

Abstract
In 1959 Hervey hypothesized that a circulating feedback signal informed the hypothalamus of the size of fat stores and initiated appropriate corrections to energy balance. The hypothesis resulted from a parabiosis study in which one animal became obese following lesioning of the ventromedial hypothalamus. The partner of the lesioned rat was hypophagic and lost a large amount of body fat. Similar results came from parabiosis studies with obese Zucker rats and rats that overate due to stimulation of the lateral hypothalamus. In studies in which one parabiont was made obese by overfeeding the non-overfed partners lost substantial amounts of fat with a minimal reduction in food intake and no loss of lean tissue. The loss of fat was due to inhibition of adipose lipogenesis and other metabolic adjustments typical of food restriction. Parabiosis with genetically obese mice implied that ob/ob mice did not produce the feedback signal and subsequently the mutant ob protein, leptin, was identified. This paper provides a review and interpretation of parabiosis work that preceded the discovery of leptin, an evaluation of leptin in relation to its function as the circulating feedback signal and evidence for additional circulating factors involved in the control of adipose tissue mass.
Keywords: Parabiosis, obesity, body composition, hypothalamus, leptin
Introduction
In 1977 I was recruited as a graduate student by G. Romaine Hervey in the Physiology Department at the University of Leeds. I had completed an undergraduate combined degree in Food Science and Physiology with the expectation that ultimately I would help to run my parents health food company. By the time I was ready to graduate this was no longer appealing and the only other jobs I had been offered were to demonstrate gas stoves in showrooms for Exxon or to join a new software company in London on a salary that they admitted was less than a living wage. Being totally uninformed of the long-term consequences of my decision, a PhD seemed like a good idea. My mother was not as enthusiastic and it was several decades before she believed that I finally had a “real job”. During my first meeting with Romaine Hervey (Prof) he told me that I would have to come to the laboratory on weekends to feed the rats. Little did I realize that over 30 years later I would still be feeding rats on weekends. I joined the laboratory when there was significant disagreement between Hervey and Stock and Rothwell regarding diet induced thermogenesis (Rothwell & Stock, 1979). Gary Armitage, another graduate student in the lab, spent years tube-feeding rats and doing calorimetry studies to determine the incremental gains in energy expenditure associated with overfeeding and weight gain (Armitage, Harris, Hervey, & Tobin, 1984; Armitage, Hervey, & Tobin, 1979). Helping Gary with his studies provided me with experience that was critical to most of my subsequent research. Sharing a beer after work at the Faversham Hotel (The Fav) made us fast friends. In addition, Prof always double checked all of my data, re-calculating daily food intake and body composition numbers for all of the studies. This taught me how important it is to pay attention to the details of a study and is probably the reason why I insist on seeing the raw data from experiments before papers are published.
My graduate research focused on the phenotype of genetically obese, fatty (fa/fa) Zucker rats. At the time little was known about the cause of obesity in these animals, which are hyperphagic (Zucker & Zucker, 1961) do not show the normal diurnal pattern of feeding (Becker & Grinker, 1977), control protein intake less efficiently than their lean littermates (Pullar & Webster, 1974; Radcliffe & Webster, 1976) and maintain a relatively low body temperature (Godbole, York, & Bloxham, 1978; Trayhurn & James, 1978). Unlike rats with lesions of the ventromedial nucleus of the hypothalamus (VMH) they partially compensate for diet dilution (Bray & York, 1972), will work to obtain food (Greenwood, Quartermain, Johnson, Cruce, & Hirsch, 1974) and eat food containing quinine (Cruce, Greenwood, Johnson, & Quartermain, 1974), suggesting that the hyperphagia is not totally uncontrolled. Prof had established a breeding colony of Zucker rats and we completed a series of studies that demonstrated that both lean and fatty Zucker rats control food intake to regulate energy intake and found no evidence that food intake of fatty rats was related to nitrogen retention (Harris, Hervey, & Tobin, 1979). A calorimetry study demonstrated that the fatty Zucker rats were capable of an appropriate thermogenic response to step-wise decreases in environmental temperature and that they maintained a stable, but reduced body temperature when room temperature was decreased either in a step-wise manner or acutely (Armitage, Harris, Hervey, & Tobin, 1984). Of more relevance to this article, we investigated the changes in body composition of lean and fatty Zucker rats joined in parabiosis (Harris, Hervey, Hervey, & Tobin, 1987) and this is described in more detail below.
The Parabiotic Model
Parabiosis is the surgical union of two animals to produce a common blood supply and allows the investigation of circulating factors in the regulation of physiologic systems. A limitation of the preparation is that if outbred strains of rats or mice are used, then there can be a significant loss of pairs when blood exchange is established (11 – 14 days after surgery) due to “disharmony”. Parabiotic “disharmony” or “intoxication” (Finerty & Panos, 1951) represents the immune rejection of one animal by the other (Nakic, Nakic, & Silobrcic, 1960) and is easily identified by hyperemia of the ears, eyes and feet of one member of the pair (Binhammer, Epstein, & Whitehouse, 1963). By contrast, if the bones of the partners do not fuse, then blood exchange is not established and the two animals gradually separate over time as the sites of surgical union grow apart.
