

Abstract
A tight balance between anti- and pro-apoptotic members of the Bcl-2 family controls cell survival and death. Exposure to hyperoxia shifts this balance towards a pro-death state that ultimately activates Bak and Bax-dependent cell death. Mechanisms underlying this shift are undefined; however, the cell cycle inhibitor p21 delays the loss of anti-apoptotic Mcl-1 and Bcl-XL, and protects against hyperoxia. Here, H1299 human lung adenocarcinoma cells are used to investigate how these and other members of the Bcl-2 family cooperate with p21 to protect against hyperoxia. Expression of anti-apoptotic Mcl-1 and Bcl-XL, but not Bcl-2 or A1 declined during hyperoxia while pro-apoptotic Bak, but not Bax increased. Conditional over-expression of p21 selectively delayed the loss of Mcl-1 and Bcl-XL, without affecting expression of the other members. SiRNA knockdown of Mcl-1 and Bcl-XL sensitized cells to hyperoxia, but only the loss of Bcl-XL ablated the protective effects of p21. Conversely, over-expression of Mcl-1 and Bcl-XL protected against hyperoxia, but only Bcl-XL bound Bak and Bax. Altogether, our data suggest Bcl-XL is the primary mediator by which p21 protects against hyperoxia-induced Bak/Bax-dependent cell death.
Keywords: apoptosis, Bcl-2 proteins, cell cycle inhibitor p21, hyperoxia, necrosis
INTRODUCTION
Hyperoxia is frequently used to treat patients in respiratory distress; however, elevated levels of reactive oxygen species (ROS) and cell death occurs with prolonged exposure [1]. While hyperoxia induces mixed apoptotic and necrotic death in cell lines [2, 3] and adult mice [4], both forms of death involve members of the Bcl-2 family [5–7]. Members of the Bcl-2 family can be classified into three categories: anti-apoptotic Bcl-2, Bcl-XL, Bcl-w, Mcl-1 and A1; pro-apoptotic members with multiple BH3 domains including Bak and Bax, and another pro-apoptotic subfamily with members containing only one BH3 domain: Bad, Bid, Bim, Nbk (also known as Bik or Blk), Noxa and Puma. Increased resistance of mouse embryo fibroblasts lacking Bak and Bax was the first evidence Bcl-2 related proteins participated in hyperoxia-induced cell death [5]. Other studies with mice and various cell lines have shown pro-apoptotic Bid, Bim, and anti-apoptotic Bcl-2, Bcl-XL, Mcl-1 and A1 can promote or antagonize hyperoxia-induced cell death [6, 7, 10–12]. Yet how hyperoxia stimulates a pro-death state with these proteins is presently not known.
As hyperoxia promotes cell death, it also activates MAP kinases [11, 13] and cell cycle checkpoints [14–16] that promote cell survival. One of the most cytoprotective pathways is the p53-dependent activation of the cell cycle inhibitor p21 Cip1/Waf1/Sdi1 that exerts both G1 growth arrest and protection against hyperoxia [17–19]. While p21 was thought to protect against genotoxic stress by inhibiting replication of damaged DNA [20–22], accumulating evidence indicate that it can inhibit cell death in a growth arrest-independent manner. For instance, cytoplasmic forms of p21 can inhibit prostaglandin A2-induced apoptosis [23], inhibit apoptosis signal-regulating kinase 1 (MAPKKK) during monocyte differentiation [24], and caspase 3 and 8 activation [25, 26]. Although cytoplasmic targeted p21 also protects against hyperoxia [27], it does not modify MAP kinase signaling or caspase 3 activation (unpublished observations). Instead, p21 delayed the loss of anti-apoptotic Mcl-1 and Bcl-XL, and protects a variety of epithelial cell lines against hyperoxia [7, 27, 28].
The concomitant activation of cell survival and cell death pathways during hyperoxia seems counter-productive. Hence, understanding how these opposing pathways ultimately dictate cell fate is of great interest and could provide new opportunities for controlling cellular response to hyperoxia or oxidative stress in general. Here, we investigate how p21, Mcl-1, Bcl-XL, and other members of the anti-apoptotic Bcl-2 family cooperate to modulate cell fate during hyperoxia.
MATERIAL AND METHODS
Cell lines and exposure conditions
Human lung adenocarcinoma H1299 cells were cultured in high glucose Dulbecco's Modified Eagle Medium (DMEM) (GIBCO, Carlsbad, CA) with 10% fetal bovine serum (GIBCO), 100U/ml penicillin, 100μg/ml streptomycin (Cellgro, Manassas, VA), and 20μg/ml gentamicin (GIBCO). Stable clones that conditionally over-express p21 fused to enhanced green fluorescence protein (EGFp21), Flag-tagged Mcl-1, or Flag-tagged Bcl-XL were generated and cultured as previously described [7, 27, 28]. Doxycycline (Sigma, St. Louis, MO) was added to the medium to induce the expression of desired genes. All cells were maintained in tissue culture flasks and exposed to room air (with 5% CO2) or hyperoxia (95% O2 and 5% CO2) (Airgas, Salem, NH).
