

Abstract
Over the past decade, comprehensive sequencing efforts have revealed the genomic landscapes of common forms of human cancer. For most cancer types, this landscape consists of a small number of “mountains” (genes altered in a high percentage of tumors) and a much larger number of “hills” (genes altered infrequently). To date, these studies have revealed ~140 genes that, when altered by intragenic mutations, can promote or “drive” tumorigenesis. A typical tumor contains two to eight of these “driver gene” mutations; the remaining mutations are passengers that confer no selective growth advantage. Driver genes can be classified into 12 signaling pathways that regulate three core cellular processes: cell fate, cell survival, and genome maintenance. A better understanding of these pathways is one of the most pressing needs in basic cancer research. Even now, however, our knowledge of cancer genomes is sufficient to guide the development of more effective approaches for reducing cancer morbidity and mortality.
Ten years ago, the idea that all of the genes altered in cancer could be identified at base-pair resolution would have seemed like science fiction. Today, such genome-wide analysis, through sequencing of the exome (see Box 1, Glossary, for definitions of terms used in this Review) or of the whole genome, is routine.
Box 1. Glossary.
- Adenoma
A benign tumor composed of epithelial cells.
- Alternative lengthening of telomeres (ALT)
A process of maintaining telomeres independent of telomerase, the enzyme normally responsible for telomere replication.
- Amplification
A genetic alteration producing a large number of copies of a small segment (less than a few megabases) of the genome.
- Angiogenesis
the process of forming vascular conduits, including veins, arteries, and lymphatics.
- Benign tumor
An abnormal proliferation of cells driven by at least one mutation in an oncogene or tumor suppressor gene. These cells are not invasive (i.e., they cannot penetrate the basement membrane lining them), which distinguishes them from malignant cells.
- Carcinoma
A type of malignant tumor composed of epithelial cells.
- Clonal mutation
A mutation that exists in the vast majority of the neoplastic cells within a tumor.
- Driver gene mutation (driver)
A mutation that directly or indirectly confers a selective growth advantage to the cell in which it occurs.
- Driver gene
A gene that contains driver gene mutations (Mut-Driver gene) or is expressed aberrantly in a fashion that confers a selective growth advantage (Epi-Driver gene).
- Epi-driver gene
A gene that is expressed aberrantly in cancers in a fashion that confers a selective growth advantage.
- Epigenetic
Changes in gene expression or cellular phenotype caused by mechanisms other than changes in the DNA sequence.
- Exome
The collection of exons in the human genome. Exome sequencing generally refers to the collection of exons that encode proteins.
- Gatekeeper
A gene that, when mutated, initiates tumorigenesis. Examples include RB, mutations of which initiate retinoblastomas, and VHL, whose mutations initiate renal cell carcinomas.
- Germline genome
An individual’s genome, as inherited from their parents.
- Germline variants
Variations in sequences observed in different individuals. Two randomly chosen individuals differ by ~20,000 genetic variations distributed throughout the exome.
- Human leukocyte antigen (HLA)
A protein encoded by genes that determine an individual’s capacity to respond to specific antigens or reject transplants from other individuals.
- Homozygous deletion
Deletion of both copies of a gene segment (the one inherited from the mother, as well as that inherited from the father).
- Indel
A mutation due to small insertion or deletion of one or a few nucleotides.
- Karyotype
Display of the chromosomes of a cell on a microscopic slide, used to evaluate changes in chromosome number as well as structural alterations of chromosomes.
- Kinase
A protein that catalyzes the addition of phosphate groups to other molecules, such as proteins or lipids. These proteins are essential to nearly all signal transduction pathways.
- Liquid tumors
Tumors composed of hematopoietic (blood) cells, such as leukemias. Though lymphomas generally form solid masses in lymph nodes, they are often classified as liquid tumors because of their derivation from hematopoietic cells and ability to travel through lymphatics.
- Malignant tumor
An abnormal proliferation of cells driven by mutations in oncogenes or tumor suppressor genes that has already invaded their surrounding stroma. It is impossible to distinguish an isolated benign tumor cell from an isolated malignant tumor cell. This distinction can be made only through examination of tissue architecture.
- Metastatic tumor
A malignant tumor that has migrated away from its primary site, such as to draining lymph nodes or another organ.
- Methylation
Covalent addition of a methyl group to a protein, DNA, or other molecule.
- Missense mutation
A single-nucleotide substitution (e.g., C to T) that results in an amino acid substitution (e.g., histidine to arginine).
- Mut-driver gene
A gene that contains driver gene mutations.
- Nonsense mutation
A single-nucleotide substitution (e.g., C to T) that results in the production of a stop codon.
- Nonsynonymous mutation
A mutation that alters the encoded amino acid sequence of a protein. These include missense, nonsense, splice site, translation start, translation stop, and indel mutations.
- Oncogene
A gene that, when activated by mutation, increases the selective growth advantage of the cell in which it resides.
- Passenger mutation (passenger)
A mutation that has no direct or indirect effect on the selective growth advantage of the cell in which it occurred.
- Primary tumor
The original tumor at the site where tumor growth was initiated. This can be defined for solid tumors, but not for liquid tumors.
- Promoter
A region within or near the gene that helps regulate its expression.
- Rearrangement
A mutation that juxtaposes nucleotides that are normally separated, such as those on two different chromosomes.
- Selective growth advantage (s)
The difference between birth and death in a cell population. In normal adult cells in the absence of injury, s = 0.000000.
- Self-renewing tissues
Tissues whose cells normally repopulate themselves, such as those lining the gastrointestinal or urogenital tracts, as well as blood cells.
- Single-base substitution (SBS)
A single-nucleotide substitution (e.g., C to T) relative to a reference sequence or, in the case of somatic mutations, relative to the germline genome of the person with a tumor.
- Solid tumors
Tumors that form discrete masses, such as carcinomas or sarcomas.
- Somatic mutations
Mutations that occur in any non–germ cell of the body after conception, such as those that initiate tumorigenesis.
- Splice sites
Small regions of genes that are juxtaposed to the exons and direct exon splicing.
- Stem cell
An immortal cell that can repopulate a particular cell type.
- Subclonal mutation
A mutation that exists in only a subset of the neoplastic cells within a tumor.
- Translocation
A specific type of rearrangement where regions from two nonhomologous chromosomes are joined.
- Tumor suppressor gene
A gene that, when inactivated by mutation, increases the selective growth advantage of the cell in which it resides.
- Untranslated regions
Regions within the exons at the 5′ and 3′ ends of the gene that do not encode amino acids.
The prototypical exomic studies of cancer evaluated ~20 tumors at a cost of >$100,000 per case (1–3). Today, the cost of this sequencing has been reduced 100-fold, and studies reporting the sequencing of more than 100 tumors of a given type are the norm (table S1A). Although vast amounts of data can now be readily obtained, deciphering this information in meaningful terms is still challenging. Here, we review what has been learned about cancer genomes from these sequencing studies—and, more importantly, what this information has taught us about cancer biology and future cancer management strategies.
How Many Genes Are Subtly Mutated in a Typical Human Cancer?
In common solid tumors such as those derived from the colon, breast, brain, or pancreas, an average of 33 to 66 genes display subtle somatic mutations that would be expected to alter their protein products (Fig. 1A). About 95% of these mutations are single-base substitutions (such as C>G), whereas the remainder are deletions or insertions of one or a few bases (such as CTT>CT) (table S1B). Of the base substitutions, 90.7% result in missense changes, 7.6% result in nonsense changes, and 1.7% result in alterations of splice sites or untranslated regions immediately adjacent to the start and stop codons (table S1B).
Fig. 1. Number of somatic mutations in representative human cancers, detected by genome-wide sequencing studies.

(A) The genomes of a diverse group of adult (right) and pediatric (left) cancers have been analyzed. Numbers in parentheses indicate the median number of nonsynonymous mutations per tumor. (B) The median number of nonsynonymous mutations per tumor in a variety of tumor types. Horizontal bars indicate the 25 and 75% quartiles. MSI, microsatellite instability; SCLC, small cell lung cancers; NSCLC, non–small cell lung cancers; ESCC, esophageal squamous cell carcinomas; MSS, microsatellite stable; EAC, esophageal adenocarcinomas. The published data on which this figure is based are provided in table S1C.
Certain tumor types display many more or many fewer mutations than average (Fig. 1B). Notable among these outliers are melanomas and lung tumors, which contain ~200 nonsynonymous mutations per tumor (table S1C). These larger numbers reflect the involvement of potent mutagens (ultraviolet light and cigarette smoke, respectively) in the pathogenesis of these tumor types. Accordingly, lung cancers from smokers have 10 times as many somatic mutations as those from nonsmokers (4). Tumors with defects in DNA repair form another group of outliers (5). For example, tumors with mismatch repair defects can harbor thousands of mutations (Fig. 1B), even more than lung tumors or melanomas. Recent studies have shown that high numbers of mutations are also found in tumors with genetic alterations of the proofreading domain of DNA polymerases POLE or POLD1 (6, 7). At the other end of the spectrum, pediatric tumors and leukemias harbor far fewer point mutations: on average, 9.6 per tumor (table S1C). The basis for this observation is considered below.