It is important to note that the nature of exchange between parabiotic rats is relatively slow, with total blood volume exchanging approximately ten times per day (Huff, Trautman, & Van Dyke, 1950). For a factor to reach equilibrium across the parabiotic union it has to have a half life that is longer than the rate of removal from the circulation by two animals. The size of the factor is not an issue because Huff et al (Huff, Trautman, & Van Dyke, 1950) demonstrated that radiolabeled red blood cells exchanged between parabiosed rats and this has recently been confirmed using mice expressing green fluorescent protein (Gibney, et al., 2012). In 1950 Van Dyke et al (Van Dyke, Simpson, Li, & Evans, 1950) showed that growth hormone, which was estimated to have a half life of 9 hours in the circulation, reached approximately half the concentration in a hypophysectomized partner as that of the intact parabiont. Interestingly, the combined amount of growth hormone present in the two animals was equal to that normally found in one rat. By contrast, adrenocorticotrophic hormone, which was estimated to have a half life of approximately 17 minutes, had almost no biological activity in the hypophysectomized partner.
Parabiosis provides an opportunity to test whether a system involves a circulating negative feedback signal on condition that the signal is relatively stable in the circulation. For example, involvement of pituitary factors in the control of gonadal hormone secretion was demonstrated when one member of a parabiosed pair of rats of was ovariectomized (Meyer, Biddulph, & Finerty, 1946). The resultant loss of negative feedback control stimulated release of the relatively stable gonadotrophin which was carried into the intact partner. The ovaries of the partner enlarged in response to the stimulation, but estrogen from this animal had a short half-life in the circulation and failed to suppress secretion of the gonadotrophin in the ovariectomized rat. Estradiol injections into the ovariectomized rat led to the normalization of ovaries in the intact partner (Meyer, Biddulph, & Finerty, 1946). Thus, the presence of a feedback control system was demonstrated by the system “running-away” once there was a break in the feedback loop. The results from parabiosis experiments with VMH lesioned rats led Hervey (Hervey, 1959) to hypothesize that a circulating signal originating in body fat fed back to the hypothalamus to regulate energy balance. In this situation the break in the feedback loop was caused by hypothalamic lesions. The hypothesized “satiety” signal must have a relatively long half-life, both to be effective in parabiosed rats and because energy balance is usually achieved over periods of days rather than hours.
Parabiosis in Zucker Rats
Although fatty Zucker rats do not have any obvious physical lesions of the hypothalamus (Zucker, 1967), a parabiosis study was conducted to test for functional impairments. If the lean partners of fatty rats responded in a similar manner to lean partners of VMH lesioned rats, then this would imply that the fatty rats were insensitive to the circulating satiety factor. By contrast, if the fatty partners of lean rats lost weight, then this would imply that they were unable to produce the circulating factor. Fatty rats parabiosed to wild type littermates were similar to rats with VMH lesions in that there was no response to parabiosis with a lean rat (Harris, Hervey, Hervey, & Tobin, 1987). Over a period of 14 weeks lean partners of obese Zucker rats reduced their body fat content by almost 50% compared with members of lean-lean pairs. It is impossible to accurately measure food intake of individual parabionts when both animals are eating, but gut content is a reliable index of food consumed during the previous 24 hours (Harris, Zhou, Weigle, & Kuijper, 1997). In the Zucker parabiosis study (Harris, Hervey, Hervey, & Tobin, 1987) gut content implied that the food intake of partners of obese rats was reduced by 25–30 % compared with lean-lean parabiotic controls. The data were interpreted as evidence that obese Zucker rats produced the hypothesized circulating satiety factor, but were unable to respond to it. In retrospect, this conclusion is fully consistent with the current knowledge that Zucker rats have a single gene mutation of the extracellular domain of the leptin receptor (Chua, et al., 1996; Truett, Bahary, Friedman, & Leibel, 1991).
Parabiosis in Overfed Obese Rats
On completion of my graduate work I moved to the laboratory of Dr Roy Martin in the Department of Nutrition at the University of Georgia, where I joined his team of graduate students and post-docs, many of whom had recently moved with Roy from Pennsylvania State University. Roy met Prof at a meeting in Cypress and was looking for a post-doc who could overfeed rats. I was recruited sight unseen and arrived at Atlanta airport with one large suitcase expecting to stay for 2 years and then return to England. Thirty years later I am still here. My initial responsibility was to establish a rat model of overfeeding, but I also was given the freedom to develop my own project and, with Roy’s encouragement, started to characterize the metabolic and endocrine phenotype of parabiotic partners of rats made obese by overfeeding. Although previous parabiosis studies had provided evidence for a circulating factor in the regulation of energy balance, very little had been done to either characterize the hormonal profile of parabiotic rats or to identify the circulating factor.
