

Abstract
Snyder–Robinson syndrome (SRS, OMIM: 309583) is an X-linked intellectual disability (XLID) syndrome, characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. It is caused by a significant decrease or loss of spermine synthase (SMS) activity. Here, we report a new missense mutation, p.Y328C (c.1084A>G), in SMS in a family with XLID. The affected males available for evaluation had mild ID, speech and global delay, an asthenic build, short stature with long fingers and mild kyphosis. The spermine/spermidine ratio in lymphoblasts was 0.53, significantly reduced compared with normal (1.87 average). Activity analysis of SMS in the index patient failed to detect any activity above background. In silico modeling demonstrated that the Y328C mutation has a significant effect on SMS stability, resulting in decreased folding free energy and larger structural fluctuations compared with those of wild-type SMS. The loss of activity was attributed to the increase in conformational dynamics in the mutant which affects the active site geometry, rather than preventing dimer formation. Taken together, the biochemical and in silico studies confirm the p.Y328C mutation in SMS is responsible for the patients having a mild form of SRS and reveal yet another molecular mechanism resulting in a non-functional SMS causing SRS.
INTRODUCTION
Snyder–Robinson syndrome (SRS, OMIM 309583) is an X-linked intellectual disability (XLID) syndrome characterized by facial asymmetry, marfanoid habitus and an unsteady gait in association with mild-moderate intellectual disability (1). This original clinical description was expanded to include a thickened lower lip, nasal dysarthic speech, narrow or cleft palate, diminished muscle mass, osteoporosis, kyphoscoliosis and long great toes upon re-evaluation of the original family (2). The causative X-linked gene was identified by Cason et al. (3), when a splice mutation was found in the spermine synthase (SMS) gene (OMIM 300105) resulting in the truncation of the protein. Biochemical studies confirmed low levels of intracellular spermine (SPM), a decreased SPM/spermidine (SPD) ratio and significantly decreased SMS activity. Recently, two additional mutations in SMS have been identified in families whose phenotypes are consistent with SRS (4,5). These additional families expanded the SRS phenotype to include profound ID, seizures, short stature, pectus carinatum and myopia.
The recently solved 3D structure of SMS (6) provides critical insights into the structural features involved in its function. It was shown that the biological unit of SMS is a dimer, an observation which was confirmed by biochemical experiments (7,8). Structural analysis suggested that SMS must function as a dimer instead of a monomer. Each monomer is composed of N- and C-terminal domains, having distinct roles for the catalysis of SPD to SPM. It was demonstrated that the deletion of the N-domain disables dimerization and results in the lack of activity (7,8). This finding indicates that the N-terminal domain plays a crucial role for the dimerization, which in turn is essential for the function of SMS. The active site of the enzyme is located in the C-terminal domain and, therefore, any mutation in the region could affect the function directly.
The availability of the 3D structure of SMS allowed a series of in silico investigations of some missense mutations on SMS stability, function and domain–domain interactions (7–10). It was demonstrated that some of the missense mutations affect the function of SMS directly by disturbing wild-type properties of the active site, while other alter the function indirectly by preventing dimer formation (7). Furthermore, an investigation was carried out to probe if the disease-causing mutation sites could accommodate harmless mutations (8). It was shown that some of the missense mutation sites can tolerate almost any other substitution except for the disease-causing mutation. In contrast, any alteration at other mutation sites results in a profound effect and is expected to cause SRS. Clearly, these sites are essential for either dimerization or catalytic function of the dimer.
Here, we describe a novel missense mutation in SMS (p.Y328C; c.1084A>G), in a small XLID family in which the affected males present with a mild phenotype. We have employed molecular, biochemical and in silico analyses to assess the effect of the p.Y328C mutation on SMS function.
RESULTS
Clinical description
The index patient, IV-3 (Fig. 1), was born as the third child of healthy and unrelated parents. His older brother and sister are healthy. Before birth, intra-uterine growth retardation was noted, as well as a single umbilical artery. He was born at 36 weeks of gestation with weight of 2000 g (10th centile) and length of 42 cm (below 3rd centile). He was transferred to the neonatal unit because of recurrent hypoglycemia, which resolved spontaneously. At that time, a sacral dimple was noted. At the age of 2 years and a half, the boy was seen by a clinical geneticist because of global developmental delay, with especially delayed language development. He learned to walk at age 18 months. Despite speech therapy, his active and passive language development remained delayed with echolalia and poor understanding. An IQ test (Wechsler preschool and primary scale of intelligence-revised) at the age of 6 years showed a borderline to mild intellectual disability (total IQ 74) with a discrepancy between verbal (VIQ 72) and performal IQ (PIQ 86). He went to a special school for children with mild ID. In the following years, he was seen at several occasions at the pediatric consultation because of growth delay and especially low weight gain. A metabolic and gastroenterologic work-up was, however, normal. At recent re-evaluation at the age of 13 years, we saw a friendly slender boy with weight (24.7 kg) and length (138.7 cm) both below the third centile. Head circumference is 52 cm which is at the 10th centile (Table 1). He has a frontal upsweep and mild frontal bossing, pronounced nasal bridge, small mouth and low-set ears. His build is slender with mild kyphoscoliosis and long fingers. We did not observe facial asymmetry, nor abnormal gait or other neurological symptoms. He shows a good social interaction with peers and has good eye contact. Because of rather rigid and perfectionistic behavior, a neuropsychological examination was performed, but no diagnosis of autism spectrum disorder could be retained.
Figure 1.

Pedigree of family L091. Males with proven mutation in SMS are indicated with an asterisk. Males II-5 and II-6 are presumed to have SRS because they had intellectual disability. Individuals available for molecular analysis are indicated by ‘DNA’.
Table 1.