siRNA transfections
Cells were plated at 3×104 in 6-well plates (Falcon, Franklin Lakes, NJ) with or without doxycycline in antibiotic-free medium. Lipofectamine2000 (Invitrogen, Carlsbad, CA) was used to transfect siRNAs targeting Bak (1nM), Bax (50nM), Mcl-1 (100nM) (all SmartPool, Dharmacon, Lafayette, CO), Bcl-XL (100nM from I nvitrogen), or luciferase (Dharmacon) [27]. Cells were washed with HBSS (Cellgro, Manassas, VA), supplemented with normal medium, and exposed to room air or hyperoxia 24 hours later.
Cell death assays
AnnexinV/7AAD and trypan blue dye exclusion assays were used to assess cell viability. For annexinV and 7AAD assay, cells were washed twice with 1X phosphate buffered saline (PBS, Cellgro), trypsinized and then centrifuged at 5,000 rpm for 5 minutes at 4°C. Cell pellets were washed twice with 1X PBS, then resuspended in 1X binding buffer (BD Biosciences, Franklin Lakes, NJ) and incubated with annexinV and 7AAD at room temperature for 15 minutes in the dark. Samples were then analyzed using BD FACSCalibur system (BD Biosciences) set to collect 10,000 events. The percentage of dead cells (including both apoptosis and necrosis) was determined using CellQuest v3.3 software (BD Biosciences). For trypan blue dye exclusion, cells were washed and collected as described above. Cell pellets were washed twice with 1X PBS then resuspended in 0.2% trypan blue solution (Sigma, 0.4% diluted with 1X PBS at 1:1 ratio) and counted on a hemocytometer. A second investigator who was blinded to the sample conditions confirmed the counts.
Immunoblotting and immunoprecipitation
Cells were lysed and proteins were collected as previously described [7]. Lysates were cleared by centrifugation at 14,000 rpm for 5 minutes at 4°C and protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). Lysates were boiled for 5 minutes in 3X Laemmli buffer (50mM Tris, pH 6.8, 1% β-mercaptoethanol, 2% SDS, 0.1% bromophenol blue and 10% glycerol) and then separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA). Membranes were then probed with anti-A1 (1:500 Santa Cruz Biotechnology, Santa Cruz, CA), anti-actin (1:1000 Sigma), anti-Bak clone Ab-1 (FL-175) (1:200 Calbiochem, Gibbstown, NJ), anti-Bax clone N-20 (1:1000 Santa Cruz Biotechnology), anti-Bcl-2 (1:1000 BD), anti-Bcl-XL clone 2H12 (1:1000 Sigma), anti-Mcl-1 (S-19) (1:1000 Santa Cruz Biotechnology) and anti-p21 clone SX118 (1:500 Pharmingen, San Diego, CA) diluted in Tris-buffered saline plus 0.1% Tween 20 with 5% milk. After incubating with appropriate secondary antibodies, membranes were visualized by chemi-luminescence ECL Plus (GE Healthcare, Piscataway, NJ). Band intensity was then captured using Fluorochem 8900 (Alpha Innotech, San Leandro, CA) or film (LPS, Rochester, NY), quantified, and normalized to actin using ImageJ software (National Institutes of Health).
For immunoprecipitation studies, cells were cultured in doxycycline for 24 hours to induce expression of Flag-tagged Mcl-1 or Bcl-XL and then exposed to room air or hyperoxia for 4 days. Cells were lysed on ice with 200 ul non-denaturing lysis buffer (50mM Tris, pH 7.4, 150mM NaCl, 1% Triton X-100, 25mM sodium fluoride, 25mM β-glycerophosphate, 0.1μg/ml pepstatin A, 1.9μg/ml aprotinin, 2μg/ml leupeptin, 1mM EDTA, 0.1mM sodium vanadate and 0.1mM phenylmethylsulfonyl fluoride). After 30 minutes, lysates were then centrifuged at 10,000rpm for 15 minutes at 4°C. Protein concentration was determined and the same amount of protein was immunoprecipitated using EZviewTM Red ANTI-FLAG Affinity Gel following manufacturer's instructions (Sigma). Proteins were eluted from beads with 2X Laemmli buffer and analyzed by immunoblotting.
Statistical Analysis
Values are means ± standard deviations. Group means were compared by ANOVA using Fisher's procedure post hoc analysis with Statview software (Abacus Concepts, Piscataway, NJ). P ≤ 0.05 was considered significant.
