockdown affects cellular phenotypes during differentiation of NSCs into neurons, by delaying the neurodevelopmental process and by compromising neurite formation and the establishment of cell–cell interactions.
Figure 3.

Differences in cellular phenotypes between cells treated with three shRNAmir vectors during differentiation of NSCs at multiple time points. (A) Stereo image on NSCs at Day 0 post differentiation, demonstrating a similar cell morphology in three culture dishes treated with sh7, sh8 and shCon, respectively. (B) RFP image on NSCs 3 days, 7 days and 14 days post differentiation. We observed increasingly clear trends that fewer axons and synaptic connections formed in differentiated neurons treated with sh7 and sh8.
Alteration of gene expression due to NLGN4X knockdown during neurodevelopment
Using whole-genome gene expression data, we next compared transcriptome alterations between three types of cells (sh7, sh8 and shCon) at different time points: Weeks 0, 0.5, 1, 2, 4 and 6 (Fig. 3). For each time point and each sample, two to three replicates were used in expression profiling. We found that the NLGN4X gene expression level has decreased 20–80% at all of the time points, similar to what we observed in a qPCR in Figure 2. Hierarchical clustering showed that samples from the same time points tightly clustered with each other, suggesting that cells with or without NLGN4X knockdown have relatively small differences, compared with differences due to developmental time course (Fig. 4). Additionally, within each time point, cells treated with sh7 and sh8 tended to cluster together and were separated from cells treated with shCon, clearly indicating that NLGN4X knockdown induced similar whole-genome expression changes in the two cell populations treated with shRNAmir.
Figure 4.
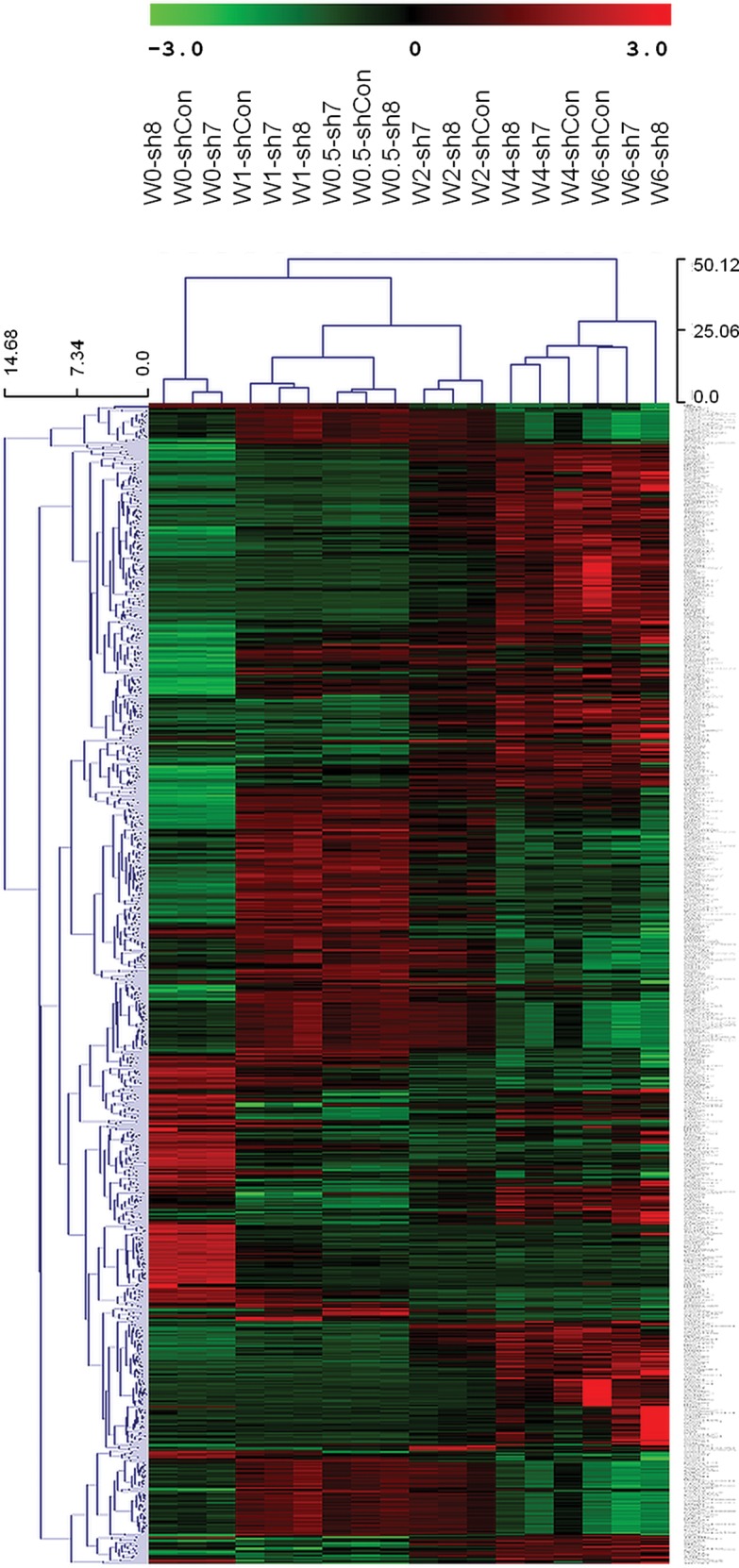
Hierarchical clustering of gene expression levels across multiple time points for two cell populations with NLGN4X knockdown (sh7 and sh8) and one cell population without knockdown (shCon). For each time point, the average expression values from two to three replicate samples were used.