Clinical presentation of affected males in family L091
| Clinical features | Proband (IV-3) | Uncle (II-3) | Publisheda |
|---|---|---|---|
| Age | 13 | 42 | – |
| Intellectual disability | + (mild) | + (mild–moderate) | 10/10 (5 profound/severe, 5 mild/moderate) |
| Asthenic body build | + | +b | 10/10 |
| Diminished muscle mass | + | − | 10/10 |
| Prominent lower lip | − | + | 10/10 |
| Speech abnormalities | Echolalia | Slow | 10/10 |
| Osteoporosis | ND | ND | 7/7 |
| Long hands | + | − | 8/10 |
| Kyphscoliosis | + | − | 8/10 |
| High narrow or cleft palate | + | + | 6/8 |
| Facial asymmetry | − | − | 6/10 |
| Unsteady gait | − | − | 6/10 |
| Long great toes | + | − | 8/10 |
The younger brother of the mother, individual III-3, also followed special school and works now in a sheltered environment. As a child and at young adult age, this man was also extremely slender. Apparently, the maternal grandmother was never able to read or to write. She also had two brothers with intellectual disability. These individuals were not available for clinical examination or testing.
Molecular and biochemical analyses
Mutation analysis
Analysis of the data obtained by RainDance amplification and sequencing by the Illumina/Solexa system identified a mutation, c.1084A>G (p.Y328C), in the SMS gene in the index patient. This mutation was subsequently confirmed by Sanger sequencing and was also present in the carrier mother and her affected brother. The mutation was absent in the healthy brother of the patient.
The SPM/SPD ratio and SMS activity
In order to confirm the pathogenicity of the p.Y328C mutation in the SMS gene, the SPM/SPD ratio was determined utilizing lymphoblastoid cells prepared from the proband. The patient's ratio of 0.529 was significantly different from the average ratio of two controls, 1.87 (Table 2). This low ratio is similar to that reported for the three previously published patients with SRS (3–5). Next, the SMS activity was measured to insure the reduced SPM/SPD ratio was due to a deficiency in the activity of SMS. The activity of the proband's SMS was 364 units, not much different from background (294 units) and significantly below the average of 7293 units found in two control samples (Table 1). Again, this was consistent with results published previously for other SMS mutations (3–5).
Table 2.
SMS activity and the SPM/SPD ratio
| Sample | SMS activitya | SPM/SPD |
|---|---|---|
| Normal control 1 | 7221 | 2.25 |
| Normal control 2 | 7365 | 1.50 |
| Patient IV-3 | 364 | 0.53 |
aActivity is measured in counts of SPM d8 generated per mg lysate per hour from SPD d8. Baseline, without any lysate, was 294.
Western blot analysis
As shown above, the p.Y328C missense mutation in SMS results in SRS due to the complete reduction in enzymatic activity. This could be a result of many things, one of which would be the absence of protein. This possibility was investigated by western blot analysis. As shown in Figure 2, the protein level is indeed reduced but only to ∼20% of normal. This level would likely result in 80% reduction of activity but not in complete reduction. One possibility is that the mutation partially prevents dimer formation which is required for activity.
Figure 2.

Western blot analysis of SMS levels in lymphoblast cell lines. (A) 10 µg of lymphoblast lysate was prepared in Lamelli sample buffer, separated on a 4–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and blotted for SMS and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (n = 3). GAPDH was used as a loading control. (B) Blots were analyzed by NIH Image J and the ratio of SMS to GAPDH is reported. The average of the three experiments with bars representing the mean standard error is reported. A Student's t-test was done to determine significance. *Not significant, P > 0.05 and **significant, P < 0.05.
Monomer/dimer analysis
Native gel analysis of patient lymphoblastoid cells was conducted in order to determine the level of the dimer form of the Y328C SMS protein since SMS functions only when it exists as a dimer. As shown in Figure 3, the SMS protein in the patient exists almost completely as a dimer. The level is ∼20% that of controls consistent with the total protein analysis. However, the finding does not account for the lack of SMS activity.
Figure 3.

Native gel analysis of SMS in lymphoblast cell lines. (A) 10 µg of lymphoblast lysate was prepared in native sample buffer, separated on a 7.5% native gel and probed for SMS. An equivalent amount of lysate was prepared in Lamelli sample buffer, separated on a 4–20% SDS–PAGE gel and blotted for GAPDH (n = 3). GAPDH was used as a loading control. (B) Blots were analyzed by NIH Image J and the ratio of SMS to GAPDH protein reported. The average of the three experiments with bars representing the mean standard error is reported. Student's t-test was done to determine significance. *Not significant P > 0.05 and **significant P < 0.05.
Neurite assay and Sholl analysis
Although the low SPM/SPD ratio and loss SMS activity are consistent with the males having SRS, their presentation is mild relative to that observed in other patients reported with SRS (Table 1) (2–5). In an attempt to better understand this variability, we examined the neurite length of PC12 cells transfected with constructs of SMS. A form of this assay has been utilized to study the effect of polyamines on nerve growth factor (NGF)-stimulated PC12 cells (11) and the effect of SSRI inhibitors on neurite outgrowth in NGF-induced PC12 cells (12). As shown in Figure 4, the Y328C mutation in SMS did not result in a significant reduction in the percentage of cells which had a neurite length at least the same as its cell body width. However, the p.89 I_ins21X mutation identified in the original SRS family caused a significant decrease in this number when compared with cells transfected with WT SMS. In addition, we performed branching analysis to look at dendritic arborization in these transfected PC12 cells. Whereas no difference is seen between the Y328C mutation and WT transfected cells, we observe a significant decrease in dendritic branching and hence complexity in the cells transfected with the p.89 I_ins21X mutation construct (Fig. 4 and Supplementary Material). This graph also confirms the shorter length of the neurites in the latter. This finding might correlate with the milder SRS presentation in family L091.