By the time Roy and I started to evaluate the phenotype of partners of overfed rats several investigators had reported substantial concentration gradients in insulin and glucose between members of parabiotic pairs in which one animal was diabetic. In a study in which one animal in a pair of two diabetic rats was injected with insulin, then the hyperglycemia was corrected in the injected rat, but was only partially corrected in the non-injected partner with a delay of several hours, indicating that the insulin did not equilibrate across the union. Parameswaran et al (Parameswaran, Steffens, Hervey, & de Ruiter, 1977) measured blood glucose, glucagon and insulin in parabiotic pairs in which one rat was driven to eat by electrical stimulation of the lateral hypothalamus (LH). Blood glucose and insulin showed substantial elevations in the stimulated rat, but only small increases in the non-stimulated partners. Glucagon was suppressed in the stimulated rat, but unchanged in the partner. Similarly, Nishizawa and Bray (Nishizawa & Bray, 1980) found only small increases in blood glucose concentrations of parabiotic partners of rats tubefed meals of high-fat or high-carbohydrate diet. Taken together these studies made it unlikely that insulin, glucose or glucagon was functioning as the parabiotic “satiety” factor.
We designed a series of experiments to further elucidate the metabolic and endocrine profile of ad libitum fed parabiotic partners of rats made obese by overfeeding (Figure 1). Female rats were used in most of the experiments because they stop growing and maintain a stable body weight at about 12 weeks of age, unlike male rats that continue to grow until late adulthood. Overfeeding was used to induce weight gain to ensure that the obesity could be directly attributed to overeating. Overfeeding required tubefeeding the rats three times a day at 8 hour intervals with a liquid diet. Over a period of 7 to 10 days the food intake of one member of a parabiotic pair was increased from 100 to 200 or 250% of the amount of food eaten voluntarily during a pre-experimental period. Tube-feeding results in both a sleep-deprived investigator and a very uniform, reproducible increase in body fat content of the overfed rats (Harris, Kasser, & Martin, 1986). Overfeeding is possible because rats do not vomit. This increases the risk of bloating which may be lethal, but can be avoided by providing the rats with small pieces of paper towel in the cage. The paper provides both fiber and oral stimulation in rats that are not consuming any food. Spacing the meals at 8 hour intervals also reduces the risk of bloating by allowing as much time as possible for the stomach to empty between meals and requires that at least one meal is given during the dark period when animals would normally eat. Because the food is delivered directly into the stomach the tube-fed rats do not experience the normal orosensory properties of the diet. In the parabiosis studies food was always available in the cage, but the overfed rats chose not to eat it. They may, however, have received some sensory stimulation from the smell of the food. Tube-feeding also limits cephalic phase responses to food, but tube-fed rats show an anticipatory response to meals because their energy expenditure increases approximately 30 minutes before each meal [Figure 6A in (Harris, Kelso, Flatt, Bartness, & Grill, 2006)].
Figure 1.
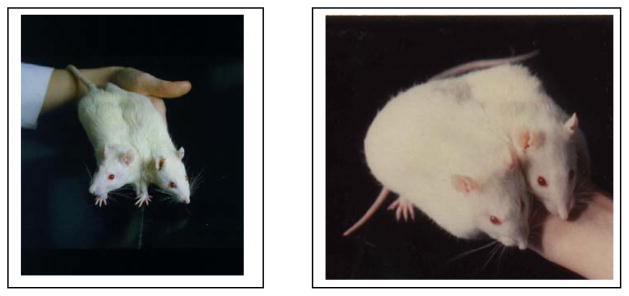
A parabiosed pair of female Sprague Dawley rats in which both animals ate ad libitum (left) and a pair in which one parabiont was tube-fed twice its daily food intake for 50 days (right).
In the parabiosis studies the ad libitum-fed partner of the overfed rat had free access to the tube-fed diet, but it was offered as a dry powder in a dish on the cage floor. Because the overfed rat did not eat any food voluntarily, which was confirmed by monitoring the color of feces when chromium oxide was added to the dry diet, it was possible to measure the daily food intake of the ad libitum partners throughout each experiment. The overfed rats did not appear to physically inhibit food intake of its partner, evidenced by the intake of the ad libitum partner being very similar to that of members of control pairs and showing very little daily variation, which would have been expected if the ad libitum animal was eating on an opportunistic basis. The controls for the experiments were members of parabiotic pairs in which both animals ate the dry diet ad libitum. Members of these control pairs had significantly less body fat than a single, non-parabiosed animal (Harris & Martin, 1984). Hervey had previously hypothesized that this was because each animal was able to monitor the total amount of fat present in the pair and regulated fat mass at a level that would be appropriate for one animal (Hervey, 1959).