We are also interested in whether NLGN4X knockdown affects the temporal patterns of gene expression during the developmental time course, that is, whether the ‘trends’ of time-dependent gene expression are different between the cells. For this purpose, we compared the overall patterns of gene expression between cells treated with sh8 or shCon using ‘timecourse’ (31), a statistical package specifically designed for this purpose. All genes are ranked by the Hotelling's T2 values, representing the magnitude of changes in temporal patterns over the time course. In this list, NLGN4X ranks as the 192nd most altered gene (Fig. 5). In previous analysis, we found that several synaptic genes, such as NRXN1, NRXN2, NLGN1, NLGN2, HOMER1, HOMER2, GRM3 and GRIA2, are differentially expressed between Weeks 0 and 4 in cells without knockdown; among these genes, only GRM3 appears to have clearly altered temporal patterns due to NLGN4X knockdown (Fig. 5), yet most other genes show weak trends of altered temporal patterns (ranked between 1000th and 2000th). HOMER1 is an exception that ranked at 23493rd. In comparison, two commonly used housekeeping genes, GAPDH and ACTB, ranked at 4460th and 9489th, respectively. We repeated the above analysis by comparing cells treated with sh7 and shCon, and obtained similar results. This analysis indicated that NLGN4X knockdown may have weak effects in impacting the temporal patterns of gene expression for synaptic genes during the developmental time course.
Figure 5.

Alteration of temporal patterns of gene expression as a result of NLGN4X knockdown during the timecourse (Weeks 0.5, 1, 2, 4 and 6). Several synaptic genes are depicted, yet only NLGN4X and GRM3 have significantly altered temporal patterns.
Biological pathways and networks affected by NLGN4X knockdown
To examine the biological pathways that were affected by NLGN4X knockdown, we identified genes that were differentially expressed at each time point between NSCs with or without NLGN4X knockdown. For Weeks 0, 0.5 and 1, we did not identify any genes that are differentially expressed, but an increasing number of genes have differential expression after Week 2. An enrichment analysis using the DAVID web server (32) identified multiple Gene Ontology (GO) categories that were significantly altered (Tables 1 and 2). We listed the top 10 most enriched genetic pathways at Week 6 for the down-regulated and upregulated genes in sh8-treated NSCs, respectively, and then compared the significance of the selected pathways at different time points. The top 10 down-regulated pathways were most related to the nervous system and development, such as ‘nervous system development’ (GO:0007399, ranked as first), ‘neurogenesis’ (GO:0022008, ranked as third), ‘neuron differentiation’ (GO:0030182, ranked as fifth) and ‘organ development’ (GO:0048513, ranked as second), ‘cell development’ (GO: 0048468, ranked as seventh), ‘tissue development’ (GO: 0009888, ranked as ninth). However, two muscle-related pathways have also been altered as a result of NLGN4X knockdown, including ‘muscle organ development’ (GO:0007517, ranked as fourth) and ‘muscle contraction’ (GO:0006936, ranked as sixth). For the down-regulated pathways in Week 6, all of them were also enriched in Weeks 2 and 4, but with an obvious increase of statistical significance with time. Though less pronounced than down-regulated pathways, the upregulated list was also enriched with several development-related pathways, such as ‘organ development’, ‘tissue development’ and ‘nervous system development’. These results suggested that NLGN4X knockdown has significant impacts on other genes that are involved in the neurodevelopmental process. Similar observations were made on cells treated with sh7 (Supplementary Material, Table S1), although it does not have as high knockdown efficiency as sh8 (Supplementary Material, Fig. S1, Fig. 2). We found that muscle-related and development-related pathways are also significantly enriched, yet the former has even stronger statistical significance. These results corroborate previous studies showing that neurexins and neuroligins play important roles in smooth muscle development and promote angiogenesis (33), in addition to their well-known roles in nervous system development.
Table 1.