Mutation Timing
When do these mutations occur? Tumors evolve from benign to malignant lesions by acquiring a series of mutations over time, a process that has been particularly well studied in colorectal tumors (8, 9). The first, or “gatekeeping,” mutation provides a selective growth advantage to a normal epithelial cell, allowing it to outgrow the cells that surround it and become a microscopic clone (Fig. 2). Gatekeeping mutations in the colon most often occur in the APC gene (10). The small adenoma that results from this mutation grows slowly, but a second mutation in another gene, such as KRAS, unleashes a second round of clonal growth that allows an expansion of cell number (9). The cells with only the APC mutation may persist, but their cell numbers are small compared with the cells that have mutations in both genes. This process of mutation followed by clonal expansion continues, with mutations in genes such as PIK3CA, SMAD4, and TP53, eventually generating a malignant tumor that can invade through the underlying basement membrane and metastasize to lymph nodes and distant organs such as the liver (11). The mutations that confer a selective growth advantage to the tumor cell are called “driver” mutations. It has been estimated (12) that each driver mutation provides only a small selective growth advantage to the cell, on the order of a 0.4% increase in the difference between cell birth and cell death. Over many years, however, this slight increase, compounded once or twice per week, can result in a large mass, containing billions of cells.
Fig. 2. Genetic alterations and the progression of colorectal cancer.

The major signaling pathways that drive tumorigenesis are shown at the transitions between each tumor stage. One of several driver genes that encode components of these pathways can be altered in any individual tumor. Patient age indicates the time intervals during which the driver genes are usually mutated. Note that this model may not apply to all tumor types. TGF-β, transforming growth factor–β.
The number of mutations in certain tumors of self-renewing tissues is directly correlated with age (13). When evaluated through linear regression, this correlation implies that more than half of the somatic mutations identified in these tumors occur during the preneoplastic phase; that is, during the growth of normal cells that continuously replenish gastrointestinal and genito-urinary epithelium and other tissues. All of these pre-neoplastic mutations are “passenger” mutations that have no effect on the neoplastic process. This result explains why a colorectal tumor in a 90-year-old patient has nearly twice as many mutations as a morphologically identical colorectal tumor in a 45-year-old patient. This finding also partly explains why advanced brain tumors (glioblastomas) and pancreatic cancers (pancreatic ductal adenocarcinomas) have fewer mutations than colorectal tumors; glial cells of the brain and epithelial cells of the pancreatic ducts do not replicate, unlike the epithelial cells lining the crypts of the colon. Therefore, the gatekeeping mutation in a pancreatic or brain cancer is predicted to occur in a precursor cell that contains many fewer mutations than are present in a colorectal precursor cell. This line of reasoning also helps to explain why pediatric cancers have fewer mutations than adult tumors. Pediatric cancers often occur in non–self-renewing tissues, and those that arise in renewing tissues (such as leukemias) originate from precursor cells that have not renewed themselves as often as in adults. In addition, pediatric tumors, as well as adult leukemias and lymphomas, may require fewer rounds of clonal expansion than adult solid tumors (8, 14). Genome sequencing studies of leukemia patients support the idea that mutations occur as random events in normal precursor cells before these cells acquire an initiating mutation (15).
When during tumorigenesis do the remaining somatic mutations occur? Because mutations in tumors occur at predictable and calculable rates (see below), the number of somatic mutations in tumors provides a clock, much like the clock used in evolutionary biology to determine species divergence time. The number of mutations has been measured in tumors representing progressive stages of colorectal and pancreatic cancers (11, 16). Applying the evolutionary clock model to these data leads to two unambiguous conclusions: First, it takes decades to develop a full-blown, metastatic cancer. Second, virtually all of the mutations in metastatic lesions were already present in a large number of cells in the primary tumors.
The timing of mutations is relevant to our understanding of metastasis, which is responsible for the death of most patients with cancer. The primary tumor can be surgically removed, but the residual metastatic lesions—often undetectable and widespread—remain and eventually enlarge, compromising the function of the lungs, liver, or other organs. From a genetics perspective, it would seem that there must be mutations that convert a primary cancer to a metastatic one, just as there are mutations that convert a normal cell to a benign tumor, or a benign tumor to a malignant one (Fig. 2). Despite intensive effort, however, consistent genetic alterations that distinguish cancers that metastasize from cancers that have not yet metastasized remain to be identified.
One potential explanation invokes mutations or epigenetic changes that are difficult to identify with current technologies (see section on “dark matter” below). Another explanation is that meta-static lesions have not yet been studied in sufficient detail to identify these genetic alterations, particularly if the mutations are heterogeneous in nature. But another possible explanation is that there are no metastasis genes. A malignant primary tumor can take many years to metastasize, but this process is, in principle, explicable by stochastic processes alone (17, 18). Advanced tumors release millions of cells into the circulation each day, but these cells have short half-lives, and only a miniscule fraction establish metastatic lesions (19). Conceivably, these circulating cells may, in a nondeterministic manner, infrequently and randomly lodge in a capillary bed in an organ that provides a favorable microenvironment for growth. The bigger the primary tumor mass, the more likely that this process will occur. In this scenario, the continual evolution of the primary tumor would reflect local selective advantages rather than future selective advantages. The idea that growth at metastatic sites is not dependent on additional genetic alterations is also supported by recent results showing that even normal cells, when placed in suitable environments such as lymph nodes, can grow into organoids, complete with a functioning vasculature (20).
Other Types of Genetic Alterations in Tumors
Though the rate of point mutations in tumors is similar to that of normal cells, the rate of chromosomal changes in cancer is elevated (21). Therefore, most solid tumors display widespread changes in chromosome number (aneuploidy), as well as deletions, inversions, translocations, and other genetic abnormalities. When a large part of a chromosome is duplicated or deleted, it is difficult to identify the specific “target” gene(s) on the chromosome whose gain or loss confers a growth advantage to the tumor cell. Target genes are more easily identified in the case of chromosome translocations, homozygous deletions, and gene amplifications. Translocations generally fuse two genes to create an oncogene (such as BCR-ABL in chronic myelogenous leukemia) but, in a small number of cases, can inactivate a tumor suppressor gene by truncating it or separating it from its promoter. Homozygous deletions often involve just one or a few genes, and the target is always a tumor suppressor gene. Amplifications contain an oncogene whose protein product is abnormally active simply because the tumor cell contains 10 to 100 copies of the gene per cell, compared with the two copies present in normal cells.
Most solid tumors have dozens of translocations; however, as with point mutations, the majority of translocations appear to be passengers rather than drivers. The breakpoints of the translocations are often in “gene deserts” devoid of known genes, and many of the translocations and homozygous deletions are adjacent to fragile sites that are prone to breakage. Cancer cells can, perhaps, survive such chromosome breaks more easily than normal cells because they contain mutations that incapacitate genes like TP53, which would normally respond to DNA damage by triggering cell death. Studies to date indicate that there are roughly 10 times fewer genes affected by chromosomal changes than by point mutations. Figure 3 shows the types and distribution of genetic alterations that affect protein-coding genes in five representative tumor types. Protein-coding genes account for only ~1.5% of the total genome, and the number of alterations in noncoding regions is proportionately higher than the number affecting coding regions. The vast majority of the alterations in noncoding regions are presumably passengers. These noncoding mutations, as well as the numerous epigenetic changes found in cancers, will be discussed later.
Fig. 3. Total alterations affecting protein-coding genes in selected tumors.

Average number and types of genomic alterations per tumor, including single-base substitutions (SBS), small insertions and deletions (indels), amplifications, and homozygous deletions, as determined by genome-wide sequencing studies. For colorectal, breast, and pancreatic ductal cancer, and medulloblastomas, translocations are also included. The published data on which this figure is based are provided in table S1D.
Drivers Versus Passenger Mutations
Though it is easy to define a “driver gene mutation” in physiologic terms (as one conferring a selective growth advantage), it is more difficult to identify which somatic mutations are drivers and which are passengers. Moreover, it is important to point out that there is a fundamental difference between a driver gene and a driver gene mutation. A driver gene is one that contains driver gene mutations. But driver genes may also contain passenger gene mutations. For example, APC is a large driver gene, but only those mutations that truncate the encoded protein within its N-terminal 1600 amino acids are driver gene mutations. Missense mutations throughout the gene, as well as protein-truncating mutations in the C-terminal 1200 amino acids, are passenger gene mutations.
Numerous statistical methods to identify driver genes have been described. Some are based on the frequency of mutations in an individual gene compared with the mutation frequency of other genes in the same or related tumors after correction for sequence context and gene size (22, 23). Other methods are based on the predicted effects of mutation on the encoded protein, as inferred from biophysical studies (24–26). All of these methods are useful for prioritizing genes that are most likely to promote a selective growth advantage when mutated. When the number of mutations in a gene is very high, as with TP53 or KRAS, any reasonable statistic will indicate that the gene is extremely likely to be a driver gene. These highly mutated genes have been termed “mountains” (1). Unfortunately, however, genes with more than one, but still relatively few mutations (so called “hills”) numerically dominate cancer genome landscapes (1). In these cases, methods based on mutation frequency and context alone cannot reliably indicate which genes are drivers, because the background rates of mutation vary so much among different patients and regions of the genome. Recent studies of normal cells have indicated that the rate of mutation varies by more than 100-fold within the genome (27). In tumor cells, this variation can be higher and may affect whole regions of the genome in an apparently random fashion (28). Thus, at best, methods based on mutation frequency can only prioritize genes for further analysis but cannot unambiguously identify driver genes that are mutated at relatively low frequencies.