Figure 4.
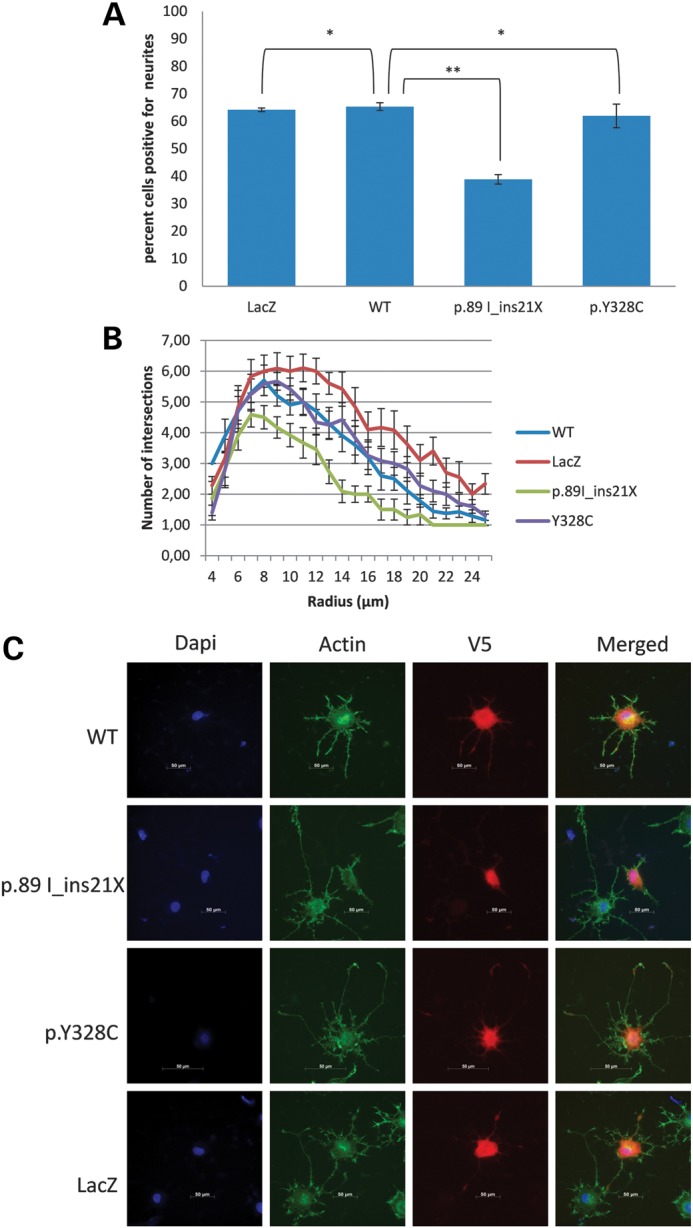
Neurite analysis of PC12 cells transfected with plasmids containing different constructs of SMS. The plasmids have a V5 tag which allowed the identification of transfected cells. LacZ, negative control; WT, wild-type SMS; p.89 I_ins21X, mutation in proband from the original SRS family; p.Y328C, mutation in patient IV-3, family L091. (A) Analysis of PC12 cells forming neurites. Three replicates of 50 cells were counted per set. A Student's t-test was done to determine significance. *Not significant, P > 0.05; **significant, P < 0.001. The average of three experiments is reported with bars representing the mean standard error. (B) Sholl analysis and branch length analysis of PC12 cells transfected with plasmids containing different constructs of SMS, as described in Materials and Methods. Three sets of 15 cells were analyzed for each sample. The number of intersections is plotted against the increasing radius (distance to the centre of the soma), including bars representing the mean standard error. Results of Student's t-test for this analysis are shown in Supplementary Material, Table S4. (C) Representative images of cells analyzed for neurite formation and Sholl analysis.
In silico modeling
Since the SMS protein exists as a dimer in the patient and is present at 20% of control levels, the lack of enzymatic activity is difficult to understand. The computational results indicate that the Y328C mutation has little effect on dimer affinity. The predicted ΔΔΔG(binding_Mut) is 1.25 kcal/mol, which is small effect from computational stand point (Supplementary Material, Table S1). Our previous work indicates that the results from Molecular Mechanics Generalized Born (MMGB) calculations overestimate the experimental folding free energy changes by a factor of ∼10 (13). Similar effect is expected for binding free energy. Thus, the expected dimer affinity change can be estimated to be ∼0.125 kcal/mol. Such minor effect on dimer affinity upon Y328C is not surprising, since the mutation site Y328 is far away from the dimer interface (Fig. 7). At the same time, it should be mentioned that this small energy change favors the dimer formation. This confirms the experimental observation that although the amount of protein is reduced to 20%, all remaining proteins form dimers.
Figure 7.
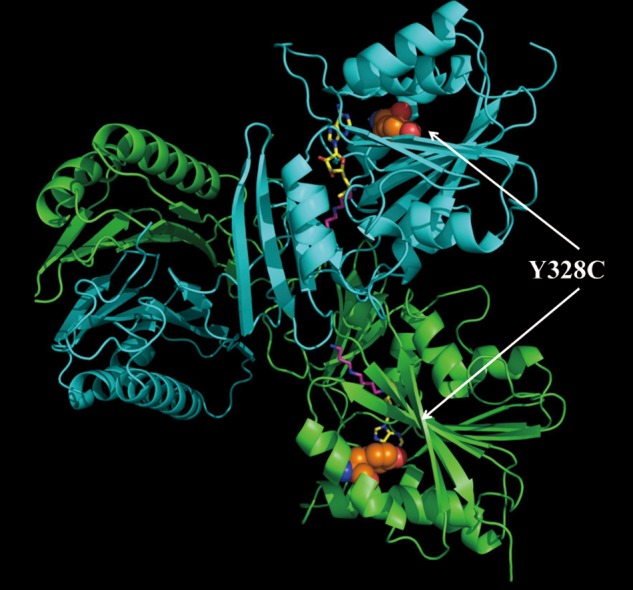
The 3D structure of the SMS dimer. The green one is the C chain, while the cyan one is the D chain. The disease-causing mutation site is shown with colored balls (orange, carbon atom; red, oxygen atom; and blue, nitrogen atom); SPD and MTA are represented with magenta stick and yellow stick, respectively.