RESULTS
Bak and Bax are essential for necrotic death of H1299 cells
As defined by trypan blue dye exclusion assays, H1299 cells die by necrosis when exposed to hyperoxia [27]. Annexin V and 7AAD staining confirmed most cells exposed to hyperoxia were necrotic (35±5%, double positive) with a small proportion (5±1.5%, annexin V positive only) being apoptotic (Figure 1A). Since necrosis predominated even with shorter exposures of 2 days, trypan blue dye exclusion was used to assess cell death in subsequent studies. To determine the contributions of Bak and Bax in hyperoxia-induced necrosis, H1299 cells were transfected with siRNA targeting their respective mRNAs and then exposed to hyperoxia for 5 days. Western blot analysis confirmed >75% knockdown of Bak and Bax (Figure 1B). Loss of either Bak or Bax significantly decreased cell death to 15±1.3% compared to mock-transfected or cells transfecting with an irrelevant siRNA targeting luciferase (P<0.005 for both) (Figure 1C).
FIGURE 1.

Bak and Bax are important regulators of cell death during hyperoxia. (A) H1299 cells were exposed to hyperoxia for 4 days and cell death was analyzed by flow cytometry with annexinV and 7AAD. Values within boxes represent percentage of 10,000 cells gated. Figures shown here are representative of 3 separate experiments with similar results. (B) Representative western blot of H1299 cells mock transfected or transfected with siRNA oligonucleotides targeting luciferase, Bak or Bax. (C) H1299 cells were transfected with siRNA oligonucleotides targeting luciferase, Bak, or Bax, and exposed to hyperoxia for 5 days. The proportion of tyrpan blue positive cells was quantified and graphed. Values represent mean +/− SD of triplicate experiments (* P<0.05).
Effect of hyperoxia and p21 on Bak and Bax expression
Since H1299 cells lack p53 required to induce p21, we previously created stable lines of that conditionally express enhanced green fluorescence protein (EGFP) fused to p21 (EGFp21) in response to doxycycline [31]. In the absence of doxycycline, exposure to hyperoxia for 2 or 4 days significantly increased the expression of Bak 3.7±1.3 fold (P=0.03), but not Bax (P=0.82) (Figure 2). Conditional over-expression of EGFp21 did not prevent the induction of Bak nor did it affect expression of Bax (P=0.81 and P=0.74, respectively).
FIGURE 2.
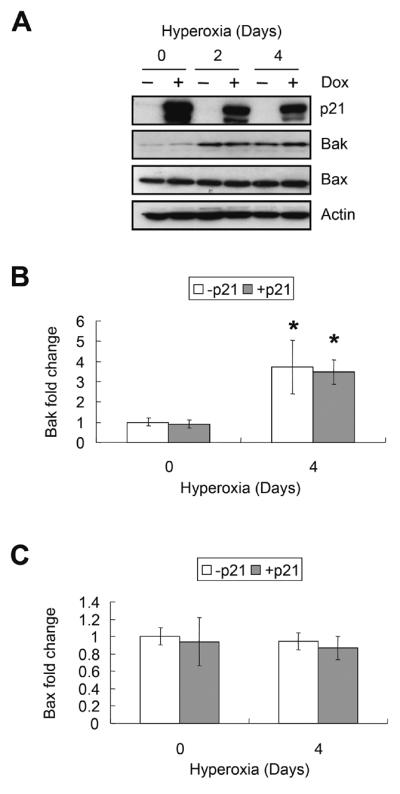
Effect of hyperoxia and p21 on Bak and Bax expression. EGFp21 cells were cultured with or without 2μg/ml doxycycline (Dox) for 24 hours (day 0) then exposed to hyperoxia for 2 and 4 days. (A) Representative western blot of p21, Bak, Bax, and actin. The results of 3 separate experiments were quantified, normalized to actin and graphed for (B) Bak and (C) Bax. Values represent mean +/− SD of three experiments (* P<0.05).
Effect of hyperoxia and p21 on anti-apoptotic Bcl-2 proteins
The effect of hyperoxia and p21 on expression of anti-apoptotic Bcl-2 members was also investigated. Hyperoxia significantly suppressed the expression of Mcl-1 to 15±9% (P<0.0001) and Bcl-XL to 45±6% (P=0.003) (Figure 3A–C). Although a 25 kd fragment of Mcl-1 was also detected, its abundance decreased in hyperoxia as well (P<0.0001) (Figure 3A). Hyperoxia did not affect expression of Bcl-2 or A1 (P=0.19 and P=0.16, respectively), and Bcl-w was not detected. Conditional over-expression of EGFp21 significantly delayed the oxygen-dependent loss of Mcl-1 and Bcl-XL (P<0.05 for both at day 4) (Figure 3B and C), and inhibited expression of Bcl-2 (P=0.003) (Figure 3D). Over-expression of p21 did not affect expression of A1 (P=0.69). To determine whether these changes were specific for p21 or related to growth arrest in G1, the expression of Mcl-1, Bcl-XL, and Bcl-2 was studied in cells conditionally over-expressing p27, a related cell cycle inhibitor that does not protect against hyperoxia [31]. Over-expression of p27 did not prevent loss of of Mcl-1 or Bcl-XL (P=0.88 and P=0.79, respectively) during hyperoxia; however, it did suppress expression of Bcl-2 (P=0.04) (Figure 4).