Further complicating matters, there are two distinct meanings of the term “driver gene” that are used in the cancer literature. The driver-versus-passenger concept was originally used to distinguish mutations that caused a selective growth advantage from those that did not (29). According to this definition, a gene that does not harbor driver gene mutations cannot be a driver gene. But many genes that contain few or no driver gene mutations have been labeled driver genes in the literature. These include genes that are overexpressed, underexpressed, or epigenetically altered in tumors, or those that enhance or inhibit some aspect of tumorigenicity when their expression is experimentally manipulated. Though a subset of these genes may indeed play an important role in the neoplastic process, it is confusing to lump them all together as driver genes.
To reconcile the two connotations of driver genes, we suggest that genes suspected of increasing the selective growth advantage of tumor cells be categorized as either “Mut-driver genes” or “Epi-driver genes.” Mut-driver genes contain a sufficient number or type of driver gene mutations to unambiguously distinguish them from other genes. Epi-driver genes are expressed aberrantly in tumors but not frequently mutated; they are altered through changes in DNA methylation or chromatin modification that persist as the tumor cell divides.
A Ratiometric Method to Identify and Classify Mut-Driver Genes
If mutation frequency, corrected for mutation context, gene length, and other parameters, cannot reliably identify modestly mutated driver genes, what can? In our experience, the best way to identify Mut-driver genes is through their pattern of mutation rather than through their mutation frequency. The patterns of mutations in well-studied oncogenes and tumor suppressor genes are highly characteristic and nonrandom. Oncogenes are recurrently mutated at the same amino acid positions, whereas tumor suppressor genes are mutated through protein-truncating alterations throughout their length (Fig. 4 and table S2A).
Fig. 4. Distribution of mutations in two oncogenes (PIK3CA and IDH1) and two tumor suppressor genes (RB1 and VHL).
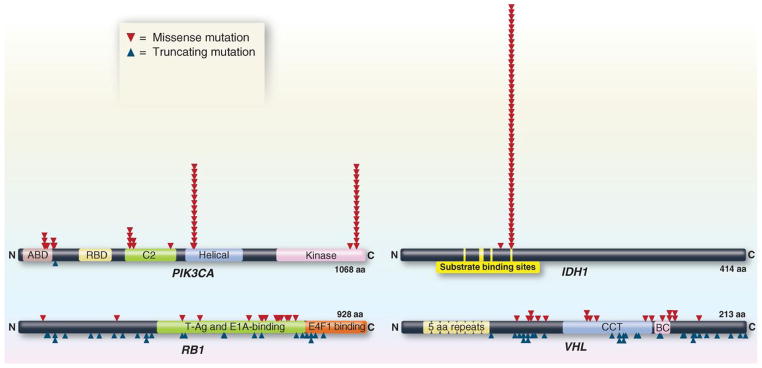
The distribution of missense mutations (red arrowheads) and truncating mutations (blue arrowheads) in representative oncogenes and tumor suppressor genes are shown. The data were collected from genome-wide studies annotated in the COSMIC database (release version 61). For PIK3CA and IDH1, mutations obtained from the COSMIC database were randomized by the Excel RAND function, and the first 50 are shown. For RB1 and VHL, all mutations recorded in COSMIC are plotted. aa, amino acids.
On the basis of these mutation patterns rather than frequencies, we can determine which of the 18,306 mutated genes containing a total of 404,863 subtle mutations that have been recorded in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (30) are Mut-driver genes and whether they are likely to function as oncogenes or tumor suppressor genes. To be classified as an oncogene, we simply require that >20% of the recorded mutations in the gene are at recurrent positions and are missense (see legend to table S2A). To be classified as a tumor suppressor gene, we analogously require that >20% of the recorded mutations in the gene are inactivating. This “20/20 rule” is lenient in that all well-documented cancer genes far surpass these criteria (table S2A).
The following examples illustrate the value of the 20/20 rule. When IDH1 mutations were first identified in brain tumors, their role in tumorigenesis was unknown (2, 31). Initial functional studies suggested that IDH1 was a tumor suppressor gene and that mutations inactivated this gene (32). However, nearly all of the mutations in IDH1 were at the identical amino acid, codon 132 (Fig. 4). As assessed by the 20/20 rule, this distribution unambiguously indicated that IDH1 was an oncogene rather than a tumor suppressor gene, and this conclusion was eventually supported by biochemical experiments (33, 34). Another example is provided by mutations in NOTCH1. In this case, some functional studies suggested that NOTCH1 was an oncogene, whereas others suggested it was a tumor suppressor gene (35, 36). The situation could be clarified through the application of the 20/20 rule to NOTCH1 mutations in cancers. In “liquid tumors” such as lymphomas and leukemias, the mutations were often recurrent and did not truncate the predicted protein (37). In squamous cell carcinomas, the mutations were not recurrent and were usually inactivating (38–40). Thus, the genetic data clearly indicated that NOTCH1 functions differently in different tumor types. The idea that the same gene can function in completely opposite ways in different cell types is important for understanding cell signaling pathways.
How Many Mut-Driver Genes Exist?
Though all 20,000 protein-coding genes have been evaluated in the genome-wide sequencing studies of 3284 tumors, with a total of 294,881 mutations reported, only 125 Mut-driver genes, as defined by the 20/20 rule, have been discovered to date (table S2A). Of these, 71 are tumor suppressor genes and 54 are oncogenes. An important but relatively small fraction (29%) of these genes was discovered to be mutated through unbiased genome-wide sequencing; most of these genes had already been identified by previous, more directed investigations.
How many more Mut-driver genes are yet to be discovered? We believe that a plateau is being reached, because the same Mut-driver genes keep being “rediscovered” in different tumor types. For example, MLL2 and MLL3 mutations were originally discovered in medulloblastomas (41) and were subsequently discovered to be mutated in non-Hodgkin lymphomas, prostate cancers, breast cancers, and other tumor types (42–45). Similarly, ARID1A mutations were first discovered to be mutated in clear-cell ovarian cancers (46, 47) and were subsequently shown to be mutated in tumors of several other organs, including those of the stomach and liver (48–50). In recent studies of several types of lung cancer (4, 51, 52), nearly all genes found to be mutated at significant frequencies had already been identified in tumors of other organs. In other words, the number of frequently altered Mut-driver genes (mountains) is nearing saturation. More mountains will undoubtedly be discovered, but these will likely be in uncommon tumor types that have not yet been studied in depth.
The newly discovered Mut-driver genes that have been detected through genome-wide sequencing have often proved illuminating. For example, nearly half of these genes encode proteins that directly regulate chromatin through modification of histones or DNA. Examples include the histones HIST1H3B and H3F3A, as well as the proteins DNMT1 and TET1, which covalently modify DNA, EZH2, SETD2, and KDM6A, which, in turn, methylate or demethylate histones (53–57). These discoveries have profound implications for understanding the mechanistic basis of the epigenetic changes that are rampant in tumors (58). The discovery of genetic alterations in genes encoding mRNA splicing factors, such as SF3B1 and U2AF1 (59–61), was similarly stunning, as mutations in these genes would be expected to lead to a plethora of nonspecific cellular stresses rather than to promote specific tumor types. Another example is provided by mutations in the cooperating proteins ATRX and DAXX (62). Tumors with mutations in these genes all have a specific type of telomere elongation process termed “ALT” (for “alternative lengthening of telomeres”) (63). Though the ALT phenotype had been recognized for more than a decade, its genetic basis was mysterious before the discovery of mutations of these genes and their perfect correlation with the ALT phenotype (64). A final example is provided by IDH1 and IDH2, whose mutations have stimulated the burgeoning field of tumor metabolism (65) and have had fascinating implications for epigenetics (66, 67).
The Mut-driver genes listed in table S2A are affected by subtle mutations: base substitutions, intragenic insertions, or deletions. As noted above, Mut-driver genes can also be altered by less subtle changes, such as translocations, amplifications, and large-scale deletions. As with point mutations, it can be difficult to distinguish Mut-driver genes that are altered by these types of changes from genes that contain only passenger mutations. Genes that are not point-mutated, but are recurrently amplified (e.g., MYC family genes) or homozygously deleted (e.g., MAP2K4) and that meet other criteria (e.g., being the only gene in the amplicon or homozygously deleted region) are listed in table S2B. This adds 13 Mut-driver genes—10 oncogenes that are amplified and 3 tumor suppressor genes that are homozygously deleted—to the 125 driver genes that are affected by subtle mutations, for a total of 138 driver genes discovered to date (table S2).
Translocations provide similar challenges for driver classification. An important discovery related to this point is chromothripsis (68), a rare cataclysmic event involving one or a small number of chromosomes that results in a large number of chromosomal rearrangements. This complicates any inferences about causality, in the same way that mismatch repair deficiency compromises the interpretation of point mutations. However, for completeness, all fusion genes that have been identified in at least three independent tumors are listed in table S3. Virtually all of these genes were discovered through conventional approaches before the advent of genome-wide DNA sequencing studies, with some notable exceptions such as those described in (6) and (69). The great majority of these translocations are found in liquid tumors (leukemias and lymphomas) (table S3C) or mesenchymal tumors (table S3B) and were initially identified through karyotypic analyses. A relatively small number of recurrent fusions, the most important of which include ERG in prostate cancers (70) and ALK in lung cancers (71), have been described in more common tumors (table S3A).
Genes exist that predispose to cancer when inherited in mutant form in the germ line, but are not somatically mutated in cancer to a substantial degree. These genes generally do not confer an increase in selective growth advantage when they are abnormal, but they stimulate tumorigenesis in indirect ways (such as by increasing genetic instability, as discussed later in this Review). For completeness, these genes and the hereditary syndromes for which they are responsible are listed in table S4.