The calculated folding free energy change, averaged over three force field parameters, is about three times larger (ΔΔG(mut) = −3.40 kcal/mol; Supplementary Material, Table S2). The negative sign indicates that the mutation makes the monomers less stable than the wild-type protein. Having in mind the close proximity of the mutation site to the active pocket of SMS, such a destabilization may have significant effect on the catalysis. However, the observations made by in vitro and in silico investigations indicate that the monomer destabilization does not affect the dimer formation. Thus, the effect on the catalysis should stem from perturbations of the active site wild-type properties.
Both the wild-type and the mutant C-terminal domains were subjected to MD simulations as described in the Materials and methods section. Figure 5 shows the root mean square deviation (RMSD) as a function of simulation time. It can be seen that RMSD fluctuations in wild-type structure quickly reach saturation and remain within 1.4 Å, while the mutant fluctuations do not completely saturate and are much larger (>2 Å) than those of the wild-type. This is consistent with the results of the energy calculations, indicating that the mutant is less stable than the wild-type. The increase in the conformational dynamics of the Y328C mutant is so significant that perhaps it is the major factor contributing to the loss of activity.
Figure 5.

RMSD of the wild-type (WT) and the mutant (Y328C) as a function of the simulation time.
Structural analysis of the effect of the Y328C mutant was carried out to reveal the changes in the hydrogen bond network in the vicinity of the mutations site (Fig. 6). It can be seen that in the wild-type, Tyr328 is involved in two hydrogen bonds. In both cases, Tyr328 oxygen serves as a hydrogen acceptor accepting the hydrogen bond from Tyr312 and Thr314. In contrast, the mutant, Cys328, plays the role of hydrogen donor providing a hydrogen bond to the neighboring Tyr258. Such a dramatic change of the hydrogen bond pattern around the active site of SMS definitely should have a large impact on the catalysis.
Figure 6.
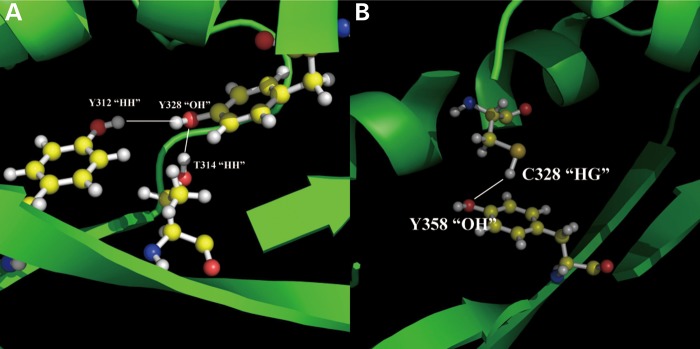
The effects on H-bond network due to the missense mutation Y328C. The yellow balls represented carbon atoms; the red balls represented oxygen atoms and the white balls represented the hydrogen atoms. (A) The energy minimized WT structure with optimized potentials for liquid simulations (OPLS); (B) the energy minimized mutant structure with OPLS.
DISCUSSION
The clinical findings observed in patients III-3 and IV-3 are very similar to those reported for other patients with SRS (Table 1) (2–5). Originally, this rare XLID syndrome was characterized by tall stature, a thin marfanoid habitus, some facial asymmetry, osteoporosis and kyphoscoliosis (1). Re-evaluation identified additional features such as an unsteady gait, nasal speech, pectus carinatum, long toes and seizures (2,3). The identification of a mutation in the SMS gene as the cause of SRS allowed additional families to be reported (4,5), expanding the associated phenotype. Although the asthenic build was always present, not all males had a tall stature. The severity of ID now included severe to profound along with mild to moderate. A short philtrum, mandibula prognatheism and pectus carinatum were also noted, and seizures are not always present.
The Belgium patients reported here present with a milder phenotype, the oldest affected man being able to live and work in a sheltered environment. Both show similar physical characteristics and have speech problems. They do not, however, have any gait problems. It is unclear why they are more mildly affected, although their SPM/SPD ratio is not lower than that reported for other affected males (Table 1) (3–5) and the SMS activity is as low as more severely affected males (Table 1). We noted, however, differences in a neurite assay and branching analysis involving transfected PC12 cells. The percent of cells with neurites longer than the width of the cell body was not statistically different from cells transfected with wild-type SMS (Fig. 4). This is in contrast to the significantly reduced number of neurite outgrowth in cells transfected with truncated SMS caused by the splice mutation in the original SRS family. We also observed a significant decrease in branching and dendritic arborization in these cells transfected with the truncated SMS variant. This would seem to indicate that the p.Y328C mutation does not interfere with neurite outgrowth and branching and might suggest that the SMS protein has an additional function beyond its enzymatic activity.
In vitro experiments showed that the newly discovered SRS causing mutation Y328C abolishes SMS activity, while retaining ∼20% of the protein and having no effect on dimer formation. This is in contrast with previous in vitro and in silico investigations, showing that another missense mutation, the G56S mutation, causing SRS (7) abolishes the function of SMS by disrupting dimer formation. Instead, the Y328C mutation has little effect on the dimerization but huge effect on conformational dynamics and hydrogen bond network around the active site (Fig. 6) and thus greatly reducing the activity.