FIGURE 3.

p21 selectively delays the loss of Mcl-1 and Bcl-XL during hyperoxia. EGFp21 cells were cultured with or without 2μg/ml doxycycline (Dox) for 24 hours (day 0) then exposed to hyperoxia for 2 and 4 days. (A) Repersentative western blot of p21, Mcl-1, Bcl-XL, Bcl-2, A1, and actin. Band intensities were quantified, normalized to actin, and graphed for (B) Mcl-1, (C) Bcl-XL and (D) Bcl-2. Values represent mean +/− SD of triplicate experiments (* P<0.05 relative to day 0, # P<0.05 between − p21 and + p21).
FIGURE 4.

P27 does not delay the loss of Mcl-1 or Bcl-XL. EGFp27 cells were cultured with or without 2μg/ml doxycycline (Dox) for 24 hours (day 0) then exposed to hyperoxia for 2 and 4 days. (A) Representative western blot of p21, Mcl-1, Bcl-XL, Bcl-2 and actin. The results of 3 separate experiments were quantified, normalized to actin and graphed for (B) Mcl-1, (C) Bcl-XL and (D) Bcl-2. Values represent mean +/− SD of three experiments (* P<0.05 relative to day 0, # P<0.05 between − p27 and + p27).
Bcl-XL is the predominant mediator of p21 cytoprotection
Since p21 selectively inhibited the loss of anti-apoptotic Mcl-1 and Bcl-XL, three approaches were used to define whether these proteins mediate p21 protection against hyperoxia. First, the ability of p21 to protect against hyperoxia was assessed in cells with Mcl-1 and Bcl-XL knocked down by siRNA oligonucleotides. Each siRNA was able to specifically reduce expression of the target protein, however, knockdown of Bcl-XL was less effective than knockdown of Mcl-1 (Figure 5A). As expected, knockdown of Mcl-1 or Bcl-XL both significantly increased cell death during hyperoxia (P=0.03 and P=0.005, respectively) (Figure 5B). Although siRNA knockdown of Bcl-XL was less efficient than Mcl-1, it completely removed the protective effects of p21.
FIGURE 5.

Loss of Bcl-XL abolishes p21 protection. EGFp21 cells were cultured with or without 2μg/ml doxycycline (Dox), transfected with mock, luciferase, Mcl-1 or Bcl-XL siRNA and then exposed to hyperoxia for 4 days. (A) Representative western blot of p21, Mcl-1, Bcl-XL, and actin. (B) Cell survival was determined by trypan blue dye exclusion. Values represent mean +/− SD of three experiments (* P<0.05 relative to mock − p21, # P<0.05 between − p21 and + p21).
Second, the ability of each protein to protect against hyperoxia in the absence of p21 was investigated using cell lines that conditionally over-express Flag-tagged Mcl-1 or Bcl-XL. Cells that conditionally express Flag-tagged Mcl-1 or Bcl-XL were treated with increasing doses of doxycycline, cultured in hyperoxia for 4 days, and the expressions of Bak and Bax were determined by western blot analysis. Surprisingly, the up-regulation of Bak was significantly inhibited (P<0.05 for all) by doses of doxycycline greater than 0.5μg/ml, which induced supra-physiological levels of Mcl-1 (Figure 6A) or Bcl-XL (Figure 6C). High levels of Bcl-XL, but not Mcl-1 also suppressed expression of Bax (Figure 6C). Interestingly, restoring endogenous levels (using 0.05 μg/ml doxycycline) that did not inhibit Bak or Bax expression provided the same degree of protection against hyperoxia as supra-physiologic levels (using 2 μg/ml doxycycline) that did suppress expression (P<0.05 for both) (Figures 6B and 6D). Moreover, over-expression of Bcl-XL provided better protection at both doses than Mcl-1. Studies using annexin V and 7AAD indicated Bcl-XL reduced apoptotic and necrotic cell death by 8.4% and 22.1%, respectively (data not shown).
FIGURE 6.

Over-expression of Mcl-1 or Bcl-XL protect against hyperoxia. Cells with conditional expression of Flag-tagged Mcl-1 (A and B) or Bcl-XL (C and D) were treated with 0 or 2μg/ml doxycyclin (Dox) and cultured in room air for 24 hours, or with increasing doses of Dox (0, 0.01, 0.05, 0.1, 0.5, 1, 2μg/ml) and exposed to hyperoxia for 4 days. (A and C) Representative western blot of Mcl-1, Bcl-XL, Bak, Bax and actin. (B and D) Cell death was assayed by trypan blue dye exclusion. Values represent mean +/− SD of three experiments (* P<0.05).