Dark Matter
Classic epidemiologic studies have suggested that solid tumors ordinarily require five to eight “hits,” now interpreted as alterations in driver genes, to develop (72). Is this number compatible with the molecular genetic data? In pediatric tumors such as medulloblastomas, the number of driver gene mutations is low (zero to two), as expected from the discussion above (Fig. 5). In common adult tumors—such as pancreatic, colorectal, breast, and brain cancers—the number of mutated driver genes is often three to six, but several tumors have only one or two driver gene mutations (Fig. 5). How can this be explained, given the widely accepted notion that tumor development and progression require multiple, sequential genetic alterations acquired over decades?
Fig. 5. Number and distribution of driver gene mutations in five tumor types.

The total number of driver gene mutations [in oncogenes and tumor suppressor genes (TSGs)] is shown, as well as the number of oncogene mutations alone. The driver genes are listed in tables S2A and S2B. Translocations are not included in this figure, because few studies report translocations along with the other types of genetic alterations on a per-case basis. In the tumor types shown here, translocations affecting driver genes occur in less than 10% of samples. The published data on which this figure is based are provided in table S1E.
First, technical issues explain some of the “missing mutations.” Genome-wide sequencing is far from perfect, at least with the technologies available today. Some regions of the genome are not well represented because their sequences are difficult to amplify, capture, or unambiguously map to the genome (73–76). Second, there is usually a wide distribution in the number of times that a specific nucleotide in a given gene is observed in the sequence data, so some regions will not be well represented by chance factors alone (77). Finally, primary tumors contain not only neoplastic cells, but also stromal cells that dilute the signal from the mutated base, further reducing the probability of finding a mutation (78).
What fraction of mutations are missed by these three technical issues? A recent study of pancreatic cancers is informative in this regard. Biankin et al. used immunohistochemical and genetic analyses to select a set of primary tumor samples enriched in neoplastic cells (79). They used massively parallel sequencing to analyze the exomes of these samples, then compared their mutational data with a set of pancreatic cancer cell lines and xenografts in which mutations had previously been identified, using conventional Sanger sequencing, and confirmed to be present in the primary tumors (3, 16). Only 159 (63%) of the expected 251 driver gene mutations were identified in the primary tumors studied by next-generation sequencing alone, indicating a false-negative rate of 37%. Genome-wide studies in which the proportion of neoplastic cells within tumors is not as carefully evaluated as in (79) will have higher false-negative rates. Moreover, these technical problems are exacerbated in whole-genome studies compared with exomic analyses, because the sequence coverage of the former is often lower than that of the latter (generally 30-fold in whole-genome studies versus more than 100-fold in exomic studies).
Conceptual issues also limit the number of detectable drivers. Virtually all studies, either at the whole-genome or whole-exome level, have focused on the coding regions. The reason for this is practical; it is difficult enough to identify driver gene mutations when they qualitatively alter the sequence of the encoded protein. Trying to make sense of intergenic or intronic mutations is much more difficult. Based on analogous studies of the identifiable mutations in patients with monogenic diseases, more than 80% of mutations should be detectable through analysis of the coding regions (80). However, this still leaves some mutations as unidentifiable “dark matter,” even in the germline genomes of heritable cases, which are usually easier to interpret than the somatic mutations in cancers. The first examples of light coming to such dark matter have recently been published: Recurrent mutations in the promoter of the TERT gene, encoding the catalytic subunit of telomerase, have been identified and shown to activate its transcription (81, 82).
Mut-driver genes other than those listed in table S2 will undoubtedly be discovered as genome-wide sequencing continues. However, based on the trends noted above, most of the Mut-driver genes will likely be mountains in rare tumor types or small hills in common tumor types; thus, these genes are unlikely to account for the bulk of the presumptive dark matter. Other types of dark matter can be envisioned, however. Copy-number alterations are ubiquitous in cancers, at either the whole-chromosome or subchromosomal levels. These alterations could subtly change the expression of their driver genes. Recent studies have suggested that the loss of one copy of chromosomes containing several tumor suppressor genes, each plausibly connected to neoplasia but not altered by mutation, may confer a selective growth advantage (83, 84).
The most obvious source of dark matter is in Epi-driver genes. Human tumors contain large numbers of epigenetic changes affecting DNA or chromatin proteins. For example, a recent study of colorectal cancers showed that more than 10% of the protein-coding genes were differentially methylated when compared with normal colorectal epithelial cells (85). Some of these changes (i.e., those in Epi-driver genes) are likely to provide a selective growth advantage (86, 87). For example, epigenetic silencing of CDK2NA and MLH1 is much more common than mutational inactivation of either of these two well-recognized driver genes (85) However, there is a critical difference between a genetic and an epigenetic change in a gene. Unlike the sequence of a gene in a given individual, methylation is plastic, varying with cell type, developmental stage, and patient age (21). The methylation state of the normal precursor cells that initiate tumorigenesis is unknown; these cells, such as normal stem cells, may represent only a tiny fraction of the cells in a normal organ. This plasticity also means that methylation can change under microenvironmental cues, such as those associated with low nutrient concentrations or abnormal cell contacts. It is therefore difficult to know whether specific epigenetic changes observed in cancer cells reflect, rather than contribute to, the neoplastic state. Criteria for distinguishing epigenetic changes that exert a selective growth advantage from those that do not (passenger epigenetic changes) have not yet been formulated. Given that Epi-driver genes are likely to compose a major component of the dark matter, further research on this topic is essential (58).
Genetic Heterogeneity
The mutations depicted in Fig. 1 are clonal; that is, they are present in the majority of the neoplastic cells in the tumors. But additional, subclonal (i.e., heterogeneous within the tumor) mutations are important for understanding tumor evolution. Four types of genetic heterogeneity are relevant to tumorigenesis (Fig. 6):
Fig. 6. Four types of genetic heterogeneity in tumors, illustrated by a primary tumor in the pancreas and its metastatic lesions in the liver.

Mutations introduced during primary tumor cell growth result in clonal heterogeneity. At the top left, a typical tumor is represented by cells with a large fraction of the total mutations (founder cells) from which subclones are derived. The differently colored regions in the subclones represent stages of evolution within a subclone. (A) Intratumoral: heterogeneity among the cells of the primary tumor. (B) Intermetastatic: heterogeneity among different metastatic lesions in the same patient. In the case illustrated here, each metastasis was derived from a different subclone. (C) Intrametastatic: heterogeneity among the cells of each metastasis develops as the metastases grow. (D) Interpatient: heterogeneity among the tumors of different patients. The mutations in the founder cells of the tumors of these two patients are almost completely distinct (see text).
-
Intratumoral: heterogeneity among the cells of one tumor. This type of heterogeneity has been recognized for decades. For example, it is rare to see a cytogenetic study of a solid tumor in which all of the tumor cells display the same karyotype (88). The same phenomenon has been noted for individual genes [e.g., (89)] and more recently has been observed throughout the genome (16, 90–96). This kind of heterogeneity must exist: Every time a normal (or tumor) cell divides, it acquires a few mutations, and the number of mutations that distinguish any two cells simply marks the time from their last common ancestor (their founder cell). Cells at the opposite ends of large tumors will be spatially distinct and, in general, will display more differences than neighboring cells (16). This phenomenon is analogous to speciation, wherein organisms on different islands are more likely to diverge from one another than are organisms on the same island.
In studies that have evaluated intratumoral heterogeneity by genome-wide sequencing, the majority of somatic mutations are present in all tumor cells. These mutations form the trunk of the somatic evolutionary tree. What is the importance of the mutations in the branches (i.e., those that are not shared by all tumor cells)? From a medical perspective, these mutations are often meaningless because the primary tumors are surgically removed. How much heterogeneity existed in the various branches before surgery is not important. However, this heterogeneity provides the seeds for intermetastastic heterogeneity, which is of great clinical importance.
-
Intermetastatic: heterogeneity among different metastatic lesions of the same patient. The vast majority of cancer patients die because their tumors were not removed before metastasis to surgically inaccessible sites, such as the liver, brain, lung, or bone. Patients who relapse with a single metastatic lesion can often still be cured by surgery or radiotherapy, but single metastases are the exception rather than the rule. A typical patient on a clinical trial has a dozen or more metastatic lesions large enough to be visualized by imaging, and many more that are smaller. If each of the metastatic lesions in a single patient was founded by a cell with a very different genetic constitution, then chemotherapeutic cures would be nearly impossible to achieve: Eradicating a subset of the metastatic lesions in a patient will not be adequate for long-term survival.
How much heterogeneity is there among different metastatic lesions? In short, a lot. It is not uncommon for one metastatic lesion to have 20 clonal genetic alterations not shared by other metastases in the same patient (16, 97). Because they are clonal, these mutations occurred in the founder cell of the metastasis; that is, the cell that escaped from the primary tumor and multiplied to form the metastasis. The founder cell for each metastasis is present in different, geographically distinct areas of the primary tumors, as expected (16).
This potentially disastrous situation is tempered by the fact that the heterogeneity appears largely confined to passenger gene mutations. In most of the studies documenting heterogeneity in malignancies, the Mut-driver genes are present in the trunks of the trees, though exceptions have been noted (95). These findings are consistent with the idea, discussed above, that the genetic alterations required for metastasis were present (i.e., selected for) before metastasis actually occurred. The data are also consistent with the observation that in patients responsive to targeted agents, the response is often seen in all metastatic lesions rather than just a small subset (98).