The molecular effect of this mutation (Y328C) is quite similar to the suggested molecular mechanism of another SRS causing mutation I150T. They are both situated in the vicinity of the active site and are far away from the dimer interface. However, in contrast to I150T, the Y328C mutant affects the hydrogen bond network of residues involved in the geometry of SPD/SPM binding pocket, rather than the MTA pocket. At the same time, these two sites, 150 and 328, were predicted to have similar mutability (Supplementary Material, Tables S1 and S2) (8). Almost any substitution is expected to have dramatic effect on the mutant SMS stability, dynamics and hydrogen bond network around the site of mutation. Other known to date SRS causing mutations are G56S and I132V, which are located at the dimer interface. Previous work (7) showed that they do not affect the active site, but disrupt the SMS function by affecting dimer formation or stability of the SMS. Thus, comparison of the current work with previous investigations (7) demonstrates that SRS is caused by various molecular mechanisms. However, despite of the different molecular mechanisms, the result at the cellular level is a deviation away from the wild-type SPM/SPD ratio resulting in SRS.
MATERIALS AND METHODS
Mutation analysis
Family L091 is part of a large cohort of families and patients with XLID that were collected at the Center for Human Genetics of the University Hospitals Leuven in order to identify causes of intellectual disability (14). The protocol was approved by the Institutional Review Board of the University Hospitals of Leuven (Belgium), and informed consent was obtained. Genomic DNA from patients as well as from family members and healthy controls was isolated from peripheral blood according to the standard procedures and stored at 4°C.
In a study to elucidate the genetic defects in families with apparent XLID, we performed the enrichment of the coding and flanking intronic sequences of 86 known XLID genes followed by next-generation sequencing using the Illumina/Solexa system. Details of this method were described in Hu et al. (15). Confirmation of the mutation in SMS and segregation analysis was done by Sanger sequencing (primer sequences available on request). The mutation was not found in the 1000 Genomes cohort, neither in the NHLBI Exome Sequencing Project which includes more than 4300 European-American exomes. The mutation was also not found in the internal Leuven exome database that consists of more than 400 exomes to date.
SPM/SPD ratio determination
Patient lymphoblast cells were pelleted by centrifugation at 450g for 3 min. The cells were resuspended in phosphate buffered saline (PBS) and again pelleted by centrifugation. PBS was removed from the cell pellet and the pellet was frozen at −80°C. The cell pellet was thawed on ice, resuspended in 50 mm phosphate buffer, pH 7.5, sonicated 3× for 10 s each and centrifuged at 10 000g for 3 min. The supernatant was removed to a fresh tube and quantified using the Lowry method. A 100µl suspension was prepared containing 35 µg of cell lysate and 2.5 µm of SPD d8 as a loading control in a 50 mm phosphate buffer. 100 µl of acetronitrile mixture containing 0.1% formic acid was added to the sample and assayed on Waters Quattro Micro tandem mass spectrometer using Waters C8 column. A standard curve for SPD and SPM was generated and the results were analyzed using Quanlynx program as described by Sowell et al. (16).
SPM activity assay
Patient lymphoblast cells were pelleted by centrifugation at 450g for 3 min. The cells were washed twice in ice-cold PBS and pelleted by centrifugation. PBS was removed from the cell pellet. The pellet was resuspended in ice-cold Buffer A (50 mm sodium phosphate buffer, pH 7.2, 0.3 m ethylenediaminetetraacetic acid) plus 1× protease inhibitor cocktail (Sigma catalogue # P2714) and frozen at −80°C. The cell pellet was thawed on ice and refrozen at −80°C again. The pellets was thawed on ice again and centrifuged at 10 000g for 5 min at 4°C. The supernatant was removed to a chilled tube and quantified using the Bradford method. 70 µg of cell lysate was used in an assay containing 100 µm sodium phosphate, pH 7.5, 100 µm SPD d8 (Sigma catalogue #709891), 1× protease inhibitor cocktail, 100 µm decarboxylated S-adenosylmethionine (provided by Dr. Ikeguci) and 50 µm 4-MCHA (Sigma catalogue #177466). After 24 h, the reaction was stopped. SPD D8 and SPM D8 were measured using tandem mass spectrometry as described by Sowell et al. (16).
Plasmid construction
cDNA was prepared with Superscipt First Strand Synthesis Kit for reverse transcription polymerase chain reaction (Invitrogen catalogue nubmer11904-018) from 2 mg of RNA prepared from lymphoblast cell lines. The gene-specific oligo SMS-RT (5′-GAA GGC TAT TTG CAG CAC ATG TGA-3′) was used to generate the first-strand cDNA for the control and patient 1. SMSmut-RT (5′-CAC TTC ATC TAT GTC ATA TTC AAC-3′) was used to generate the first-strand of cDNA for the original patient carrying a truncating mutation (3). A polymerase chain reaction with the oligos SMS-F (5′-CAC CAT GGC AGC AGC ACG GCA CAG CAG G-3′) and SMS-R (5′-GGG TTT AGC TTT CTT CCA AAC AGT-3′) and PFU Turbo (Stratagene catalogue number 600250) were employed to generate the insert for the control and patient 1. For the original patient, SMS-F and the oligo SMSmut-R (5′-ACC AGG CGC CCG TCG GCG GTG GGC-3′) were employed to generate the insert. The fragments were run on a 1% tris-acetate-EDTA agarose gel and purified with a Gel Extraction Kit (Qiagen catalogue number 287040). The purified product was cloned in the pcDNA3.1D/V5-His-Topo vector using the pcDNA3.1 Directional Topo Expression Kit (Invitrogen catalogue number K4900-01). All plasmids generated from this have a V5 tag on the C terminus. The LacZ V5 plasmid was included as a positive control with the kit. Plasmids were sequenced to confirm the insert with the vector specific primers PC (5′-GGG AGA CCC AAG CTG GCT AGT-3′) and BGH (5′-TAG AAG GCA CAG TCG AGG-3′) and insert specific primers SMS285For (5′-GAG AAT TTA CCC ACA TGG ATT-3′), SMS330Rev (5′-GTC GGC CAG TAT CTG TCG AT-3′), SMS660Rev (5′-GTC TCC ACC TCC CAG AAT GA-3′) and SMS960Rev (5′-GGA GAC GTG GAG ATT GGA ACA-3′).