Third, the ability of Mcl-1 and Bcl-XL to bind Bak or Bax in cells exposed to room air or hyperoxia was investigated. Cells with conditional over-expression of Flag-tagged Mcl-1 or Bcl-XL were cultured in 0, 0.05 or 2μg/ml of doxycycline and exposed to room air or hyperoxia for 4 days. Flag-tagged Mcl-1 or Bcl-XL was immunoprecipitated with anti-Flag antibody and co-immunoprecipitating proteins were investigated by western blot. Calnexin, an endoplasmic reticulum (ER) membrane protein, was blotted as a negative control to show binding specificity. Although Bak has been reported to bind both Mcl-1 and Bcl-XL [32], minimal binding between Flag-tagged Mcl-1 and Bak or Bax was observed (Figure 7A). Exposure to hyperoxia did not stimulate binding (Figure 7B). In contrast, Flag-tagged Bcl-XL bound Bak and Bax in cells exposed to room air or hyperoxia (Figure 7C and D). The binding seen in room air treated cells lacking doxycycline (Figure 7D) was most likely due to leaky expression of Flag-tagged Bcl-XL and the increased binding to Bak seen in hyperoxic cells (Figure 7D) was most likely due to hyperoxia increasing expression of Bak.
FIGURE 7.

Bak and Bax predominantly bind Bcl-XL and not Mcl-1. Cells with conditional expression of Flag-tagged Mcl-1 (A and B) or Flag-tagged Bcl-XL (C and D) were cultured with 0, 0.05 or 2μg/ml doxycycline (Dox) for 4 days in room air (A and C) or hyperoxia (B and D). Proteins were collected with Triton X-100 buffer and immunoprecipitated with anti-Flag antibody, and potential interactions with Mcl-1, Bcl-XL, Bak, Bax, or the ER membrane protein calnexin were detected by immunoblotting with specific antibodies. The asterisk indicates a non-specific band seen in immunoprecipitations with Bak and Bax indicated by an arrow. Blots are representative of 3 individual experiments with similar results.
DISCUSSION
It is known that Bcl-2 proteins and p21 play important roles in regulating cell survival under oxidative stress. While p21 has been shown to promote survival against genotoxic damage via its ability to inhibit DNA replication, modify MAP kinase signaling, or block caspase activation, it delays the loss of Mcl-1 and Bcl-XL during hyperoxia [7]. While this implies p21 protects through Mcl-1 and Bcl-XL, the current study provides conclusive evidence that Bcl-XL, which binds both Bak and Bax, is the primary mediator of p21-mediated protection against hyperoxia. Therefore, the selective maintenance of Bcl-XL by p21 is not simply a consequence of pro-survival proteins remaining high in healthy cells, but rather involves cooperation between these proteins that antagonizes hyperoxia-induced death signaling.
Using siRNA approaches to deplete Bak and Bax, we found that both proteins contribute to hyperoxia-induced necrosis of H1299 cells. This agrees with our previous finding that siRNA knockdown of Bak in Bax-deficient HCT116 colon carcinoma cells increased survival [29], as well as another study where Bak and Bax double knockout mouse embryonic fibroblasts were resistant to hyperoxia [5]. It appears hyperoxia shifts the balance towards death by stimulating expression of these two proteins through p53-dependent and independent pathways. P53-dependent upregulation of Bax has been reported during retinal ganglion cell degeneration, UV-irradiated melanocytes, and in adult mice exposed to hyperoxia [33–36]. Hyperoxia also increased Bax in wildtype but not p53-deficient HCT116 colon carcinoma cells [28], or the p53-deficient H1299 cells used in the current study. On the other hand, p53-independent induction of Bak has been reported during butyrate [37] or tumor suppressor WT1-induced apoptosis [38], and in HCT116 cells exposed to hyperoxia [29]. In the current study, hyperoxia stimulated p53-independent expression of Bak at a pre-translational level (data not shown). Altogether, these findings indicate hyperoxia stimulates p53-dependent Bax and p53-independent Bak, both of which shift cells towards a pro-death state presumably through the mitochondria (Figure 8).
FIGURE 8.

Hypothetical model for how p21 protects against hyperoxia. Hyperoxia shifts the balance towards cell death by stimulating (arrows) p53-dependent expression of Bax and p53-independent induction of Bak that presumably activate mitochondrial cell death. It also inhibits (T-bar) expression of anti-apoptotic Mcl-1 and Bcl-XL and this loss is inhibited by the p53-dependent expression of p21, with Bcl-XL being the predominant inhibitor of Bak/Bax-dependent cell death.