Intrametastatic: heterogeneity among the cells of an individual metastasis. Each metastasis is established by a single cell (or small group of cells) with a set of founder mutations. As it grows, the metastasis acquires new mutations with each cell division. Though the founder mutations may make the lesion susceptible to antitumor agents, the new mutations provide the seeds for drug resistance. Unlike primary tumors, the metastatic lesions generally cannot be removed by surgery and must be treated with systemic therapies. Patients with complete responses to targeted therapies invariably relapse. Most of the initial lesions generally recur, and the time frame at which they recur is notably similar. This time course can be explained by the presence of resistance mutations that existed within each metastasis before the onset of the targeted therapy (99–102). Calculations show that any metastatic lesion of a size visible on medical imaging has thousands of cells (among the billions present) that are already resistant to virtually any drug that can be imagined (99, 101, 102). Thus, recurrence is simply a matter of time, entirely predictable on the basis of known mutation frequencies and tumor cell growth rates. This “fait accompli” can be circumvented, in principle, by treatment with multiple agents, as it is unlikely that a single tumor cell will be resistant to multiple drugs that act on different targets.
Interpatient: heterogeneity among the tumors of different patients. This type of heterogeneity has been observed by every oncologist; no two cancer patients have identical clinical courses, with or without therapy. Some of these differences could be related to host factors, such as germline variants that determine drug half-life or vascular permeability to drugs or cells, and some could be related to nongenetic factors (103). However, much of this interpatient heterogeneity is probably related to somatic mutations within tumors. Though several dozen somatic mutations may be present in the breast cancers from two patients, only a small number are in the same genes, and in the vast majority of cases, these are the Mut-driver genes (1, 104, 105). Even in these driver genes, the actual mutations are often different. Mutations altering different domains of a protein would certainly not be expected to have identical effects on cellular properties, as experimentally confirmed (106). Though it may seem that different mutations in adjacent codons would have identical effects, detailed studies of large numbers of patients have shown that this need not be the case. For example, a Gly12→Asp12 (G12D) mutation of KRAS does not have the same clinical implications as a G13D mutation of the same gene (107). Interpatient heterogeneity has always been one of the major obstacles to designing uniformly effective treatments for cancer. Efforts to individualize treatments based on knowledge of the genomes of cancer patients are largely based on an appreciation of this heterogeneity.
Signaling Pathways in Tumors
The immense complexity of cancer genomes that could be inferred from the data described above is somewhat misleading. After all, even advanced tumors are not completely out of control, as evidenced by the dramatic responses to agents that target mutant BRAF in melanomas (108) or mutant ALK in lung cancers (109). Albeit transient, these responses mean that interference with even a single mutant gene product is sufficient to stop cancer in its tracks, at least transiently. How can the genomic complexity of cancer be reconciled with these clinical observations?
Two concepts bear on this point. The first, mentioned above, is that >99.9% of the alterations in tumors (including point mutations, copy-number alterations, translocations, and epigenetic changes distributed throughout the genome, not just in the coding regions) are immaterial to neoplasia. They are simply passenger changes that mark the time that has elapsed between successive clonal expansions. Normal cells also undergo genetic alterations as they divide, both at the nucleotide and chromosomal levels. However, normal cells are programmed to undergo cell death in response to such alterations, perhaps as a protective mechanism against cancer. In contrast, cancer cells have evolved to tolerate genome complexity by acquiring mutations in genes such as TP53 (110). Thus, genomic complexity is, in part, the result of cancer, rather than the cause.
To appreciate the second concept, one must take the 30,000-foot view. A jungle might look chaotic at ground level, but the aerial view shows a clear order, with all the animals gathering at the streams at certain points in the day, and all the streams converging at a river. There is order in cancer, too. Mutations in all of the 138 driver genes listed in table S2 do one thing: cause a selective growth advantage, either directly or indirectly. Moreover, there appears to be only a limited number of cellular signaling pathways through which a growth advantage can be incurred (Fig. 7 and table S5).
Fig. 7. Cancer cell signaling pathways and the cellular processes they regulate.

All of the driver genes listed in table S2 can be classified into one or more of 12 pathways (middle ring) that confer a selective growth advantage (inner circle; see main text). These pathways can themselves be further organized into three core cellular processes (outer ring). The publications on which this figure is based are provided in table S5.
All of the known driver genes can be classified into one or more of 12 pathways (Fig. 7). The discovery of the molecular components of these pathways is one of the greatest achievements of biomedical research, a tribute to investigators working in fields that encompass biochemistry, cell biology, and development, as well as cancer. These pathways can themselves be further organized into three core cellular processes:
Cell fate: Numerous studies have demonstrated the opposing relationship between cell division and differentiation, the arbiters of cell fate. Dividing cells that are responsible for populating normal tissues (stem cells) do not differentiate, and vice versa. Regenerative medicine is based on this distinction, predicated on ways to get differentiated cells to de-differentiate into stem cells, then forcing the stem cells to differentiate into useful cell types for transplantation back into the patient. Many of the genetic alterations in cancer abrogate the precise balance between differentiation and division, favoring the latter. This causes a selective growth advantage, because differentiating cells eventually die or become quiescent. Pathways that function through this process include APC, HH, and NOTCH, all of which are well known to control cell fate in organisms ranging from worms to mammals (111). Genes encoding chromatin-modifying enzymes can also be included in this category. In normal development, the heritable switch from division to differentiation is not determined by mutation, as it is in cancer, but rather by epigenetic alterations affecting DNA and chromatin proteins. What better way to subvert this normal mechanism for controlling tissue architecture than to debilitate the epigenetic modifying apparatus itself?
Cell survival: Though cancer cells divide abnormally because of cell-autonomous alterations, such as those controlling cell fate, their surrounding stromal cells are perfectly normal and do not keep pace. The most obvious ramification of this asymmetry is the abnormal vasculature of tumors. As opposed to the well-ordered network of arteries, veins, and lymphatics that control nutrient concentrations in normal tissues, the vascular system in cancers is tortuous and lacks uniformity of structure (112, 113). Normal cells are always within 100 μm of a capillary, but this is not true for cancer cells (114). As a result, a cancer cell acquiring a mutation that allows it to proliferate under limiting nutrient concentrations will have a selective growth advantage, thriving in environments in which its sister cells cannot. Mutations of this sort occur, for example, in the EGFR, HER2, FGFR2, PDGFR, TGFbR2, MET, KIT, RAS, RAF, PIK3CA, and PTEN genes (table S2A). Some of these genes encode receptors for the growth factors themselves, whereas others relay the signal from the growth factor to the interior of the cell, stimulating growth when activated (115, 116). For instance, mutations in KRAS or BRAF genes confer on cancer cells the ability to grow in glucose concentrations that are lower than those required for the growth of normal cells or of cancer cells that do not have mutations in these genes (117, 118). Progression through the cell cycle (and its antithesis, apoptosis) can be directly controlled by intracellular metabolites, and driver genes that directly regulate the cell cycle or apoptosis, such as CDKN2A, MYC, and BCL2, are often mutated in cancers. Another gene whose mutations enhance cell survival is VHL, the product of which stimulates angiogenesis through the secretion of vascular endothelial growth factor. What better way to provision growth factors to a rogue tumor than to lure the unsuspecting vasculature to its hideout?
Genome maintenance: As a result of the exotic microenvironments in which they reside, cancer cells are exposed to a variety of toxic substances, such as reactive oxygen species. Even without microenvironmental poisons, cells make mistakes while replicating their DNA or during division (119, 120), and checkpoints exist to either slow down such cells or make them commit suicide (apoptosis) under such circumstances (110, 121, 122). Although it is good for the organism to remove these damaged cells, tumor cells that can survive the damage will, by definition, have a selective growth advantage. Therefore, it is not surprising that genes whose mutations abrogate these checkpoints, such as TP53 and ATM, are mutated in cancers (123). Defects in these genes can also indirectly confer a selective growth advantage by allowing cells that have a gross chromosomal change favoring growth, such as a translocation or an extra chromosome, to survive and divide. Analogously, genes that control point mutation rates, such as MLH1 or MSH2, are mutated in cancers (table S2A) or in the germ line of patients predisposed to cancers (table S4) because they accelerate the acquisition of mutations that function through processes that regulate cell fate or survival. What better way to promote cancer than by increasing the rate of occurrence of the mutations that drive the process?
Because the protein products of genes regulating cell fate, cell survival, and genome maintenance often interact with one another, the pathways within them overlap; they are not as discrete as might be inferred from the description above. However, grouping genes into pathways makes perfect sense from a genetics standpoint. Given that cancer is a genetic disease, the principles of genetics should apply to its pathogenesis. When performing a conventional mutagenesis screen in bacteria, yeast, fruit flies, or worms, one expects to discover mutations in several different genes that confer similar phenotypes. The products of these genes often interact with one another and define a biochemical or developmental pathway. Therefore, it should not be surprising that several different genes can result in the same selective growth advantage for cancer cells and that the products of these genes interact. The analogy between cancer pathways and biochemical or developmental pathways in other organisms goes even deeper: The vast majority of our knowledge of the function of driver genes has been derived from the study of the pathways through which their homologs work in nonhuman organisms. Though the functions are not identical to those in human cells, they are highly related and have provided the starting point for analogous studies in human cells.