The initial experiments with overfed obese rats established that the ad libitum fed partners lost a significant amount of their body fat in response to the development of obesity in the tube-fed partner (Harris & Martin, 1984). These changes in body composition of each member of the pair were fully reversible once overfeeding stopped (Harris & Martin, 1990). The partners of overfed rats were not identical to parabiotic partners of VMH-lesioned obese rats because the food intake of partners of overfed rats was not different from members of control pairs (Harris & Martin, 1984). Hervey had reported that the gut content of partners of VMH lesioned rats was reduced by approximately 25% compared with their controls (Hervey, 1959), a similar decrease was indicated in the Zucker parabiosis study (Harris, Hervey, Hervey, & Tobin, 1987) and Parameswaran et al (Parameswaran, Steffens, Hervey, & de Ruiter, 1977) found a gradual decline in the voluntary food intake of partners of rats that became obese due to electrical stimulation of the LH. Secondly, despite a substantial loss of body fat, the partners of tube-fed obese rats maintained a normal lean body mass compared with their controls (Harris & Martin, 1984). Again, this was in contrast to the partners of VMH lesioned (Hervey, 1959), Zucker (Harris, Hervey, Hervey, & Tobin, 1987) and LH stimulated rats (Parameswaran, Steffens, Hervey, & de Ruiter, 1977), all of which lost lean body mass in addition to body fat (see Table 1). This loss of lean tissue may be explained by the reduced food intake and suggests either that there is a potent inhibitor of feeding in the circulation of hypothalamic and genetically obese animals that is not present in overfed rats, or that these obese animals interfere with the normal feeding of their partners. This second possibility may be supported by observations by Nishizawa and Bray (1980) who reported a significant reduction in food intake of partners of rats that were overfed through gastric fistulas. The intubated rats received 160% of their voluntary intake through the fistula and the voluntary chow consumption of the non-intubated partner fell by up to 30%. There are no details of the feeding schedule of the intubated rats, but if all of the food was given in a single meal, then this could cause malaise in the overfed animal and if the chow was not available on floor of the cage, then the overfed animal could have inhibited the chow consumption of its partner. In the same paper Nishizawa and Bray (1980) confirmed the results from Hervey’s (1959) study using VMH lesioned rats. In this experiment they found that the food intake of the lesioned rat increased by about 50% and all of this increase was attributable to daytime feeding, suggesting that the normal feeding pattern of the rats had been substantially disrupted. The weight of the retroperitoneal pad of the partners of intubated rats and of VMH lesioned rats was reduced by about 50% within 9 or 10 days, which is much faster than fat was lost in the partner of tube-fed rats in our studies with tube-feeding. No measures of lean tissue mass were made.
Table 1.
Body composition and serum insulin in members of leptin-injected or vehicle-injected pairs of ob/ob mice.
| Leptin-injected Parabiont | Partner of leptin-injected | Member of vehicle injected pair | |
|---|---|---|---|
| Preoperative wt (g) | 43 ± 2 | 42 ± 2 | 42 ± 1 |
| Carcass wt (g) | 33 ± 2A | 37 ± 1B | 40 ± 1C |
| Carcass fat (g) | 16.0 ± 1.3A | 19.0 ± 0.9B | 23.3 ± 0.7C |
| Calculated food intake (mg/24 hr) | 443 ± 299A | 882 ± 260A | 1101 ± 162B |
| Serum insulin (ng/ml) | 1.1 ± 0.3A | 1.6 ± 0.3A | 14.0 ± 1.7B |
Data are means ± sem for mice from 5 pairs of leptin-injected ob/ob mice or 4 pairs of vehicle injected ob/ob mice. Values for a specific parameter that do not share a common superscript are significantly different at P<0.05. Mice received twice daily i.p. injections of 50 ug human recombinant leptin or vehicle for 11 days (Harris, Zhou, Weigle, & Kuijper, 1997)
A time course study with tube-fed overfed parabionts determined that loss of fat in their lean partners did not occur until 23 to 39 days after initiation of overfeeding (Harris & Martin, 1986). This implies that the factor that inhibited accumulation of body fat was not present at significant concentrations in the partners of overfed rats until body fat in the overfed rats was 10-fold greater than in the ad libitum partner (Harris & Martin, 1986). The failure of the circulating factor to reach bioactive concentrations could be due to it either having a short half-life so that there was a large concentration gradient between partners, or that it had to reach substantial levels to inhibit body fat accumulation. In studies that were completed after leptin had been identified we found that tube-feeding alone caused a rapid three-fold increase in adipose tissue leptin mRNA expression. Overfeeding increased leptin expression further and by 31 days it was about five-fold higher in overfed than control tube-fed rats (Harris, Ramsay, Smith, & Bruch, 1996). Serum leptin was increased 10-fold after 30 days of overfeeding (24 + 2 vs. 2 + 0.2 ng/ml: unpublished data) in 200%-fed single rats compared with their ad libitum fed controls. These observations are consistent with the possibility that it takes 23 to 39 days of overfeeding for leptin concentrations to increase by appreciable amounts in the partners of overfed, obese rats. In an effort to identify, or exclude, known hormones as potential “satiety” factors we found no reliable changes in thyroid hormones, insulin, corticosterone, growth hormone, free fatty acids or ketone bodies of lean partners of overfed rats compared with members of control pairs (Harris & Martin, 1984). Hypoinsulinemia in partners of overfed rats (Harris & Martin, 1990) also argued against basal insulin acting as a feedback signal of adiposity (Woods, et al., 1985).
In the parabiosis studies with overfed rats whole body glucose tolerance tests indicated that partners of obese rats were hypoinsulinemic, but glucose clearance was normal (Harris & Martin, 1990). The overfed rat was hyperglycemic and hyperinsulinemic, but glucose clearance was faster than in members of ad libitum-fed pairs (Harris & Martin, 1990). This may have been due to tube-feeding inducing metabolic pathways that would facilitate glucose clearance after each of the three large meals that were fed each day. Measurement of hepatic and adipose tissue glucose and fatty acid metabolism revealed that the loss of fat in partners of overfed rats was preceded by a drop in adipose tissue lipogenesis and hepatic fatty acid esterification. There was no increase in lipolysis, which decreased once fat depots were lipid deplete (Harris & Martin, 1986). There also was a significant decrease in glucose metabolism through the pentose phosphate pathway in both liver and adipose tissue (Harris & Martin, 1990).