Biological pathways that were significantly down-regulated in differentiated NSCs with NLGN4X knockdown at Week 6, and their significance levels in Weeks 1, 2 and 4
| Pathway |
W1 | W2 | W4 | W6 | ||
|---|---|---|---|---|---|---|
| GO:0007399 | Nervous system development | Count | 0 | 9 | 62 | 143 |
| P-value | NA | 2.382E − 03 | 1.174E − 14 | 5.523E − 35 | ||
| FDR | NA | 2.975E − 02 | 1.807E − 13 | 9.073E − 34 | ||
| GO:0048513 | Organ development | Count | 0 | 16 | 77 | 166 |
| P-value | NA | 1.327E − 06 | 1.506E − 12 | 2.284E − 24 | ||
| FDR | NA | 1.681E − 05 | 2.314E − 01 | 3.752E − 23 | ||
| GO:0022008 | Neurogenesis | Count | 0 | 6 | 36 | 84 |
| P-value | NA | 1.074E − 02 | 4.548E − 09 | 1.003E − 21 | ||
| FDR | NA | 1.278E − 01 | 6.987E − 08 | 1.647E − 20 | ||
| GO:0007517 | Muscle organ development | Count | 0 | 7 | 25 | 45 |
| P-value | NA | 6.772E − 06 | 2.414E − 12 | 1.185E − 18 | ||
| FDR | NA | 8.575E − 05 | 3.709E − 11 | 1.947E − 17 | ||
| GO:0030182 | Neuron differentiation | Count | 0 | 4 | 28 | 65 |
| P-value | NA | 7.646E − 02 | 9.730E − 08 | 3.824E − 18 | ||
| FDR | NA | 6.348E − 01 | 1.495E − 06 | 6.283E − 17 | ||
| GO:0006936 | Muscle contraction | Count | 0 | 7 | 23 | 38 |
| P-value | NA | 1.049E − 06 | 1.685E − 13 | 4.012E − 18 | ||
| FDR | NA | 1.328E − 05 | 2.589E − 12 | 6.591E − 17 | ||
| GO:0048468 | Cell development | Count | 0 | 6 | 34 | 77 |
| P-value | NA | 1.418E − 02 | 2.708E − 07 | 7.333E − 16 | ||
| FDR | NA | 1.655E − 01 | 4.160E − 06 | 1.277E − 14 | ||
| GO:0060284 | Regulation of cell development | Count | 0 | 3 | 20 | 39 |
| P-value | NA | 7.889E − 02 | 1.586E − 08 | 1.908E − 14 | ||
| FDR | NA | 6.468E − 01 | 2.437E − 07 | 3.137E − 13 | ||
| GO:0009888 | Tissue development | Count | 0 | 7 | 32 | 73 |
| P-value | NA | 3.396E − 03 | 4.648E − 06 | 3.782E − 13 | ||
| FDR | NA | 4.216E − 02 | 7.140E − 05 | 6.212E − 12 | ||
| GO:0032989 | Cellular component morphogenesis | Count | 0 | 4 | 20 | 52 |
| P-value | NA | 6.051E − 02 | 2.547E − 04 | 2.507E − 12 | ||
| FDR | NA | 5.464E − 01 | 3.905E − 03 | 4.119E − 11 | ||
| Total count of down-regulated genes | 11 | 36 | 375 | 842 | ||
Table 2.
Biological pathways that are significantly up-regulated in differentiated NSCs with NLGN4X knockdown at Week 6, and their significance levels in Weeks 1, 2 and 4
| Pathway |
W1 | W2 | W4 | W6 | ||
|---|---|---|---|---|---|---|
| GO:0048513 | Organ development | Count | 13 | 3 | 64 | 114 |
| P-value | 4.036E − 02 | 8.694E − 02 | 4.654E − 06 | 1.290E − 07 | ||
| FDR | 4.461E − 01 | 6.932E − 01 | 7.249E − 05 | 2.109E − 06 | ||
| GO:0009888 | Tissue development | Count | 0 | 3 | 27 | 52 |
| P-value | NA | 1.426E − 02 | 1.410E − 03 | 8.219E − 06 | ||
| FDR | NA | 1.702E − 01 | 2.174E − 01 | 1.343E − 04 | ||
| GO:0042060 | Wound healing | Count | 0 | 0 | 0 | 23 |
| P-value | NA | NA | NA | 9.860E − 06 | ||
| FDR | NA | NA | NA | 1.611E − 04 | ||
| GO:0060429 | Epithelium development | Count | 0 | 0 | 0 | 25 |
| P-value | NA | NA | NA | 1.698E − 05 | ||
| FDR | NA | NA | NA | 2.774E − 04 | ||
| GO:0044255 | Cellular lipid metabolic process | Count | 0 | 0 | 0 | 43 |
| P-value | NA | NA | NA | 2.070E − 05 | ||
| FDR | NA | NA | NA | 3.382E − 04 | ||
| GO:0070011 | Peptidase activity, acting on L-amino acid peptides | Count | 0 | 0 | 0 | 46 |
| P-value | NA | NA | NA | 2.229E − 05 | ||
| FDR | NA | NA | NA | 2.825E − 04 | ||
| GO:0022603 | Regulation of anatomical structure morphogenesis | Count | 0 | 0 | 11 | 24 |
| P-value | NA | NA | 1.618E − 02 | 2.821E − 05 | ||
| FDR | NA | NA | 2.244E − 01 | 4.608E − 04 | ||
| GO:0007399 | Nervous system development | Count | 0 | 0 | 37 | 71 |
| P-value | NA | NA | 3.405E − 03 | 6.675E − 05 | ||
| FDR | NA | NA | 5.173E − 02 | 1.090E − 03 | ||
| GO:0022604 | Regulation of cell morphogenesis | Count | 0 | 0 | 8 | 17 |
| P-value | NA | NA | 1.93E − 02 | 7.863E − 05 | ||
| FDR | NA | NA | 2.62E − 01 | 1.284E − 03 | ||
| GO:0030855 | Epithelial cell differentiation | Count | 0 | 0 | 0 | 17 |
| P-value | NA | NA | NA | 1.30E − 04 | ||
| FDR | NA | NA | NA | 1.70E − 02 | ||
| Total count of upregulated genes | 100 | 7 | 399 | 824 | ||