Transfection
PC12 cells were cultured on poly-l-lysine (Sigma catalogue number P4707) coated 24-well tissue culture dishes in growth media 18–24 h priors to transfection. A transfection complex containing Dulbecco's modified Eagle's medium (Sigma catalogue number D5796), 1 mg of plasmid and 3.5 µl of NeuroMag (Bocca Scientific catalogue number NM50500) was prepared and added to each well. The 24-well plate was placed on the Super Magnetic Plate (Bocca Scientific catalogue number MF10000). After 20 min on the Super Magnetic Plate, the 24-well plate was removed and placed in a 5% CO2 humidified 37°C incubator. After 5 h, the transfection complexes were removed, the cells washed one time with PBS and growth media added back to the cells. Twenty-four hours post-transfection, the cells were moved to a poly-l-lysine coated glass slip and cultured in growth media with 100 ng of NGF for 36–40 h.
Immunofluorescense
Forty-eight hours after NGF treatment, the cells were washed twice with PBS, fixed with 4% paraformaldehyde/PBS, permeabilized with 0.1% Triton X-100/PBS and blocked with block (2% horse sera, 0.4% bovine serum albumin (BSA) in PBS). The cells were incubated with the anti-V5 mouse monoclonal antibody (Invitrogen catalogue number R960.25), washed with block and incubated with Alexa Fluor 594 anti-Mouse IgG and Alexa Fluor 488 Phalloidin (Invitrogen catalogue numbers A21200 and A12379), washed with block, incubated with Dapi and mounted with Gold Prolong anti-fade medium (Invitrogen catalogue number P36930).
Neurite assay and Sholl analysis
PC12 cells were transfected with V5-tagged vectors containing either wild-type SMS or mutations from the original SRS family (1) or the present family and immunofluorescence was done as described above. Cells expressing the different constructs were imaged on a Zeiss Observer. A1 AX10 inverted microscope using AxioViosion software 4.8. Cells with at least one neurite process, the length of which was at least the width of the cell body, were counted as positive. Three sets of 100 cells were counted for each sample. Sholl analysis was performed using NIH ImageJ software with ‘Advanced Sholl analysis’ plug-in v1.0 to quantify neuritic complexity (available at http://biology.ucsd.edu/labs/ghosh/software/index.html). Analysis was done by specifying a center point exactly at the middle of soma and by applying a grid with concentric rings of 1 µm apart over the dendritic drawing. The number of intersections given by the program was used to estimate the total dendritic branching. We only used cells that were completely stained and that were not overlapping so that the complete arborization was visible. Three sets of 15 cells were analyzed for each sample.
Western blot
The membrane was blocked with 2% BSA/TBST (tris-buffered saline Tween), probed with an anti-SMS antibody (Abnova catalogue number H00006611-M01), rinsed in TBST, incubated with anti-Mouse IgG horseradish peroxidase (HRP) (Pierce catalogue number 31432), rinsed in TBST and detected on an X-ray film after development with West Dura ECL Kit (Pierce). The membrane was stripped with restore western blot stripper (Pierce catalogue number 21059), blocked with 2% BSA/TBST, probed with an anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Biotechnology catalogue number 32232), rinsed in TBST, incubated with anti-mouse IgG HRP (Pierce catalogue number 21059), rinsed in TBST and detected on an X-ray film after development with West Dura ECL Kit (Pierce catalogue number 34075). The films were analyzed using NIH Image J software.
Native gel electrophoresis
Patient lymphocytes were centrifuged at 450g for 3 min, resuspended in PBS and centrifuged at 450g for 3 min. The PBS wash was repeated. The cell pellet was resuspended in ice cold native gel sample buffer (0.62 mm Tris–HCL pH 6.8, 0.01% bromophenol blue, 105 glycerol), and sonicated. Equal sample volumes were separated on a 7.5% native gel and transferred to nitrocellulose. The membrane was blocked with 2% BSA/TBST, probed with anti-SMS antibody (Abnova catalogue number H00006611-M01), rinsed in TBST, incubated with anti-mouse IgG HRP (Pierce catalogue number 31432), rinsed in TBST and detected on an X-ray film after development with West Dura ECL Kit (Pierce catalogue number 34075). An equivalent amount of protein was denatured in Lamelli buffer and separated on a 4–20 sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) (Fisher) and transferred to nitrocellulose and western blotted for GAPDH as indicated above.
Protein structures
The 3D SMS structure was downloaded from the Protein Data Bank (PDB), PDB ID: 3C6K (17). Following our previous work (7,8), we used chains C and D in our analysis. The structural defects as missing residues and atoms were fixed with ‘profix’ from Jackal package (http://wiki.c2b2.columbia.edu/honiglab_public/index.php/Software:Jackal_General_Description). The missense mutation and any other mutation were introduced in silico utilizing side chain confirmation prediction (SCAP) in Jackal package as well (18). Figure 7 shows the 3D structure of the human native SMS with the ligands of SPD and MTA. And the corresponding mutation site Y328 is represented by the colored balls.