The pentose phosphate pathway normally provides reducing equivalents for de novo fatty acid synthesis. When lipogenesis was measured on isolated adipocytes and liver tissue slices we found an inhibition of lipogenesis only in adipose tissue of the partners of obese rats (Harris & Martin, 1986). In vivo measures of de novo lipid synthesis, however, indicated a significant inhibition of hepatic fatty acid synthesis and esterification in addition to the inhibition in adipose tissue (Harris, Martin, & Bruch, 1995). The difference between in vivo and in vitro measures may be explained either by the requirement of acute exposure of liver tissue to endogenous neural, endocrine and/or paracrine factors, or to partial loss of viability of the liver slices used for the in vitro measures (Ross, 1979). We did not determine whether the decline in activity of the pentose phosphate shunt inhibited lipogenesis or whether it was secondary to a limited requirement for reducing equivalents.
The change in metabolite utilization by the tissues was accompanied by a decrease in glucose 6 phosphate dehydrogenase, malic enzyme and fatty acid synthetase in both the liver and adipose tissue of ad libitum fed partners of overfed rats (Harris & Martin, 1990). Similar changes in glucose and fatty acid utilization were found in single rats that had been food restricted to 60% voluntary intake for 21 days and then 50% of voluntary intake for 10 days (Harris, Martin, & Bruch, 1995). Thus the parabiosed partners of obese overfed rats represent a model in which food intake is almost normal (324 ± 17 vs 331 ± 13 g/29 days in control pairs), but body fat is substantially reduced (3.2 ± 0.3 vs 11.8 ± 1.0 g in control pairs) and tissue metabolism is identical to that in rats that are in a state of severe negative energy balance. These results correlate with studies in which chronic peripheral infusion of a physiological dose of leptin from a miniosmotic pump reduces body fat, but causes no significant change in food intake or lean body mass (Harris, Bowen, & Mitchell, 2003). By contrast, central infusions of leptin cause a substantial, but transient inhibition of food intake and loss of all visible fat (Halaas, et al., 1997).
One of my collaborators at University of Georgia was Thomas Kasser who was interested in how glucose and fatty acid metabolism in the hypothalamus changed in response to shifts in whole animal energy balance. He previously had shown that glucose flux through the pentose phosphate pathway was increased in the VMH, but that flux through the gamma aminobutyric acid shunt and fatty acid oxidation decreased in the lateral hypothalamus (LH) of rats made obese by overfeeding (Kasser, Harris, & Martin, 1985b). The level of LH fatty acid oxidation declined and VMH glucose oxidation rose with increasing levels of body fat and this pattern of change paralleled that in liver metabolism (Kasser, Harris, & Martin, 1985a). We subsequently found that LH fatty acid oxidation was decreased not only in overfed rats, but also in their ad lib fed parabiotic partners (Kasser, Harris, & Martin, 1989). Similarly, glucose flux through the pentose phosphate pathway in the VMH was increased in both the overfed parabiont and its ad libitum fed partner, indicative of increased production of energy intermediates for fatty acid synthesis. The changes occurred in both members of a pair despite large difference in body fat content (40% fat in the overfed partner, 4.8% fat in the ad lib partner) (Kasser, Harris, & Martin, 1989). These data were interpreted as evidence that the putative circulating satiety factor might regulate energy balance by acting directly on hypothalamic metabolic pathways. It is important to note that although we found changes in fatty acid and glucose utilization in the VMH and LH of overfed and parabiotic rats this was associated with a significant loss of body fat, but a minimal change in food intake of the ad libitum fed partner.
As noted above, the difference in food intake of partners of obese rats and their controls was not statistically significant, even when considered over a period of 60 days (Harris & Martin, 1984). Calculations indicated that a decline in intake of only 1 g/day over the course of a study would be enough energy to account for the difference in fat content of partners of obese rats and their controls (Harris & Martin, 1984). In a typical experiment the members of control pairs were eating 11.0 ± 0.3 g/day, whereas partners of obese rats ate 10.2 ± 0.3 g. Therefore, we tested the importance of the contribution of energy intake to loss of fat in the partners of obese rats by tube-feeding them their normal daily food intake. Controls were members of pairs in which both rats were tube-fed 100% of their ad libitum food intake. In this study the difference in daily food intake of ad libitum partners of obese rats and 100%-fed partners of obese rats was only 0.3 g/d, but this was sufficient to prevent loss of body fat in the 100% tube-fed animals (Harris & Martin, 1990). We did not determine whether the very small decrease in food intake of ad libitum partners of obese rats was driving the inhibition of lipogenesis or whether the fall in intake was secondary to a change in metabolism.