We next examined the overlap of the differentially expressed (DE) genes at different time points (Table 3). At Week 2, 25 of 43 (58.1%) DE genes are also differentially expressed at Week 4, and 31 of 43 (72.1%) are also identified at Week 6. In addition, 522 of 774 (71.3%) DE genes at Week 4 are also identified at Week 6. However, the percentage of overlap between Week 1 and the following weeks are much smaller (3.6–22.5%), suggesting that genes differentially expressed at Week 2 are more likely to be continuously differentially expressed in later weeks. This observation is consistent with the pathway enrichment analysis (Tables 1 and 2), where the same down-regulated pathways at Week 6 also tend to have down-regulation in Weeks 2 and 4, but not Week 1.
Table 3.
Overlap of the DE genes with and without NLGN4X knockdown at different time points post differentiation.
| Time point | # DE genes | Overlap with DE genes at week 2 |
Overlap with DE genes at week 4 |
Overlap with DE genes at week 6 |
|||
|---|---|---|---|---|---|---|---|
| # Genes | Fraction | # Genes | Fraction | # Genes | Fraction | ||
| Week 0 | 0 | 0 | NA | 0 | NA | 0 | NA |
| Week 0.5 | 0 | 0 | NA | 0 | NA | 0 | NA |
| Week 1 | 111 | 4 | 3.6% | 25 | 22.5% | 18 | 16.2% |
| Week 2 | 43 | 25 | 58.1% | 31 | 72.1% | ||
| Week 4 | 774 | 522 | 71.3% | ||||
| Week 6 | 1666 | ||||||
To further evaluated the biological networks that are perturbed, we compiled a list of 60 well-known proteins involved in a postsynaptic complex (Supplementary Material, Table S2) and mapped them onto the human protein–protein interaction (PPI) network (34). We extracted their directly connected gene neighbors to construct a network, which contains 537 nodes and 2601 edges. Although it is difficult to discern details of the network visually, it is clear that all sub-modules of the network contain differentially expressed genes, especially down-regulated genes (Fig. 6A). Among these 60 postsynaptic proteins, 11 have differential expression, so we constructed a sub-network on these 11 genes and their first-degree interaction partners (Fig. 6B). In this sub-network, the NLGN4X gene has two directly connected genes, DLG4 and DLGAP2. DLG4 is significantly down-regulated, and it serves as a hub protein in this sub-network that connects with ∼100 proteins including NLGN1, NLGN2 and NLGN3. NLGN1 and NLGN3 are significantly down-regulated as a result of NLGN4X knockdown. We also noted that NRXN2, but not NRXN1, has down regulation; this is consistent with the previous report that NRXN1 knockdown did not affect gene expression for neuroligins (29). In summary, the sub-network demonstrated that a considerable number of important synaptic genes, especially DLG4, NLGN1, NLGN3, NRXN2, directly or indirectly interacted with NLGN4X and form a down-regulated network driven by NLGN4X knockdown. We recognize that the current protein–protein interaction information is still incomplete, but this analysis further reconfirmed our hypothesis that NLGN4X knockdown affects gene expression for a network containing synaptic genes.
Figure 6.

Network of known protein–protein interactions that includes all first-degree neighbors of 60 well-known postsynaptic proteins. (A) Sub-networks formed by all proteins and neighbors (B) sub-network formed by of 11 DE proteins and their neighbors. Proteins involved in a postsynaptic complex are labeled in diamonds, yet their first-degree neighbors in the interaction network are labeled in circles. Upregulated genes are colored by red, yet down-regulated genes are colored in green.
DISCUSSION
In the current study, we investigated the functional significance of NLGN4X deletions in neurodevelopment, using NSCs as in vitro models. During differentiation of NSCs into mature neurons across a 6-week period, we found that knockdown of a single neuronal gene NLGN4X leads to morphological changes as well as transcriptome alterations. Altogether, our results suggested that NLGN4X deletion influences normal developmental course for neurons, and may alter several biological pathways including nervous system development and neuron differentiation. These cellular alterations could play a role in the molecular pathophysiology of several neurodevelopmental diseases affected by NLGN4X deletion, including autism and schizophrenia.