As the levels of these pro-death proteins increased during hyperoxia, the levels of anti-death Mcl-1 and Bcl-XL decreased and further depletion with siRNA enhanced cell death. Conditional over-expression of p21 delayed the loss of Mcl-1 and Bcl-XL during hyperoxia. When over-expressed, supra-physiological levels of Mcl-1 repressed Bak, while supra-physiological levels of Bcl-XL repressed both Bak and Bax. However, this suppression of Bak and Bax expression was not required for Mcl-1 and Bcl-XL to protect against hyperoxia, as restoring endogenous levels of these anti-apoptotic proteins was equally protective. This implies endogenous levels of these proteins are sufficient to maintain survival of cells even in the face of external threats such as hyperoxia. In other words and consistent with other experimental models [32, 39], endogenous levels of Bcl-XL bind and block Bak and Bax-dependent death of healthy cells. As Bak levels rise during hyperoxia, endogenous levels of Bcl-XL are theoretically sufficient to block death. However, levels of Bcl-XL decline during hyperoxia, thereby resulting in unbound Bak and Bax that promote cell death. Hence, p21 protects against hyperoxia by delaying the loss of Bcl-XL that sequesters Bak and Bax. It remains to be determined if this pathway exists in other oxygen sensitive cell types (i.e. endothelial cells). However, over-expression of epidermal growth factor-like domain 7 (EGFL7) protects human endothelial cells (EA.hy946) against hyperoxia and like the effects of p21 shown here, this was associated with elevated levels of Bcl-XL and reduced levels of Bax [40].
In contrast to Bcl-XL, p21 does not require Mcl-1 to protect against hyperoxia because siRNA depletion of Mcl-1 did not abrogate the protective effects of p21. Strong interactions between Mcl-1 and Bak or Bax were also not observed, which has been reported in other models, but is also currently controversial [41, 42]. Interestingly, siRNA knockdown of Mcl-1 did not significantly sensitize HCT116 colon carcinoma cells to hyperoxia (unpublished observations) whereas siRNA knockdown of Bcl-XL does [28]. While presently unknown, these differential effects of Mcl-1 and Bcl-XL may be attributed to the ability of these proteins to bind Bak and Bax, or related to the rate at which these proteins are lost during hyperoxia. Bcl-2 is also unlikely to be a downstream mediator of p21 since its expression declined in the presence of p21 and the related kinase p27. Because Bcl-2 can retard cell cycle entry by stimulating expression of p27 [43, 44], the over-expression of p21 or the related kinase p27 may create a negative feedback to repress Bcl-2 levels. The significance of down regulating Bcl-2 in p21-sufficient cells remains to be determined especially when considering over-expression of Bcl-2 protects fibroblasts exposed to hyperoxia [6].
The mechanisms by which p21 selectively delay the loss of Mcl-1 or Bcl-XL remains to be elucidated. Hyperoxia and p21 are likely to control the translation of Mcl-1 and Bcl-XL because their mRNA levels were unaltered (unpublished observations). Alternatively, p21 may also regulate the stability of Mcl-1 and Bcl-XL via disrupting their interactions with other proteins. For instance, interactions between Mcl-1 and Puma can stabilize Mcl-1 [45], while the over-expression of Noxa promotes Mcl-1 degradation [46]. Both Puma and Noxa have been shown to interact with Bcl-XL [47, 48], yet whether the interaction affects the stability of Bcl-XL is unknown. Puma-deficient HCT116 cells were protected against hyperoxia-induced cell death via p21/Bcl-XL pathway [29], however, it is unclear if p21 affects levels of Mcl-1 or Bcl-XL through BH3 only proteins. The current observation p21 cooperates with Bcl-XL to block Bak and Bax-dependent cell death during hyperoxia now provides a framework to define the role of various BH3-only proteins. Clarifying how pro-survival pathways such as p21 antagonize pro-death pathways during hyperoxia may identify new opportunities for treating diseases involving oxidative stress.
ACKNOWLEDGEMENTS
We thank Dr. Peter F. Vitiello and Jennifer Gewandter for their critical thinking during the course of these studies, Dr. Peter C. Keng and Rhonda J. Staversky for their generous help, and the Flow Cytometry Core at the University of Rochester.
GRANTS This work was funded in part by National Institutes of Health (NIH) Grants HL-67392 (M. A. O'Reilly). NIH Training Grant HL-66988 supported Y. M. Wu.