Recognition of these pathways also has important ramifications for our ability to understand interpatient heterogeneity. One lung cancer might have an activating mutation in a receptor for a stimulatory growth factor, making it able to grow in low concentrations of epidermal growth factor (EGF). A second lung cancer might have an activating mutation in KRAS, whose protein product normally transmits the signal from the epidermal growth factor receptor (EGFR) to other cell signaling molecules. A third lung cancer might have an inactivating mutation in NF1, a regulatory protein that normally inactivates the KRAS protein. Finally, a fourth lung cancer might have a mutation in BRAF, which transmits the signal from KRAS to downstream kinases (Fig. 8). One would predict that mutations in the various components of a single pathway would be mutually exclusive—that is, not occurring in the same tumor—and this has been experimentally confirmed (124, 125). Apart from being intellectually satisfying, knowledge of these pathways has implications for cancer therapy, as discussed in the next section.
Fig. 8. Signal transduction pathways affected by mutations in human cancer.

Two representative pathways from Fig. 7 (RAS and PI3K) are illustrated. The signal transducers are color coded: red indicates protein components encoded by the driver genes listed in table S2; yellow balls denote sites of phosphorylation. Examples of therapeutic agents that target some of the signal transducers are shown. RTK, receptor tyrosine kinase; GDP, guanosine diphosphate; MEK, MAPK kinase; ERK, extracellular signal–regulated kinase; NFkB, nuclear factor κB; mTOR, mammalian target of rapamycin.
A Perspective on Genome-Based Medicine in Oncology
Opportunities
Though cancer genome sequencing is a relatively new endeavor, it has already had an impact on the clinical care of cancer patients. The recognition that certain tumors contain activating mutations in driver genes encoding protein kinases has led to the development of small-molecule inhibitor drugs targeting those kinases.
Representative examples of this type of genome-based medicine include the use of EGFR kinase inhibitors to treat cancers with EGFR gene mutations (126), the aforementioned ana-plastic lymphoma kinase (ALK) inhibitors to treat cancers with ALK gene translocations (109), and specific inhibitors of mutant BRAF to treat cancers with BRAF mutations (108). Before instituting treatment with such agents, it is imperative to determine whether the cancer harbors the mutations that the drug targets. Only a small fraction of lung cancer patients have EGFR gene mutations or ALK gene translocations, and only these patients will respond to the drugs. Treating lung cancer patients without these particular genetic alterations would be detrimental, as such patients would develop the toxic side effects of the drugs while their tumors progressed.
A second type of genome-based medicine focuses on the side effects and metabolism of the therapeutic agents, rather than the genetic alterations they target. At present, the dose of cancer drugs given to patients is based on the patients’ size (body weight or surface area). But the therapeutic ratio of cancer drugs (ratio of the concentration that causes side effects to the concentration required to kill tumor cells) is generally low, particularly for conventional (nontargeted) therapeutic agents. Small changes in circulating concentrations of these drugs can make the difference between substantial tumor regression and intolerable side effects. Interrogation of the germline status of the genes encoding drug-metabolizing enzymes could substantially improve the outcomes of treatment by informing drug dosing (127). Optimally, this genome interrogation would be accompanied by pharmacokinetic measurements of drug concentrations in each patient. The additional cost of such analyses would be small compared with the exorbitant costs of new cancer therapies—for recently approved drugs, the cost is estimated to be $200,000 to $300,000 per quality life year produced (128).
Challenges
One challenge of genome-based medicine in oncology is already apparent from the opportunities described above: All of the clinically approved drugs that target the products of genetically altered genes are directed against kinases. One reason for this is that kinases are relatively easy to target with small molecules and have been extensively studied at the biochemical, structural, and physiologic levels (129). But another reason has far deeper ramifications. The vast majority of drugs on the market today, for cancer or other diseases, inhibit the actions of their protein targets. This inhibition occurs because the drugs interfere with the protein’s enzymatic activity (such as the phosphorylation catalyzed by kinases) or with the binding of the protein to a small ligand (such as with G protein–coupled receptors). Only 31 of the oncogenes listed in tables S2 and S3 have enzymatic activities that are targetable in this manner. Many others participate in protein complexes, involving large interfaces and numerous weak interactions. Inhibiting the function of such proteins with small drugs is notoriously difficult because small compounds can only inhibit one of these interactions (130, 131).
Though one can at least imagine the development of drugs that inhibit nonenzymatic protein functions, the second challenge evident from table S2 poses even greater difficulties: A large fraction of the Mut-driver genes encode tumor suppressors. Drugs generally interfere with protein function; they cannot, in general, replace the function of defective genes such as those resulting from mutations in tumor suppressor genes. Unfortunately, tumor suppressor gene–inactivating mutations predominate over oncogene-activating mutations in the most common solid tumors: Few individual tumors contain more than one oncogene mutation (Fig. 5).
The relatively small number of oncogene mutations in tumors is important in light of the intrametastatic heterogeneity described earlier. To circumvent the inevitable development of resistance to targeted therapies, it will likely be necessary to treat patients with two or more drugs. The probability that a single cancer cell within a large metastatic lesion will be resistant to two agents that target two independent pathways is exponentially less than the probability that the cell will be resistant to a single agent. However, if the cancer cell does not contain more than one targetable genetic alteration (i.e., an oncogene mutation), then this combination strategy is not feasible.
Given the paucity of oncogene alterations in common solid tumors and these principles, can targeted therapeutic approaches ever be expected to induce long-term remissions, even cures, rather than the short-term remissions now being achieved? The saviors are pathways; every tumor suppressor gene inactivation is expected to result in the activation of some growth-promoting signal downstream of the pathway. An example is provided by PTEN mutations: Inactivation of the tumor suppressor gene PTEN results in activation of the AKT kinase (Fig. 8). Similarly, inactivation of the tumor suppressor gene CDKN2A results in activation of kinases, such as cyclin-dependent kinase 4, that promote cell cycle traverse (132). Furthermore, inactivation of tumor suppressor gene APC results in constitutive activity of oncogenes such as CTNNB1 and CMYC (133–135).
We believe that greater knowledge of these pathways and the ways in which they function is the most pressing need in basic cancer research. Successful research on this topic should allow the development of agents that target, albeit indirectly, defective tumor suppressor genes. Indeed, there are already examples of such indirect targeting. Inactivating mutations of the tumor suppressor genes BRCA1 or BRCA2 lead to activation of downstream pathways required to repair DNA damage in the absence of BRCA function. Thus, cancer cells with defects in BRCA1 or BRCA2 are more susceptible to DNA damaging agents or to drugs that inhibit enzymes that facilitate the repair of DNA damage such as PARP [poly(adenosine diphosphate–ribose) polymerase] (136). PARP inhibitors have shown encouraging results in clinical trials when used in patients whose tumors have inactivating mutations of BRCA genes (137).
Further progress in this area will require more detailed information about the signaling pathways through which cancer genes function in human cancer cells, as well as in model organisms. One of the lessons of molecular biology over the past two decades is that pathway functions are different, depending on the organism, cell type, and precise genetic alterations in that cell (138). A pertinent example of this principle is provided by results of treatment with drugs inhibiting mutant BRAF kinase activity. In the majority of patients with melanomas harboring (V600E; V, Val; E, Glu) mutations in the BRAF gene, these drugs induce dramatic (though transient) remissions (108). But the same drugs have no therapeutic effect in colorectal cancer patients harboring the identical BRAF mutations (139). This observation has been attributed to the expression of EGFR, which occurs in some colorectal cancers but not in melanoma and is thought to circumvent the growth-inhibitory effects of the BRAF inhibitors. With this example in mind, no one should be surprised that a new drug that works well in an engineered tumor in mice fails in human trials; the organism is different, the cell type is usually different, and the precise genetic constitutions are always different. The converse of this statement—that a drug that fails in animal trials will not necessarily fail in human trials—has important practical consequences. In our view, if the biochemical and conceptual bases for a drug’s actions are solid and the drug is shown to be safe in animals, then a human trial may be warranted, even if it does not shrink tumors in mice.
Genome-Based Medicines of the Future
Cancer genomes can also be exploited for the development of more effective immunotherapies. As noted above, typical solid tumors contain 30 to 70 mutations that alter the amino acid sequences of the proteins encoded by the affected genes. Each of these alterations is foreign to the immune system, as none have been encountered during embryonic or postnatal life. Therefore, these alterations, in principle, provide a “holy grail” for tumor immunology: truly tumor-specific antigens. These antigens could be incorporated into any of the numerous platforms that already exist for the immunotherapy of cancer. These include administration of vaccines containing the mutant peptide, viruses encoding the mutant peptides on their surfaces, dendritic cells presenting the mutated peptide, and antibodies or T cells with reactivity directed against the mutant peptides (140).
To realize these sorts of therapeutics, several conditions must be met. First, the mutant protein must be expressed. As cancer cells generally express about half of the proteins that are encoded by the human genome (141), this condition is not limiting. Second, as most proteins affected by mutations are intracellular, these mutations will not be visible to the immune system unless the mutant residue is presented in the context of a human leukocyte antigen (HLA) protein. Based on in silico analyses of binding affinities, it has been estimated that a typical breast or colorectal cancer contains 7 to 10 mutant proteins that can bind to an individual patient’s HLA type (142). These theoretical predictions have recently gained experimental support. Studies of mouse tumors have identified mutant genes and shown that the corresponding peptides can induce antitumor immunity when administered as vaccines (143). Moreover, clinical trials of brain cancer patients immunized against a mutant peptide have yielded encouraging results (144).