Energy calculations
The SMS is a large protein containing 381 (only 366 amino acids are present in the X-ray structure) amino acids in each chain. Applying the same approach as in our previous study (7,8), each monomer was split into two terminal domains: N-terminal domain (residue numbers from 2 to 109) which plays an essential role for the dimerization and C-terminal domain (residue numbers from 118 to 364) which carries the reaction and where the pathogenic mutation site Y328 is. Since both the dimerization and the stability of individual domains are equally important for the SMS function, below we carry in silico analysis on binding affinity of the two subunits and domain stability.
Binding free energy (binding affinity) calculations were done using the C-terminal domain of SMS and utilizing the program ‘minimize’ in TINKER package (19). The energy minimization was carried out with the limited memory BFGS Quasi–Newton optimization algorithm. Details are described in our previous work (7,8). All minimizations were done with three force field parameters: Amber98 (20), Charmm27 (21) and Oplsaa (22), to probe the sensitivity of the results with respect to the force field parameters. Once the minimization was successfully done, the minimized structure of each subunit C chain and D chain were extracted from the minimized dimer structure. This ‘rigid body’ approach was shown to provide better results (7,8). The energy minimized structures of the dimer and two monomers were then subjected to the ‘analyze’ program in TINKER package (19) to obtain the energies. The binding free energy is calculated with the following formula:
| (1) |
where ΔΔG(binding) is the binding free energy of the dimer and ΔG(dimer) is the potential energy of the dimer state, while ΔG(C) and ΔG(D) are the potential energy of the monomeric states.
The same protocol was used for both native structure and the mutants, and then the binding free energy change due to the mutations is obtained as:
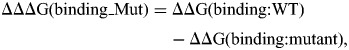 |
(2) |
where the ΔΔΔG(binding_Mut) is the binding free energy change due to the mutations and ΔΔG(binding:WT) and ΔΔG(binding:mutant) are the potential energy of the native protein and mutants, respectively. This protocol assumes that the entropies of the dimer and monomer are quite similar and that the entropy of both wild-type protein and mutated protein are very similar as well; therefore, the entropy term canceled out in Equations (1) and (2).
Folding free energy calculations (monomer stability) were done using both C and D chains, separately. The detailed procedure is described in Zhang et al. (13). Here, we provide short summary only. The structures were subjected to ‘minimize’ in TINKER to perform the energy minimization with the same protocol as the above. Then, we extracted a small structural segment of seven residues with the mutation site in the middle. The energy minimized structures of both monomers and the extracted seven residue segments were subjected to ‘analyze’ to perform the energy calculations. For the folding free energy of each subunit, the following formula was applied (13):
 |
(3) |
where ΔG(folding) is the folding free energy of the monomer; G(folded) is the potential energy of the folded state, while G(unfolded) is the potential energy of the unfolded state. Furthermore, the unfolded state is split into two terms: G0(unfolded) and G7(unfolded). G7(unfolded) is the unfolded state energy of the seven residue segments, while G0(unfolded) is the unfolded state of all the other part. We used the same formula to calculate the folding free energy of both C chain and D chain, and then results were averaged. The same protocol was used for both the wild-type protein and the mutants, and then the effects on protein stability upon the mutation are obtained as:
 |
(4) |
Entropy in both wild-type protein and mutants are assumed very similar and cancel out in formula (4). The approach of modeling seven residue segment in unfolded state instead of whole protein results that G0(unfolded) is canceled out as well (13). This method as recently tested on a set of more than 1000 experimentally determined changes of the folding free energy and was shown to provide very good results (13).
Molecular dynamics simulations
In order to address the effects of the flexibility of the active sites due to the missense mutation, molecular dynamics (MD) simulations were performed (23). The MD simulations for the wild-type and the disease-causing missense mutation Y328C structures were carried out in parallel. The temperature was set at 298 K, the time step length at 1 fs, the time between dumps at 0.1 ps and the total simulation time at 3 ns. The energy minimized structure of the C-terminal domain of both the wild-type structure and the mutant structure were then subjected to the program ‘dynamic’ in TINKER package (19).
The resulting snapshot structures were compared with the original starting structures using the Multiple Alignment with Translations and Twists (MATT) program (24). The superimposed structures were used to calculate the RMSD for each snapshot. Then, the time average of RMSD is obtained as:
| (5) |
where RMSDi stands for the RMSD of the ith snapshot and ‘n’ is the snapshot number. Such a quantity has important property to tend asymptotically to a particular value and thus to be used to assess the convergence.