The ability to differentiate NSCs into neuronal lineage in a tightly controlled manner has made NSCs as attractive in vitro model systems to study complex neurodevelopmental disorders. For many neurodevelopmental disorders, animal models are generally not available, not representative enough or not ideal to use. On the other hand, the neuronal system is also difficult to study in human models, as live neurons, especially developing neurons, are not readily available from the patients to understand the molecular pathophysiology of the diseases. Therefore, NSCs especially those derived from patients (such as NSCs generated from patient-specific fibroblasts or patient-specific iPS cells) may find more use in future genetic studies. By analyzing morphological, electrophysiological, transcriptional and functional differences of neurons derived from specific patients and control subjects, or from the same patient with and without gene knockdown, we may better understand the molecular mechanism of disease pathogenesis. Additionally, these in vitro models may also facilitate drug discovery (for example, in drug screening applications), and may facilitate pharmacogenomic studies to understand how deficiency in candidate genes impact cellular responses to pharmacological treatments.
There are several potential enhancements to the current study that we wish to elaborate here. First, we studied the neurons from the perspective of cell morphology and gene expression, but one interesting hypothesis to explore is whether there are electrophysiological consequences as a result of NLGN4X knockdown. This can be accomplished by electrophysiological recording of single neurons by patch clamp techniques (35,36), which provides a high-resolution method of observing the function of individual ionic channels in a variety of normal and pathological cell types. Second, developing neurons are highly heterogeneous, and it is possible that one subtype of neuronal cells or astrocytes is more influenced by NLGN4X knockdown than the other subtypes. However, by pooling all cells (including astrocytes) together in gene expression analysis, we lose the resolution to look at specific subtypes of neurons. This can be addressed by technical advancement in isolating specific subtypes of cells from culture dish, or even single-cell microarray analysis or single-cell RNA-Seq analysis (37). Third, there are multiple other ways in which genome expression and function can be modulated, including DNA methylation, nucleosome positioning, and post-transcriptional or post-translational modifications (38–40). Our analysis did not evaluate the impact of NLGN4X deletions on these aspects, although the prior probability that NLGN4X affects these biological processes is low, given the known functional role of NLGN4X. Finally, we used gene expression microarrays rather than more modern sequencing techniques to measure gene expression levels. Several studies have compared the performance of microarray versus RNA-Seq; these studies demonstrated that both have their advantages and limitations, although the biological interpretations (such as pathway enrichment) from both platforms are mostly consistent (41–43). Given the need for examine multiple time points in several cell lines each with two to three replicates, we elected to use gene expression microarrays as this is a more economical choice for our study.
In conclusion, by introducing targeted gene knockdown in NSCs and measuring whole-genome gene expression levels, in the context of time course series data, we developed an interesting approach for studying the functional significance of CNVs during neurodevelopment. The models that we established here would help confirm how candidate CNVs identified from previous CNV association studies on complex neuropsychiatric and neurodevelopmental disorders modulate disease risk, and also help uncover biological pathways and networks underlying disease susceptibility. We believe that these models can be further developed in a high-throughput fashion, whereas we simultaneously introduce multiple genetic knockdowns in 96-well plates, and follow-up the same batch of cells across multiple weeks, then assay the morphological changes and gene expression levels both in a high-throughput fashion, for systems understanding of candidate gene functions. These methods may ultimately represent high-throughput means to understanding the functional role of human DNA sequence variants in complex neurodevelopmental diseases.
MATERIALS AND METHODS
Neural stem cells (NSCs) induction and neuronal differentiation
We have previously generated hiPSC lines using 2.0 × 106 human fetal dermal fibroblasts (HDFf, acquired from ATCC) transfected with 4 μg CAG.OSKM-puDtk reprogramming transposon and 2 μg pCyL43 transposase plasmid through nucleofection (Amaxa Nucleofector technology). Embryoid Bodies (EBs) were formed first by splitting the hiPSCs colonies into appropriate size and seeding on 6 cm ultra-low attachment dish (BD Biosciences) with ES culture media without bFGF, and changing the medium every 2 days. On Day 5 of EBs formation, we switched to N2 media (DMED/F12 with N2 supplement (Invitrogen) and 1% penicillin/streptomycin) for targeted differentiation of EBs to neurospheres. On Day 10, we collected all the neurospheres and seeded them on prepared Matrigel coated culture dish in N2 media with 20 ng/ml bFGF. The neural rosettes were formed on Matrigel plates after 5–10 days culture. We manually dissected the neural rosettes from the Matrigel plate, and gentally digested them with 0.05% trypsin to break the rosettes to smaller pieces and then seeded on poly-ornithine and fibronectin (Sigma) double coated plate in N2/B27 culture media (50% N2 media (DMED/F12 with N2 supplement (Invitrogen) and 1% penicillin/streptomycin), 50% B27 media (DMEM/F12 with B27 supplement (Invitrogen) and 1% penicillin/streptomycin), with 20 ng/ml bFGF). Spontaneous neuronal differentiation was performed in N2/B27 culture medium without bFGF. The culture media were changed every 2 days for both NSCs culture and neuronal differentiation.