Abbreviations
- EGFP
enhanced green fluorescence protein
- p21
cyclin-dependent kinase inhibitor p21 Cip1/Waf1/Sdi1
- ROS
reactive oxygen species
Footnotes
DECLARATION OF INTEREST None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this paper. The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- [1].Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- [2].Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad Sci. 2003;1010:405–416. doi: 10.1196/annals.1299.074. [DOI] [PubMed] [Google Scholar]
- [3].Mantell LL, Horowitz S, Davis JM, Kazzaz JA. Hyperoxia-induced cell death in the lung--the correlation of apoptosis, necrosis, and inflammation. Ann N Y Acad Sci. 1999;887:171–180. doi: 10.1111/j.1749-6632.1999.tb07931.x. [DOI] [PubMed] [Google Scholar]
- [4].Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19:573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- [5].Budinger GR, Tso M, McClintock DS, Dean DA, Sznajder JI, Chandel NS. Hyperoxia-induced apoptosis does not require mitochondrial reactive oxygen species and is regulated by Bcl-2 proteins. J Biol Chem. 2002;277:15654–15660. doi: 10.1074/jbc.M109317200. [DOI] [PubMed] [Google Scholar]
- [6].Metrailler-Ruchonnet I, Pagano A, Carnesecchi S, Ody C, Donati Y, Barazzone Argiroffo C. Bcl-2 protects against hyperoxia-induced apoptosis through inhibition of the mitochondria-dependent pathway. Free Radic Biol Med. 2007;42:1062–1074. doi: 10.1016/j.freeradbiomed.2007.01.008. [DOI] [PubMed] [Google Scholar]
- [7].Vitiello PF, Wu YC, Staversky RJ, O'Reilly MA. p21(Cip1) protects against oxidative stress by suppressing ER-dependent activation of mitochondrial death pathways. Free Radic Biol Med. 2009;46:33–41. doi: 10.1016/j.freeradbiomed.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- [9].Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- [10].He CH, Waxman AB, Lee CG, Link H, Rabach ME, Ma B, Chen Q, Zhu Z, Zhong M, Nakayama K, Nakayama KI, Homer R, Elias JA. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115:1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nyunoya T, Monick MM, Powers LS, Yarovinsky TO, Hunninghake GW. Macrophages survive hyperoxia via prolonged ERK activation due to phosphatase down-regulation. J Biol Chem. 2005;280:26295–26302. doi: 10.1074/jbc.M500185200. [DOI] [PubMed] [Google Scholar]
- [12].Wang X, Ryter SW, Dai C, Tang ZL, Watkins SC, Yin XM, Song R, Choi AM. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J Biol Chem. 2003;278:29184–29191. doi: 10.1074/jbc.M301624200. [DOI] [PubMed] [Google Scholar]
- [13].Franek WR, Morrow DM, Zhu H, Vancurova I, Miskolci V, Darley-Usmar K, Simms HH, Mantell LL. NF-kappaB protects lung epithelium against hyperoxia-induced nonapoptotic cell death-oncosis. Free Radic Biol Med. 2004;37:1670–1679. doi: 10.1016/j.freeradbiomed.2004.08.007. [DOI] [PubMed] [Google Scholar]
- [14].O'Reilly MA, Staversky RJ, Finkelstein JN, Keng PC. Activation of the G2 cell cycle checkpoint enhances survival of epithelial cells exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2003;284:L368–375. doi: 10.1152/ajplung.00299.2002. [DOI] [PubMed] [Google Scholar]
- [15].Rancourt RC, Hayes DD, Chess PR, Keng PC, O'Reilly MA. Growth arrest in G1 protects against oxygen-induced DNA damage and cell death. J Cell Physiol. 2002;193:26–36. doi: 10.1002/jcp.10146. [DOI] [PubMed] [Google Scholar]
- [16].Shenberger JS, Dixon PS. Oxygen induces S-phase growth arrest and increases p53 and p21(WAF1/CIP1) expression in human bronchial smooth-muscle cells. Am J Respir Cell Mol Biol. 1999;21:395–402. doi: 10.1165/ajrcmb.21.3.3604. [DOI] [PubMed] [Google Scholar]
- [17].Helt CE, Rancourt RC, Staversky RJ, O'Reilly MA. p53-dependent induction of p21(Cip1/WAF1/Sdi1) protects against oxygen-induced toxicity. Toxicol Sci. 2001;63:214–222. doi: 10.1093/toxsci/63.2.214. [DOI] [PubMed] [Google Scholar]
- [18].Gehen SC, Staversky RJ, Bambara RA, Keng PC, O'Reilly MA. hSMG-1 and ATM sequentially and independently regulate the G(1) checkpoint during oxidative stress. Oncogene. 2008;27:4065–4074. doi: 10.1038/onc.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McGrath-Morrow SA, Cho C, Soutiere S, Mitzner W, Tuder R. The Effect of Neonatal Hyperoxia on the Lung of p21Waf1/Cip1/Sdi1-Deficient Mice. Am J Respir Cell Mol Biol. 2004;30:635–640. doi: 10.1165/rcmb.2003-0049OC. [DOI] [PubMed] [Google Scholar]
- [20].Bissonnette N, Hunting DJ. p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene. 1998;16:3461–3469. doi: 10.1038/sj.onc.1201899. [DOI] [PubMed] [Google Scholar]
- [21].Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996;10:1945–1952. doi: 10.1101/gad.10.15.1945. [DOI] [PubMed] [Google Scholar]
- [22].Rancourt RC, Keng PC, Helt CE, O'Reilly MA. The role of p21(CIP1/WAF1) in growth of epithelial cells exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2001;280:L617–626. doi: 10.1152/ajplung.2001.280.4.L617. [DOI] [PubMed] [Google Scholar]