As with all cancer therapies that are attractive in concept, obstacles abound in practice. If a tumor expresses a mutant protein that is recognizable as foreign, why has the host immune system not eradicated that tumor already? Indeed, immunoediting in cancers has been shown to exist, resulting in the down-regulation or absence of mutant epitopes that should have, and perhaps did, elicit an immune response during tumor development (145, 146). Additionally, tumors can lose immunogenicity through a variety of genetic alterations, thereby precluding the presentation of epitopes that would otherwise be recognized as foreign (147). Though these theoretical limitations are disheartening, recent studies on immune regulation in humans portend cautious optimism (148, 149).
Other Ways to Reduce Morbidity and Mortality Through Knowledge of Cancer Genomics
When we think about eradicating cancer, we generally think about curing advanced cases—those that cannot be cured by surgery alone because they have already metastasized. This is a curious way of thinking about this disease. When we think of cardiovascular or infectious diseases, we first consider ways to prevent them rather than drugs to cure their most advanced forms. Today, we are in no better position to cure polio or massive myocardial infarctions than we were a thousand years ago. But we can prevent these diseases entirely (vaccines), reduce incidence (dietary changes, statins), or mitigate severity (stents, thrombolytic agents) and thereby make a major impact on morbidity and mortality.
This focus on curing advanced cancers might have been reasonable 50 years ago, when the molecular pathogenesis of cancers was mysterious and when chemotherapeutic agents against advanced cancers were showing promise. But this mindset is no longer acceptable. We now know precisely what causes cancer: a sequential series of alterations in well-defined genes that alter the function of a limited number of pathways. Moreover, we know that this process takes decades to develop and that the incurable stage, metastasis, occurs only a few years before death. In other words, of the one million people that will die from cancer this year, the vast majority will die only because their cancers were not detected in the first 90% of the cancers’ lifetimes, when they were amenable to the surgeons’ scalpel.
This new knowledge of cancer (Box 2) has reinvigorated the search for cures for advanced cancers, but has not yet permeated other fields of applied cancer research. A common and limited set of driver genes and pathways is responsible for most common forms of cancer (table S2); these genes and pathways offer distinct potential for early diagnosis. The genes themselves, the proteins encoded by these genes, and the end products of their pathways are, in principle, detectable in many ways, including analyses of relevant body fluids, such as urine for genitourinary cancers, sputum for lung cancers, and stool for gastrointestinal cancers (150). Equally exciting are the possibilities afforded by molecular imaging, which not only indicate the presence of a cancer but also reveal its precise location and extent. Additionally, research into the relationship between particular environmental influences (diet and lifestyle) and the genetic alterations in cancer is sparse, despite its potential for preventative measures.
Box 2. Highlights.
Most human cancers are caused by two to eight sequential alterations that develop over the course of 20 to 30 years.
Each of these alterations directly or indirectly increases the ratio of cell birth to cell death; that is, each alteration causes a selective growth advantage to the cell in which it resides.
The evidence to date suggests that there are ~140 genes whose intragenic mutations contribute to cancer (so-called Mut-driver genes). There are probably other genes (Epi-driver genes) that are altered by epigenetic mechanisms and cause a selective growth advantage, but the definitive identification of these genes has been challenging.
The known driver genes function through a dozen signaling pathways that regulate three core cellular processes: cell fate determination, cell survival, and genome maintenance.
Every individual tumor, even of the same histopathologic subtype as another tumor, is distinct with respect to its genetic alterations, but the pathways affected in different tumors are similar.
Genetic heterogeneity among the cells of an individual tumor always exists and can impact the response to therapeutics.
In the future, the most appropriate management plan for a patient with cancer will be informed by an assessment of the components of the patient’s germline genome and the genome of his or her tumor.
The information from cancer genome studies can also be exploited to improve methods for prevention and early detection of cancer, which will be essential to reduce cancer morbidity and mortality.
The reasons that society invests so much more in research on cures for advanced cancers than on prevention or early detection are complex. Economic issues play a part: New drugs are far more lucrative for industry than new tests, and large individual costs for treating patients with advanced disease have become acceptable, even in developing countries (151). From a technical standpoint, the development of new and improved methods for early detection and prevention will not be easy, but there is no reason to assume that it will be more difficult than the development of new therapies aimed at treating widely metastatic disease.
Our point is not that strenuous efforts to develop new therapies for advanced cancer patients should be abandoned. These will always be required, no matter our arsenal of early detection or preventative measures. Instead, we are suggesting that “plan A” should be prevention and early detection, and “plan B” (therapy for advanced cancers) should be necessary only when plan A fails. To make plan A viable, government and philanthropic organizations must dedicate a much greater fraction of their resources to this cause, with long-term considerations in mind. We believe that cancer deaths can be reduced by more than 75% in the coming decades (152), but that this reduction will only come about if greater efforts are made toward early detection and prevention.
Supplementary Material
Acknowledgments
We thank M. Nowak and I. Bozic for critical reading of the manuscript, S. Gabelli for assisting with the production of Fig. 8, and A. Dixon, V. Ferranta, and E. Cook for artwork. This work was supported by The Virginia and D.K. Ludwig Fund for Cancer Research; The Lustgarten Foundation for Pancreatic Cancer Research; and NIH grants CA 43460, CA 47345, CA 62924, and CA 121113. All authors are Founding Scientific Advisors of Personal Genome Diagnostics (PGDx), a company focused on the identification of genetic alterations in human cancer for diagnostic and therapeutic purposes. All authors are also members of the Scientific Advisory Board of Inostics, a company that is developing technologies for the molecular diagnosis of cancer. All authors own stock in PGDx and Inostics. The terms of these arrangements are being managed by Johns Hopkins University, in accordance with their conflict-of-interest policies.
Footnotes
References and Notes
- 1.Wood LD, et al. Science. 2007;318:1108. [Google Scholar]
- 2.Parsons DW, et al. Science. 2008;321:1807. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, et al. Science. 2008;321:1801. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Govindan R, et al. Cell. 2012;150:1121. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gryfe R, Gallinger S. Surgery. 2001;130:17. doi: 10.1067/msy.2001.112738. [DOI] [PubMed] [Google Scholar]
- 6.The Cancer Genome Atlas Network. Nature. 2012;487:330. [Google Scholar]
- 7.Palles C, et al. Nat Genet. 2013;45:136. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nowell PC. Science. 1976;194:23. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 9.Fearon ER, Vogelstein B. Cell. 1990;61:759. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 10.Kinzler KW, Vogelstein B. Nature. 1997;386:761. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, et al. Proc Natl Acad Sci USA. 2008;105:4283. [Google Scholar]
- 12.Bozic I, et al. Proc Natl Acad Sci USA. 2010;107:18545. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasetti C, Vogelstein B, Parmigiani G. Proc Natl Acad Sci USA. 2013;110:1999. doi: 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laurenti E, Dick JE. Ann NY Acad Sci. 2012;1266:68. doi: 10.1111/j.1749-6632.2012.06577.x. [DOI] [PubMed] [Google Scholar]
- 15.Welch JS, et al. Cell. 2012;150:264. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yachida S, et al. Nature. 2010;467:1114. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerbel RS. Adv Cancer Res. 1990;55:87. doi: 10.1016/s0065-230x(08)60469-8. [DOI] [PubMed] [Google Scholar]
- 18.Bernards R, Weinberg RA. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 19.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. J Cell Biol. 2011;192:373. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komori J, Boone L, DeWard A, Hoppo T, Lagasse E. Nat Biotechnol. 2012;30:976. doi: 10.1038/nbt.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelizzola M, Ecker JR. FEBS Lett. 2011;585:1994. doi: 10.1016/j.febslet.2010.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parmigiani G, et al. Genomics. 2009;93:17. doi: 10.1016/j.ygeno.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyerson M, Gabriel S, Getz G. Nat Rev Genet. 2010;11:685. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 24.Carter H, et al. Cancer Res. 2009;69:6660. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Youn A, Simon R. Bioinformatics. 2011;27:175. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminker JS, Zhang Y, Watanabe C, Zhang Z. Nucleic Acids Res. 2007;35:W595. doi: 10.1093/nar/gkm405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michaelson JJ, et al. Cell. 2012;151:1431. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nik-Zainal S, et al. Cell. 2012;149:979. [Google Scholar]
- 29.Thiagalingam S, et al. Nat Genet. 1996;13:343. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 30.Forbes SA, et al. Nucleic Acids Res. 2011;39:D945. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, et al. N Engl J Med. 2009;360:765. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao S, et al. Science. 2009;324:261. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward PS, et al. Cancer Cell. 2010;17:225. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dang L, et al. Nature. 2010;465:966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pear WS, Aster JC. Curr Opin Hematol. 2004;11:426. doi: 10.1097/01.moh.0000143965.90813.70. [DOI] [PubMed] [Google Scholar]