Screening shRNAmir with high knockdown efficiency on NLGN4X
Four commercial shRNAmir targeting NLGN4X were acquired from Open Biosystems (catalog number: RHS4696-101317495, RHS4696-101318313, RHS4696-101318401, RHS4696-101324262, hereafter referred to as sh5, sh6, sh7, sh8, respectively), then expanded in LB medium with 100 μg/ml ampicillin and purified using Plasmid Maxi kit (Qiagen). We transfected these shRNAmir vectors into HEK-293T cells cultured in 10 cm dish by the method of PEI (polyethylenimine), changing medium every day. The knockdown efficiency was tested by a qPCR (see below), and we selected two most efficient shRNAmir vectors for use in downstream studies.
Lentivirus production and infection of NSCs
Human TRIPZ lentivirus inducible shRNAmir for NLGN4X plasmids stock was expanded in LB medium with 100 μg/ml ampicillin and purified using a Plasmid Maxi kit (Qiagen). The non-targeting TRIPZ lentivirus inducible shRNAmir control was made by integrating the non-silencing scrambled shRNAmir sequence at MluI and XhoI restriction sites on a pTRIPZ vector, which does not match any known mammalian genes following the Open Biosystems shRNAmir manual. The TRIPZ lentivirus plasmids were packed by lentivirus packaging vectors pMD2.g and psPAX.2, and transfected into HEK-293T cells using polyethylenimine (Sigma) as the transfection reagent. Lentivirus was collected 48–72 h after transfection by a centrifuge at 28 000 rpm for 1.5 h at 4°C using Beckman Counter Optima L-100 XP ultracentrifuge. The lentivirus particles were resuspended in PBS and stored at −80°C. The lentivirus was titered using HEK-293T cells following the Open Biosystems shRNAmir manual. For the infection of NSCs, we first cultured the NSCs in N2/B27 media. When the cells reached confluence, they were trypsinized and collected in a 1.5 ml Eppendorf tube with 150 μl media. The lentivirus was then added to the tube and incubated at 37°C for 1 h, then re-plated on a poly-ornithine and fibronectin double coated 6 cm plate with 2.5 ml media. The cells were incubated overnight and transferred to the fresh media in the morning next day. To induce the shRNAmir expression, 1 μg/μl doxycycline (Enzo Life Science) was added to the media. For long-term shRNAmir expression, doxycycline was refreshed every 2 days.
Quantitative real-time-PCR
Total RNA was extracted using RNeasy mini kit (Qiagen), in combination of RNAase-free DNAase (Qiagen) to remove the potential genomic DNA contamination. The cell lysate was homogenized by passing five times through a blunt 27-gauge needle. RNA concentration was quantified by Nanodrop 1000 Spectrophotometer (Thermo Scientific). Reverse transcription was performed with 1.5 μg RNA using a ProtoScript M-MuLV First Strand cDNA Synthesis kit using random primers (New England Biolabs). The quantitative real-time PCR was carried on with gene-specific primers and iQ SYBR Green Supermix using Bio-Rad CFX96 system (Bio-Rad). The mRNA starting quantity was determined by the relative standard curve method using Bio-Rad CFX Manager software. The sequences of primers used were below:
NLGN4X: Forward: 5′-TGAGACTCACAGGCGCCCCA-3′; Reverse: 5′- CGTGTGCCTGCAGCGACTCA -3′
GAPDH: Forward: 5′-CATGTTCGTCATGGGTGTGAA-3′; Reverse: 5′- AGTGATGGCATGGACTGTGGT-3′
The primers for NLGN4X were designed following a previous study (44), which targets exon 7 of NLGN4X and have high specificity to avoid cross-targeting NLGN4Y. The expression of genes of interest was normalized to GAPDH in all samples. After normalization, data were transformed as the target mRNA signal relative to untreated control samples, and then plotted on GraphPad Prism 5.0 (GraphPad Software, Inc).
Whole-genome gene expression analysis
The total RNAs were also subject to gene expression microarray analysis at selected time points during neuronal differentiation. Briefly, total RNAs were isolated from cell lysate by RNeasy mini kit (Qiagen), and their qualities (OD260/280, rRNA ratio, RNA integrity number) were determined by BioAnalyzer 2100 (Agilent). 250 ng of total RNA was subject to standard Illumina protocols for cRNA production, amplification, and then hybridized to Human12 Expression array. The data were imported into Illumina GenomeStudio, and quantile normalization was used to calculate gene expression values. For each time point and each cell, two to three replicate samples were used in the expression microarray analysis.