Because the parabiosis studies described above indicated that overfed obese rats produced a circulating factor that specifically inhibited adipocyte lipogenesis (Harris & Martin, 1986) it was possible to establish an in vitro bioassay that could be used to fractionate serum from obese animals in an attempt to isolate the factor. In this assay adipocytes isolated from the fat depots of young ad libitum fed Sprague Dawley rats were incubated in the presence of 2% by volume serum from control or overfed rats. De novo lipogenesis measured by glucose incorporation into triglyceride fatty acids showed a significant inhibition of lipogenesis in cells exposed to serum from overfed rats compared with those exposed to serum from ad libitum fed animals (Harris, Bruch, & Martin, 1989). We used the assay to determine that the inhibitory factor (given the acronym ALF for anti-lipogenic factor) was a glycosylated protein, molecular weight in the range of 30 – 100 kD that was released from a site other than the pituitary gland. It was not long after this that Friedman’s group at Rockefeller identified leptin as the protein that was mutated in ob/ob mice (Zhang, et al., 1994) and thus it appeared that the search for the parabiotic “satiety” signal was over. As noted below, this assumption may have been premature.
Leptin in Parabiosied Mice
The initiation of the search for the ob protein (leptin) was based on the outcome of parabiosis experiments between genetically obese diabetic (db/db) and obese (ob/ob) mice. ob/ob mice lost weight when parabiosed to a wild type or db/db animal (Coleman, 1973), which was in direct contrast to genetically obese db/db mice and Zucker rats which retained their hyperphagia and obesity when they were incorporated into a parabiotic pair (Coleman & Hummel, 1969) (Harris, Hervey, Hervey, & Tobin, 1987). Thus, it was concluded that the majority of obese models could produce the hypothesized circulating “satiety” factor, but were unable to respond effectively to the factor. By contrast, ob/ob mice were unable to produce the circulating factor, but were responsive to its’ presence in the blood received from wild type or db/db partners (Coleman, 2010).
The early parabiosis experiments reported by Coleman and Hummel (Coleman, 1973; Coleman & Hummel, 1969) were designed to investigate the causes of diabetes in the mice and focused on pancreatic histology and blood glucose and insulin concentrations. Although it was impossible to measure food intake of individual members of a pair, Coleman reported that partners of db/db mice appeared to eat little and there was little or no food in their gastrointestinal tract at the end of the study. In addition it was noted that adipose tissue was vastly decreased in ob/ob and wild type partners of db/db mice compared with their controls (Coleman, 1973; Coleman & Hummel, 1969).
Once recombinant leptin became available I completed three parabiosis studies that essentially replicated those of Coleman, but included measures of body composition and of leptin concentrations in the circulation of individual parabionts by western blot because quantitative leptin assays were not yet available. In the first experiment recombinant human leptin was injected into one member of a pair of ob/ob mice and demonstrated that leptin could cross the parabiotic union (Harris, Zhou, Weigle, & Kuijper, 1997). The half-life of leptin in the circulation of a single mouse was approximately 36 minutes and this was consistent with the observation that leptin did not reach equilibrium in pairs of ob/ob mice when one mouse received twice daily intraperitoneal injections of 50 ug leptin for 11 days. At the end of the study the weights of the carcass and carcass fat were reduced in members of leptin-treated pairs compared with their vehicle injected controls. The effect was greater in the injected mouse than its partner (see Table 1). Based on measures of gut content food intake in leptin treated mice and their partners had been inhibited by 75–85% and was equivalent to that in a single wild type mouse. Body temperature was increased in the leptin-injected mouse, but not its partner. Serum insulin was normalized in both members of the injected pairs (Harris, Zhou, Weigle, & Kuijper, 1997). Therefore, it appeared that some metabolic abnormalities in ob/ob mice were more responsive to leptin than others.
Additional studies compared the effects of parabiosis between ob/ob mice and wild type or db/db partners (Harris, 1997, 1999). The results of these studies confirmed and expanded on the observations that had been reported previously by Coleman and Hummel (Coleman, 1973; Coleman & Hummel, 1969). As expected, ob/ob partners of db/db mice lost 70% of their body fat (Harris, 1999). Measures of gut content of the mice at the end of the experiment showed that the food intake of the ob/ob partners was inhibited by 75%, whereas that of the db/db partner did not change. Carcass composition measured 18 days after surgery (7–9 days after blood exchange was established) showed no change in carcass protein of the ob/ob partner consistent with observations from parabiosis studies with overfed rats (Harris & Martin, 1984) and supporting the notion that the circulating factor exchanging between the mice acted to specifically inhibit fat accumulation. Serum leptin levels had risen to approximately 30% of those found in db/db mice which is substantially higher than would be found in a lean mouse. Serum glucose and insulin were normalized and corticosterone was significantly elevated. Surprisingly, there was a 30% increase in carcass protein and a 12% decrease in the carcass fat content of db/db partners of ob/ob mice (Harris, 1999). The implications of this observation are discussed in more detail below. When wild type mice were parabiosed to db/db mice carcass fat was decreased by 40%, gut content was decreased by 25% and carcass protein by 14% after 25 days (Harris, 1999). These results are similar to those found for parabiotic partners of Zucker rats, intubated rats and VMH obese rats.
When ob/ob mice were parabiosed to wild type mice the circulating levels of leptin reached those found in the wild type mice. These lower levels of leptin produced much less dramatic responses than were found in ob/ob partners of db/db mice (Harris, 1997). Fifty days after surgery carcass fat was decreased by 24%, gut content was reduced by approximately 30%, but remained higher than in wild type mice, body temperature did not change and insulin was reduced by 50%, but was significantly higher than that of wild type mice. The wild type partners lost approximately 30% of their body fat, but there was no change in carcass protein (Harris, 1997). Although this loss of fat was proportionally similar to the change in fat of ob/ob mice, it was represented by a much smaller weight of fat in the lean than the ob/ob animals (0.9 vs 5 g).