- 36.Nicolas M, et al. Nat Genet. 2003;33:416. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 37.Weng AP, et al. Science. 2004;306:269. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 38.Agrawal N, et al. Science. 2011;333:1154. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stransky N, et al. Science. 2011;333:1157. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agrawal N, et al. Cancer Discovery. 2012;2:899. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parsons DW, et al. Science. 2011;331:435. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasqualucci L, et al. Nat Genet. 2011;43:830. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morin RD, et al. Nature. 2011;476:298. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grasso CS, et al. Nature. 2012;487:239. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellis MJ, et al. Nature. 2012;486:353. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiegand KC, et al. N Engl J Med. 2010;363:1532. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones S, et al. Science. 2010;330:228. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones S, et al. Hum Mutat. 2012;33:100. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang K, et al. Nat Genet. 2011;43:1219. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 50.Huang J, et al. Nat Genet. 2012;44:1117. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 51.Imielinski M, et al. Cell. 2012;150:1107. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudin CM, et al. Nat Genet. 2012;44:1111. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delhommeau F, et al. N Engl J Med. 2009;360:2289. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 54.Schwartzentruber J, et al. Nature. 2012;482:226. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 55.Wu G, et al. Nat Genet. 2012;44:251. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ley TJ, et al. N Engl J Med. 2010;363:2424. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dalgliesh GL, et al. Nature. 2010;463:360. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suvà ML, Riggi N, Bernstein BE. Science. 2013;339:1567. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papaemmanuil E, et al. N Engl J Med. 2011;365:1384. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graubert TA, et al. Nat Genet. 2012;44:53. [Google Scholar]
- 61.Yoshida K, et al. Nature. 2011;478:64. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 62.Jiao Y, et al. Science. 2011;331:1199. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cesare AJ, Reddel RR. Nat Rev Genet. 2010;11:319. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 64.Heaphy CM, et al. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vander Heiden MG, Cantley LC, Thompson CB. Science. 2009;324:1029. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu C, et al. Nature. 2012;483:474. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turcan S, et al. Nature. 2012;483:479. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stephens PJ, et al. Cell. 2011;144:27. [Google Scholar]
- 69.Bass AJ, et al. Nat Genet. 2011;43:964. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomlins SA, et al. Science. 2005;310:644. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 71.Soda M, et al. Nature. 2007;448:561. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 72.Armitage P, Doll R. Br J Cancer. 1954;8:1. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Böttcher R, et al. Nucleic Acids Res. 2012;40:e125. doi: 10.1093/nar/gks393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forster M, et al. Nucleic Acids Res. 2013;41:e16. doi: 10.1093/nar/gks836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding J, et al. Bioinformatics. 2012;28:167. [Google Scholar]
- 76.Beal MA, Glenn TC, Somers CM. Mutat Res. 2012;750:96. doi: 10.1016/j.mrrev.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 77.Mertes F, et al. Brief Funct Genomics. 2011;10:374. doi: 10.1093/bfgp/elr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gundry M, Vijg J. Mutat Res. 2012;729:1. doi: 10.1016/j.mrfmmm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biankin AV, et al. Nature. 2012;491:399. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan H, Kinzler KW, Vogelstein B. Science. 2000;289:1890. doi: 10.1126/science.289.5486.1890. [DOI] [PubMed] [Google Scholar]
- 81.Huang FW, et al. Science. 2013;339:957. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Horn S, et al. Science. 2013;339:959. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 83.Xue W, et al. Proc Natl Acad Sci USA. 2012;109:8212. [Google Scholar]
- 84.Solimini NL, et al. Science. 2012;337:104. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beggs AD, et al. J Pathol. 2012 doi: 10.1002/path.4132. [DOI] [Google Scholar]
- 86.Feinberg AP, Tycko B. Nat Rev Cancer. 2004;4:143. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 87.Jones PA, Baylin SB. Cell. 2007;128:683. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Höglund M, Gisselsson D, Säll T, Mitelman F. Cancer Genet Cytogenet. 2002;135:103. doi: 10.1016/s0165-4608(01)00645-8. [DOI] [PubMed] [Google Scholar]
- 89.Shibata D, Schaeffer J, Li ZH, Capella G, Perucho M. J Natl Cancer Inst. 1993;85:1058. doi: 10.1093/jnci/85.13.1058. [DOI] [PubMed] [Google Scholar]
- 90.Sottoriva A, Spiteri I, Shibata D, Curtis C, Tavaré S. Cancer Res. 2013;73:41. doi: 10.1158/0008-5472.CAN-12-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shah SP, et al. Nature. 2009;461:809. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 92.Anderson K, et al. Nature. 2011;469:356. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 93.Navin N, et al. Nature. 2011;472:90. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nik-Zainal S, et al. Cell. 2012;149:994. [Google Scholar]
- 95.Gerlinger M, et al. N Engl J Med. 2012;366:883. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu X, et al. Cell. 2012;148:886. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campbell PJ, et al. Nature. 2010;467:1109. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wagle N, et al. J Clin Oncol. 2011;29:3085. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Komarova NL, Wodarz D. Proc Natl Acad Sci USA. 2005;102:9714. doi: 10.1073/pnas.0501870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Turke AB, et al. Cancer Cell. 2010;17:77. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Durrett R, Moseley S. Theor Popul Biol. 2010;77:42. doi: 10.1016/j.tpb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 102.Diaz LA, Jr, et al. Nature. 2012;486:537. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kreso A, et al. Science. 2013;339:543. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stephens PJ, et al. Nature. 2012;486:400. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.The Cancer Genome Atlas Network. Nature. 2012;490:61. [Google Scholar]
- 106.Pang H, et al. Cancer Res. 2009;69:8868. doi: 10.1158/0008-5472.CAN-09-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen J, Ye Y, Sun H, Shi G. Cancer Chemother Pharmacol. 2013;71:265. doi: 10.1007/s00280-012-2005-9. [DOI] [PubMed] [Google Scholar]
- 108.Chapman PB, et al. N Engl J Med. 2011;364:2507. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kwak EL, et al. N Engl J Med. 2010;363:1693. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ljungman M, Lane DP. Nat Rev Cancer. 2004;4:727. doi: 10.1038/nrc1435. [DOI] [PubMed] [Google Scholar]
- 111.Perrimon N, Pitsouli C, Shilo BZ. Cold Spring Harb Perspect Biol. 2012;4:a005975. doi: 10.1101/cshperspect.a005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kerbel RS. N Engl J Med. 2008;358:2039. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chung AS, Ferrara N. Annu Rev Cell Dev Biol. 2011;27:563. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 114.Baish JW, et al. Proc Natl Acad Sci USA. 2011;108:1799. doi: 10.1073/pnas.1018154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hynes NE, Lane HA. Nat Rev Cancer. 2005;5:341. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 116.Turner N, Grose R. Nat Rev Cancer. 2010;10:116. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 117.Yun J, et al. Science. 2009;325:1555. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ying H, et al. Cell. 2012;149:656. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Araten DJ, et al. Cancer Res. 2005;65:8111. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 120.Kunkel TA. Cold Spring Harb Symp Quant Biol. 2009;74:91. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou BBS, Elledge SJ. Nature. 2000;408:433. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 122.Medema RH, Macůrek L. Oncogene. 2012;31:2601. doi: 10.1038/onc.2011.451. [DOI] [PubMed] [Google Scholar]
- 123.Derheimer FA, Kastan MB. FEBS Lett. 2010;584:3675. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ciriello G, Cerami E, Sander C, Schultz N. Genome Res. 2012;22:398. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yeang CH, McCormick F, Levine A. FASEB J. 2008;22:2605. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 126.Sharma SV, Bell DW, Settleman J, Haber DA. Nat Rev Cancer. 2007;7:169. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 127.McLeod HL. Science. 2013;339:1563. doi: 10.1126/science.1234139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brock DW. Oncologist. 2010;15(suppl 1):36. doi: 10.1634/theoncologist.2010-S1-36. [DOI] [PubMed] [Google Scholar]
- 129.Lemmon MA, Schlessinger J. Cell. 2010;141:1117. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 131.Bienstock RJ. Curr Pharm Des. 2012;18:1240. doi: 10.2174/138161212799436449. [DOI] [PubMed] [Google Scholar]
- 132.Besson A, Dowdy SF, Roberts JM. Dev Cell. 2008;14:159. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 133.Morin PJ, et al. Science. 1997;275:1787. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 134.He TC, et al. Science. 1998;281:1509. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 135.van de Wetering M, et al. Cell. 2002;111:241. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 136.Farmer H, et al. Nature. 2005;434:917. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 137.Irshad S, Ashworth A, Tutt A. Expert Rev Anticancer Ther. 2011;11:1243. doi: 10.1586/era.11.52. [DOI] [PubMed] [Google Scholar]
- 138.Grueneberg DA, et al. Proc Natl Acad Sci USA. 2008;105:16472. doi: 10.1073/pnas.0808019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mao M, et al. Clin Cancer Res. 2013;19:657. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kirkwood JM, et al. CA Cancer J Clin. 2012;62:309. doi: 10.3322/caac.20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barrett T, et al. Nucleic Acids Res. 2011;39:D1005. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Segal NH, et al. Cancer Res. 2008;68:889. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 143.Castle JC, et al. Cancer Res. 2012;72:1081. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 144.Sampson JH, et al. J Clin Oncol. 2010;28:4722. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Matsushita H, et al. Nature. 2012;482:400. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Nature. 2012;482:405. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Challa-Malladi M, et al. Cancer Cell. 2011;20:728. doi: 10.1016/j.ccr.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hodi FS, et al. N Engl J Med. 2010;363:711. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Topalian SL, et al. N Engl J Med. 2012;366:2443. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dunn BK, Jegalian K, Greenwald P. Recent Results Cancer Res. 2011;188:21. doi: 10.1007/978-3-642-10858-7_3. [DOI] [PubMed] [Google Scholar]
- 151.Lopes LC, Barberato-Filho S, Costa AC, Osorio-de-Castro CGS. Rev Saude Publica. 2010;44:620. doi: 10.1590/s0034-89102010000400005. [DOI] [PubMed] [Google Scholar]
- 152.Colditz GA, Wolin KY, Gehlert S. Sci Transl Med. 2012;4:127rv4. doi: 10.1126/scitranslmed.3003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.