The gene expression values were loaded into the MeV software version 4.8 (45) for clustering and differential expression analysis. The HCT module was used to generate hierarchical clustering, with average linkage clustering on Euclidean distance. The DE genes across time series were identified by ‘timecourse’ (31), an R package available in Bioconductor. For each individual time point, the ‘limma’ (46) R package from Bioconductor repository was used to detect the DE genes between the samples with and without knockdown.
The lists of significantly DE genes were analyzed by the DAVID web server (32) for enrichment of GO categories. Additionally, the results of gene expression (fold change and P-values) were overlaid with known protein–protein interactions from MiML database (34) in the Cytoscape software (47) for network-based analysis and visualization.
SUPPLEMENTARY MATERIAL
FUNDING
The study is supported by start-up funds from the Zilkha Neurogenetic Institute to K.W., and in part by NIH grant P30 MH08977 (PI: Levitt) and R01 HG006465 (PI: K.W.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Wang laboratory and Lu laboratory for helpful suggestions and comments. We thank Dr Le Ma and Dan Gibson at the Zilkha Neurogenetic Institute at USC for their help and suggestions during the course of the experiments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Melom J.E., Littleton J.T. Synapse development in health and disease. Curr. Opin. Genet. Dev. 2011;21:256–261. doi: 10.1016/j.gde.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 2.van Spronsen M., Hoogenraad C.C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 2010;10:207–214. doi: 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourgeron T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Grant S.G. Synaptopathies: diseases of the synaptome. Curr. Opin. Neurobiol. 2012;22:522–529. doi: 10.1016/j.conb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Krueger D.D., Tuffy L.P., Papadopoulos T., Brose N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 2012;22:412–422. doi: 10.1016/j.conb.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Bottos A., Rissone A., Bussolino F., Arese M. Neurexins and neuroligins: synapses look out of the nervous system. Cell Mol. Life Sci. 2011;68:2655–2666. doi: 10.1007/s00018-011-0664-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sudhof T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig A.M., Kang Y. Neurexin–neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007;17:43–52. doi: 10.1016/j.conb.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolliger M.F., Frei K., Winterhalter K.H., Gloor S.M. Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem. J. 2001;356:581–588. doi: 10.1042/0264-6021:3560581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas N.S., Sharp A.J., Browne C.E., Skuse D., Hardie C., Dennis N.R. Xp deletions associated with autism in three females. Hum. Genet. 1999;104:43–48. doi: 10.1007/s004390050908. [DOI] [PubMed] [Google Scholar]
- 11.Jamain S., Quach H., Betancur C., Rastam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C., et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laumonnier F., Bonnet-Brilhault F., Gomot M., Blanc R., David A., Moizard M.P., Raynaud M., Ronce N., Lemonnier E., Calvas P., et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J., Oliveira G., Coutinho A., Yang C., Feng J., Katz C., Sram J., Bockholt A., Jones I.R., Craddock N., et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry. 2005;10:329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 14.Pampanos A., Volaki K., Kanavakis E., Papandreou O., Youroukos S., Thomaidis L., Karkelis S., Tzetis M., Kitsiou-Tzeli S. A substitution involving the NLGN4 gene associated with autistic behavior in the Greek population. Genet. Test Mol. Biomarkers. 2009;13:611–615. doi: 10.1089/gtmb.2009.0005. [DOI] [PubMed] [Google Scholar]
- 15.Gauthier J., Bonnel A., St-Onge J., Karemera L., Laurent S., Mottron L., Fombonne E., Joober R., Rouleau G.A. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005;132B:74–75. doi: 10.1002/ajmg.b.30066. [DOI] [PubMed] [Google Scholar]
- 16.Ylisaukko-oja T., Rehnstrom K., Auranen M., Vanhala R., Alen R., Kempas E., Ellonen P., Turunen J.A., Makkonen I., Riikonen R., et al. Analysis of four neuroligin genes as candidates for autism. Eur. J. Hum. Genet. 2005;13:1285–1292. doi: 10.1038/sj.ejhg.5201474. [DOI] [PubMed] [Google Scholar]
- 17.Lawson-Yuen A., Saldivar J.S., Sommer S., Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 2008;16:614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- 18.Talebizadeh Z., Lam D.Y., Theodoro M.F., Bittel D.C., Lushington G.H., Butler M.G. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J. Med. Genet. 2006;43:e21. doi: 10.1136/jmg.2005.036897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamain S., Radyushkin K., Hammerschmidt K., Granon S., Boretius S., Varoqueaux F., Ramanantsoa N., Gallego J., Ronnenberg A., Winter D., et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl Acad. Sci. USA. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Kordi A., Winkler D., Hammerschmidt K., Kastner A., Krueger D., Ronnenberg A., Ritter C., Jatho J., Radyushkin K., Bourgeron T., et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav. Brain Res. 2012 doi: 10.1016/j.bbr.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Konopka G., Wexler E., Rosen E., Mukamel Z., Osborn G.E., Chen L., Lu D., Gao F., Gao K., Lowe J.K., et al. Modeling the functional genomics of autism using human neurons. Mol. Psychiatry. 2012;17:202–214. doi: 10.1038/mp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H., Lensch M.W., Cahan P., Daley G.Q. Investigating monogenic and complex diseases with pluripotent stem cells. Nat. Rev. Genet. 2011;12:266–275. doi: 10.1038/nrg2951. [DOI] [PubMed] [Google Scholar]