The results of the studies described above clearly demonstrated that the correction of different aspects of the phenotype of ob/ob mice by leptin was dose dependent and, despite subsequent evidence that ob/ob mice are hypersensitive to exogenous leptin (Harris, et al., 1998), it appeared that the amount of leptin that would normally be found in a lean animal did not fully reverse the hyperphagia, hypothermia or hyperinsulinemia of ob/ob mice (Harris, 1997). By contrast, the high doses of leptin in ob/ob mice that were partners of db/db mice (Harris, 1999) or that were injected with leptin (Harris, Zhou, Weigle, & Kuijper, 1997) corrected each of these abnormalities. Interestingly, all of the weight loss in ob/ob partners of db/db mice was fat, whereas lean partners of db/db mice lost both fat and lean tissue. It seems likely that the large stores of adipose tissue protect lean body mass in ob/ob mice, but this remains to be confirmed. The hypersensitivity to exogenous leptin (Harris, et al., 1998) and abnormal neuronal development (Ahima, Bjorbaek, Osei, & Flier, 1999) results in leptin deficient ob/ob mice functioning as a sensitive model for testing the potential effects of leptin on a physiologic system, but they do not necessarily predict leptin action in a normal animal.
Is Leptin the Parabiotic “Satiety” Factor?
Table 2 summarizes the changes in food intake and body composition of parabiotic partners of obese rats and mice. Data from the ob/ob mouse studies provides the most convincing evidence for leptin as the parabiosis signal, because it is essentially a model for restoration of function. Leptin was not measured in any of the rat parabiosis studies, but it is now known that leptin is elevated in the obese animals (see Table 2). When all of the different parabiosis models are compared it is clear that the lean partners of obese animals showed substantial variability in their response. Loss of body fat is consistent across models, but changes in food intake and lean body mass are less consistent. This may explained by circulating factors other than leptin being produced by some obese animals and the possibility that some models of obese rats interfere with the food intake of their partners. Alternately it may result from differences in the concentration of leptin that reaches the non-obese partner, assuming that higher concentrations are required to inhibit food intake than to reduce body fat mass. Using values taken from the literature, circulating concentrations of leptin are similar in the different models of obesity (see Table 2), but leptin concentration in the non-obese partner could vary if there were differences in efficiency of blood exchange in different experiments or if there were genotypic differences in the ability of the parabionts to clear leptin from the circulation.
Table 2.
Comparision of responses in parabiotic partners of obese rats and mice.
| Model of obesity | Representative leptin values for obese animal** (ng/ml) | Body fat | Lean body mass | Food intake/gut content* |
|---|---|---|---|---|
| VMH lesioned rat | 301 | Decreased | Decreased | Decreased 10–20% |
| LH stimulated rat | Decreased | Decreased | Decreased 70% | |
| Intubated overfed rat | Decreased | Not measured | Decreased 15–30% | |
| Tubefed overfed rat | 242 | Decreased | Unchanged | Unchanged |
| Male Zucker rat | 503 | Decreased | Decreased | Unchanged |
| Female Zucker rat | 423 | Decreased | Decreased | Decreased 21% |
| ob/ob partner of db/db mouse | 254 | Decreased | Unchanged | Decreased 75% |
| Wild type partner of db/db mouse | 254 | Decreased | Decreased | Unchanged |
| Leptin injected ob/ob mouse | Decreased | Unchanged | Decreased 75% |
The change in food intake is the only parameter that is quantified because changes in body composition are influenced by the duration of the study.
Gut content shows a high correlation with the amount of food consumed during the previous 24 hours (Harris, Zhou, Weigle, & Kuijper, 1997).
Leptin values are taken from published data on single animals of the same genotype as the obese parabiont.
Harris (unpublished data),
Site of Leptin Action
Based on early lesioning studies the site of action of the parabiotic satiety signal was hypothesized to be the hypothalamus (Hervey, 1959). Consistent with this, the arcuate nucleus of the hypothalamus expresses relatively high concentrations of the leptin receptor and leptin down-regulates expression of orexigenic neuropeptides (Schwartz, Seeley, Campfield, Burn, & Baskin, 1996), but increases expression of anorexic neuropeptides (Schwartz, et al., 1997). Until recently a majority of investigation of leptin activity in the brain focused on the arcuate nucleus as the primary site at which food intake and energy balance were controlled (Baskin, Blevins, & Schwartz, 2001). Leptin receptors are expressed on arcuate neurons that project to the paraventricular nucleus of the hypothalamus, an area that integrates information from multiple sources and contributes to the control of food intake, energy expenditure, reproductive function, thermoregulation and stress responses. Restoration of leptin receptors to all neural tissues (central and peripheral) of db/db mice corrected food intake, obesity and diabetes in male mice and normalized food intake and diabetes in females, but did not change their percent body fat (Kowalski, Liu, Leibel, & Chua, 2001). Contrary to expectations, a study in which leptin receptors were selectively replaced in the arcuate nucleus of the hypothalamus of db/db mice showed that the receptors corrected abnormalities in glucose metabolism, but had relatively small effects on obesity; reducing food intake and body fat mass of the db/db mice by only about 10% (Coppari, et al., 2005). In a further study (van de Wall, et al., 2008) leptin receptors were deleted from AgRP and POMC-expressing neurons, which should prevent leptin action in the arcuate nucleus. These animals showed a transient increase in food intake, a doubling of body fat and maintained a normal insulinemia; a phenotype that is much less severe than that of ob/ob or db/db mice. It is now well established that leptin receptors are expressed in many areas of the brain (Leshan, et al., 2010; Myers, Munzberg, Leinninger, & Leshan, 2009) (Scott, et al., 2009) including those that have the potential to modify food intake or energy balance secondary to changes in food preference. For example, the ventral tegmental area (VTA) expresses leptin receptors (Figlewicz, Evans, Murphy, Hoen, & Baskin, 2003) and mediates reward aspects of stimuli including food. Therefore, although it is clear that leptin plays a critical role in mediating the metabolic responses of parabiotic partners of obese mice, it is unlikely that it is simply due to leptin acting in the hypothalamus.