- [23].Gorospe M, Wang X, Guyton KZ, Holbrook NJ. Protective role of p21(Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol Cell Biol. 1996;16:6654–6660. doi: 10.1128/mcb.16.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. Embo J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931–939. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- [26].Xu SQ, El-Deiry WS. p21(WAF1/CIP1) inhibits initiator caspase cleavage by TRAIL death receptor DR4. Biochem Biophys Res Commun. 2000;269:179–190. doi: 10.1006/bbrc.2000.2247. [DOI] [PubMed] [Google Scholar]
- [27].Vitiello PF, Staversky RJ, Gehen SC, Johnston CJ, Finkelstein JN, Wright TW, O'Reilly MA. P21Cip1 protection against hyperoxia requires Bcl-XL and is uncoupled from its ability to suppress growth. Am J Pathol. 2006;168:1838–1847. doi: 10.2353/ajpath.2006.051162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Staversky RJ, Vitiello PF, Gehen SC, Helt CE, Rahman A, Keng PC, O'Reilly MA. p21(Cip1/Waf1/Sdi1) protects against hyperoxia by maintaining expression of Bcl-X(L) Free Radic Biol Med. 2006;41:601–609. doi: 10.1016/j.freeradbiomed.2006.04.029. [DOI] [PubMed] [Google Scholar]
- [29].Vitiello PF, Staversky RJ, Keng PC, O'Reilly MA. PUMA inactivation protects against oxidative stress through p21/Bcl-XL inhibition of bax death. Free Radic Biol Med. 2008;44:367–374. doi: 10.1016/j.freeradbiomed.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:1931–1936. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Helt CE, Staversky RJ, Lee YJ, Bambara RA, Keng PC, O'Reilly MA. The Cdk and PCNA domains on p21Cip1 both function to inhibit G1/S progression during hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L506–513. doi: 10.1152/ajplung.00243.2003. [DOI] [PubMed] [Google Scholar]
- [32].Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].O'Reilly MA, Staversky RJ, Huyck HL, Watkins RH, LoMonaco MB, D'Angio CT, Baggs RB, Maniscalco WM, Pryhuber GS. Bcl-2 family gene expression during severe hyperoxia induced lung injury. Lab Invest. 2000;80:1845–1854. doi: 10.1038/labinvest.3780195. [DOI] [PubMed] [Google Scholar]
- [34].Kim YG, Kim HJ, Kim DS, Kim SD, Han WS, Kim KH, Chung JH, Park KC. Up-Regulation and redistribution of Bax in ultraviolet B-irradiated melanocytes. Pigment Cell Res. 2000;13:352–357. doi: 10.1034/j.1600-0749.2000.130508.x. [DOI] [PubMed] [Google Scholar]
- [35].Isenmann S, Wahl C, Krajewski S, Reed JC, Bahr M. Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur J Neurosci. 1997;9:1763–1772. doi: 10.1111/j.1460-9568.1997.tb01534.x. [DOI] [PubMed] [Google Scholar]
- [36].Pearson AS, Spitz FR, Swisher SG, Kataoka M, Sarkiss MG, Meyn RE, McDonnell TJ, Cristiano RJ, Roth JA. Up-regulation of the proapoptotic mediators Bax and Bak after adenovirus-mediated p53 gene transfer in lung cancer cells. Clin Cancer Res. 2000;6:887–890. [PubMed] [Google Scholar]
- [37].Chirakkal H, Leech SH, Brookes KE, Prais AL, Waby JS, Corfe BM. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene. 2006;25:7192–7200. doi: 10.1038/sj.onc.1209702. [DOI] [PubMed] [Google Scholar]
- [38].Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res. 2005;65:8174–8182. doi: 10.1158/0008-5472.CAN-04-3657. [DOI] [PubMed] [Google Scholar]
- [39].Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 2007;21:929–941. doi: 10.1101/gad.1522007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xu D, Perez RE, Ekekezie II, Navarro A, Truog WE. Epidermal growth factor-like domain 7 protects endothelial cells from hyperoxia-induced cell death. Am J Physiol Lung Cell Mol Physiol. 2008;294:L17–23. doi: 10.1152/ajplung.00178.2007. [DOI] [PubMed] [Google Scholar]
- [41].Germain M, Milburn J, Duronio V. MCL-1 inhibits BAX in the absence of MCL-1/BAX Interaction. J Biol Chem. 2008;283:6384–6392. doi: 10.1074/jbc.M707762200. [DOI] [PubMed] [Google Scholar]
- [42].Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- [43].Vairo G, Soos TJ, Upton TM, Zalvide J, DeCaprio JA, Ewen ME, Koff A, Adams JM. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol Cell Biol. 2000;20:4745–4753. doi: 10.1128/mcb.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Greider C, Chattopadhyay A, Parkhurst C, Yang E. BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21:7765–7775. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- [45].Mei Y, Du W, Yang Y, Wu M. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene. 2005;24:7224–7237. doi: 10.1038/sj.onc.1208873. [DOI] [PubMed] [Google Scholar]
- [46].Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104:6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- [48].Ming L, Wang P, Bank A, Yu J, Zhang L. PUMA Dissociates Bax and Bcl-X(L) to induce apoptosis in colon cancer cells. J Biol Chem. 2006;281:16034–16042. doi: 10.1074/jbc.M513587200. [DOI] [PubMed] [Google Scholar]