SUPPLEMENTARY MATERIAL
FUNDING
A.E., Z.Z. and L.W. were supported by award from NLM/NIH (1R03LM009748). H.V. is a clinical investigator of the Fonds voor Wetenschappelijk Onderzoek Vlaanderen. Support, in part, by a grant from the South Carolina Department of Disabilities and Special Needs. Dedicated to the memory of Ethan Francis Schwartz, 1996–1998.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the patients and their family for their cooperation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Snyder R.D., Robinson A. Recessive sex-linked mental retardation in the absence of other recognizable abnormalities. Report of a family. Clin. Pediatr. (Phila.) 1969;8:669–674. doi: 10.1177/000992286900801114. doi:10.1177/000992286900801114. [DOI] [PubMed] [Google Scholar]
- 2.Arena J.F., Schwartz C., Ouzts L., Stevenson R., Miller M., Garza J., Nance M., Lubs H. X-linked mental retardation with thin habitus, osteoporosis, and kyphoscoliosis: linkage to Xp21.3-p22.12. Am. J. Med. Genet. 1996;64:50–58. doi: 10.1002/(SICI)1096-8628(19960712)64:1<50::AID-AJMG7>3.0.CO;2-V. doi:10.1002/(SICI)1096-8628(19960712)64:1<50::AID-AJMG7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 3.Cason A.L., Ikeguchi Y., Skinner C., Wood T.C., Holden K.R., Lubs H.A., Martinez F., Simensen R.J., Stevenson R.E., Pegg A.E., et al. X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur. J. Hum. Genet. 2003;11:937–944. doi: 10.1038/sj.ejhg.5201072. doi:10.1038/sj.ejhg.5201072. [DOI] [PubMed] [Google Scholar]
- 4.de Alencastro G., McCloskey D.E., Kliemann S.E., Maranduba C.M., Pegg A.E., Wang X., Bertola D.R., Schwartz C.E., Passos-Bueno M.R., Sertie A.L. New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder-Robinson X-linked recessive mental retardation syndrome. J. Med. Genet. 2008;45:539–543. doi: 10.1136/jmg.2007.056713. doi:10.1136/jmg.2007.056713. [DOI] [PubMed] [Google Scholar]
- 5.Becerra-Solano L.E., Butler J., Castaneda-Cisneros G., McCloskey D.E., Wang X., Pegg A.E., Schwartz C.E., Sanchez-Corona J., Garcia-Ortiz J.E. A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder-Robinson syndrome. Am. J. Med. Genet. A. 2009;149:328–335. doi: 10.1002/ajmg.a.32641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H., Min J., Zeng H., McCloskey D.E., Ikeguchi Y., Loppnau P., Michael A.J., Pegg A.E., Plotnikov A.N. Crystal structure of human spermine synthase: implications of substrate binding and catalytic mechanism. J. Biol. Chem. 2008;283:16135–16146. doi: 10.1074/jbc.M710323200. doi:10.1074/jbc.M710323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z., Teng S., Wang L., Schwartz C.E., Alexov E. Computational analysis of missense mutations causing Snyder-Robinson syndrome. Hum. Mutat. 2010;31:1043–1049. doi: 10.1002/humu.21310. doi:10.1002/humu.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z., Norris J., Schwartz C., Alexov E. In silico and in vitro investigations of the mutability of disease-causing missense mutation sites in spermine synthase. PLoS One. 2011;6:e20373. doi: 10.1371/journal.pone.0020373. doi:10.1371/journal.pone.0020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teng S., Madej T., Panchenko A., Alexov E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 2009;96:2178–2188. doi: 10.1016/j.bpj.2008.12.3904. doi:10.1016/j.bpj.2008.12.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teng S., Michonova-Alexova E., Alexov E. Approaches and resources for prediction of the effects of non-synonymous single nucleotide polymorphism on protein function and interactions. Curr. Pharm. Biotechnol. 2008;9:123–133. doi: 10.2174/138920108783955164. doi:10.2174/138920108783955164. [DOI] [PubMed] [Google Scholar]
- 11.Schreiber R.C., Boeshore K.L., Laube G., Veh R.W., Zigmond R.E. Polyamines increase in sympathetic neurons and non-neuronal cells after axotomy and enhance neurite outgrowth in nerve growth factor-primed PC12 cells. Neuroscience. 2004;128:741–749. doi: 10.1016/j.neuroscience.2004.07.014. doi:10.1016/j.neuroscience.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura T., Ishima T., Iyo M., Hashimoto K. Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS One. 2008;3:e2558. doi: 10.1371/journal.pone.0002558. doi:10.1371/journal.pone.0002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z., Wang L., Gao Y., Zhang J., Zhenirovskyy M., Alexov E. Predicting folding free energy changes upon single point mutations. Bioinformatics. 2012;28:664–671. doi: 10.1093/bioinformatics/bts005. doi:10.1093/bioinformatics/bts005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Brouwer A.P., Yntema H.G., Kleefstra T., Lugtenberg D., Oudakker A.R., de Vries B.B., van Bokhoven H., Van Esch H., Frints S.G., Froyen G., et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum. Mutat. 2007;28:207–208. doi: 10.1002/humu.9482. doi:10.1002/humu.9482. [DOI] [PubMed] [Google Scholar]
- 15.Hu H., Wrogemann K., Kalscheuer V., Tzschach A., Richard H., Haas S.A., Menzel C., Bienek M., Froyen G., Raynaud M., et al. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 2009;3:41–49. doi: 10.1007/s11568-010-9137-y. doi:10.1007/s11568-010-9137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sowell J., Norris J., Jones K., Schwartz C., Wood T. Diagnostic screening for spermine synthase deficiency by liquid chromatography tandem mass spectrometry. Clin. Chim. Acta. 2011;412:655–660. doi: 10.1016/j.cca.2010.12.037. doi:10.1016/j.cca.2010.12.037. [DOI] [PubMed] [Google Scholar]
- 17.Kouranov A., Xie L., de la Cruz J., Chen L., Westbrook J., Bourne P.E., Berman H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006;34:D302–305. doi: 10.1093/nar/gkj120. doi:10.1093/nar/gkj120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiang Z., Honig B. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 2001;311:421–430. doi: 10.1006/jmbi.2001.4865. doi:10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 19.Ponder J.W. TINKER-Software Tools for Molecular Design. 3.7. St. Luis: Washington University; 1999. [Google Scholar]
- 20.Case D.A., Cheatham T.E., 3rd, Darden T., Gohlke H., Luo R., Merz K.M., Jr, Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. doi:10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr, Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. doi:10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen W.L., Tiradorives J. The Opls potential functions for proteins—energy minimizations for crystals of cyclic-peptides and crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. doi:10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z., Miteva M.A., Wang L., Alexov E. Analyzing effects of naturally occurring missense mutations. Comput. Math. Methods Med. 2012;2012:805–827. doi: 10.1155/2012/805827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menke M., Berger B., Cowen L. Matt: local flexibility aids protein multiple structure alignment. PLoS Comput. Biol. 2008;4:e10. doi: 10.1371/journal.pcbi.0040010. doi:10.1371/journal.pcbi.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.