- 23.Vaccarino F.M., Urban A.E., Stevens H.E., Szekely A., Abyzov A., Grigorenko E.L., Gerstein M., Weissman S. Annual research review: the promise of stem cell research for neuropsychiatric disorders. J. Child Psychol. Psychiatry. 2011;52:504–516. doi: 10.1111/j.1469-7610.2010.02348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross P.J., Ellis J. Modeling complex neuropsychiatric disease with induced pluripotent stem cells. F1000 Biol. Rep. 2010;2:84. doi: 10.3410/B2-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saporta M.A., Grskovic M., Dimos J.T. Induced pluripotent stem cells in the study of neurological diseases. Stem Cell Res. Ther. 2011;2:37. doi: 10.1186/scrt78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hotta A., Cheung A.Y., Farra N., Vijayaragavan K., Seguin C.A., Draper J.S., Pasceri P., Maksakova I.A., Mager D.L., Rossant J., et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods. 2009;6:370–376. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- 27.Brennand K.J., Simone A., Jou J., Gelboin-Burkhart C., Tran N., Sangar S., Li Y., Mu Y., Chen G., Yu D., et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urbach A., Bar-Nur O., Daley G.Q., Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng L., Zhang P., Shi L., Yamamoto V., Lu W., Wang K. Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS ONE. 2013;8:e59685. doi: 10.1371/journal.pone.0059685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J.E., O'Sullivan M.L., Sanchez C.A., Hwang M., Israel M.A., Brennand K., Deerinck T.J., Goldstein L.S., Gage F.H., Ellisman M.H., et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc. Natl Acad. Sci. USA. 2011;108:3005–3010. doi: 10.1073/pnas.1007753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tai Y.C. Timecourse: Statistical Analysis for Developmental Microarray Time Course Data. 2007. http://www.bioconductor.org .
- 32.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Bottos A., Destro E., Rissone A., Graziano S., Cordara G., Assenzio B., Cera M.R., Mascia L., Bussolino F., Arese M. The synaptic proteins neurexins and neuroligins are widely expressed in the vascular system and contribute to its functions. Proc. Natl. Acac. Sci. USA. 2009;106:20782–20787. doi: 10.1073/pnas.0809510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayapandian M., Chapman A., Tarcea V.G., Yu C., Elkiss A., Ianni A., Liu B., Nandi A., Santos C., Andrews P., et al. Michigan Molecular Interactions (MiMI): putting the jigsaw puzzle together. Nucleic Acids Res. 2007;35:D566–D571. doi: 10.1093/nar/gkl859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakmann B., Neher E. Patch clamp techniques for studying ionic channels in excitable membranes. Annu. Rev. Physiol. 1984;46:455–472. doi: 10.1146/annurev.ph.46.030184.002323. [DOI] [PubMed] [Google Scholar]
- 36.Bebarova M. Advances in patch clamp technique: towards higher quality and quantity. Gen. Physiol. Biophys. 2012;31:131–140. doi: 10.4149/gpb_2012_016. [DOI] [PubMed] [Google Scholar]
- 37.Qiu S., Luo S., Evgrafov O., Li R., Schroth G.P., Levitt P.K., Knowles J.A., Wang K. Single-neuron RNA-Seq: technical feasibility and reproducibility. Front in Genet. 2012;3:124. doi: 10.3389/fgene.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bell O., Tiwari V.K., Thoma N.H., Schubeler D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 40.Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su Z., Li Z., Chen T., Li Q.Z., Fang H., Ding D., Ge W., Ning B., Hong H., Perkins R.G., et al. Comparing next-generation sequencing and microarray technologies in a toxicological study of the effects of aristolochic acid on rat kidneys. Chem. Res. Toxicol. 2011;24:1486–1493. doi: 10.1021/tx200103b. [DOI] [PubMed] [Google Scholar]
- 42.Nookaew I., Papini M., Pornputtapong N., Scalcinati G., Fagerberg L., Uhlen M., Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40:10084–10097. doi: 10.1093/nar/gks804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baginsky S., Hennig L., Zimmermann P., Gruissem W. Gene expression analysis, proteomics, and network discovery. Plant Physiol. 2010;152:402–410. doi: 10.1104/pp.109.150433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saeed A.I., Bhagabati N.K., Braisted J.C., Liang W., Sharov V., Howe E.A., Li J., Thiagarajan M., White J.A., Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 46.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 47.Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.