Is there a second circulating factor?
As mentioned above, we were surprised to find that body fat was decreased in db/db and wild type parabiotic partners of ob/ob mice. If leptin is the circulating “satiety” signal identified by parabiosis studies, then reduced body fat in these animals cannot be explained by the possibility that they were sensing the total fat mass of the pair (Hervey, 1959), because ob/ob mice do not produce leptin and db/db mice do not respond to it. This led to the hypothesis that the decrease in body fat mass of leptin treated animals may not be due to a direct effect of leptin, but that leptin induces release of a second circulating factor that inhibits adipose tissue growth. We propose that activation of leptin receptors in parabiosed ob/ob mice resulted in release of this factor which was then circulated to their wild type or db/db partners where it acted to reduce body fat mass. The loss of fat in db/db partners of ob/ob mice implies that activation of leptin receptors in the ob/ob partner is required for the factor to be released, but that leptin receptors are not required for the factor to reduce the size of body fat stores.
We have two pieces of evidence to support this hypothesis. The first is that physiological concentrations of leptin have no effect on preadipocyte proliferation or differentiation in a primary culture system. By contrast, if serum from rats that have been infused with low doses for leptin is included in the culture media, then cell proliferation is inhibited (Wagoner, Hausman, & Harris, 2006). Secondly, if pieces of wild type fat are transplanted subcutaneously into db/db mice, then the transplant increases in size, consistent with early reports that fat transplants between lean and obese mice take on the characteristics of their environment (Meade, Ashwell, & Sowter, 1979). These results argue against leptin having any direct effect on adipose tissue because db/db mice have high circulating concentrations of leptin and wild type fat expresses functional leptin receptors. We have used a combination of fat transplant and parabiosis between ob/ob and db/db mice to demonstrate that leptin induces release of a circulating factor in ob/ob mice that inhibits enlargement of wild type fat transplants in db/db mice. Wild type fat that is transplanted into db/db partners of db/db mice expands, whereas wild type fat transplants in db/db partners of ob/ob mice are reduced in size (unpublished data). Further work is needed to confirm the presence and identity of this factor. In addition it would be interesting to know whether this factor is the same as ALF, which we were trying to isolate from serum of overfed rats before leptin was discovered (Harris, Bruch, & Martin, 1989).
Soon after leptin was identified several animal studies were published to show that leptin treatment corrected hyperphagia, obesity, hyperglycemia, hyperinsulinemia and hypoactivity in ob/ob mice. Leptin also increased energy expenditure, reduced food intake, body weight and body fat in wild type mice (Campfield, Smith, Guisez, Devos, & Burn, 1995; Halaas, et al., 1995; Pelleymounter, et al., 1995). By contrast db/db mice, which have a mutation of the leptin receptor (Tartaglia, et al., 1995), were unresponsive to leptin administration (Campfield, Smith, Guisez, Devos, & Burn, 1995). The loss of fat in leptin-treated animals is associated with inhibition of adipose tissue lipogenesis (Buettner, et al., 2008) and high doses of leptin also stimulate lipolysis (Fruhbeck, Aguado, Gomez-Ambrosi, & Martinez, 1998) and increase fatty acid oxidation (Wang, Lee, & Unger, 1999). These observations are consistent with the changes in metabolism observed in lean partners of obese rats (Harris, 1999; Harris & Martin, 1984, 1986), but they do not exclude the possibility that leptin is inducing the release of a second, anti-lipogenic factor because Buettner et al (Buettner, et al., 2008) report that leptin acts centrally to inhibit adipose tissue lipogenesis, whereas our bioassay experiments clearly show that serum from obese rats inhibits adipocytes lipogenesis directly (Harris, Bruch, & Martin, 1989) and the cell culture studies demonstrate that serum from leptin-treated rats inhibits adipose tissue cell proliferation directly (Wagoner, Hausman, & Harris, 2006). Further work is needed to compare the direct effects of leptin versus serum from leptin-treated animals on adipose tissue metabolism.
Summary
A parabiosis study with rats made obese by hypothalamic lesions suggested the presence of a circulating factor that functioned as a feedback signal in the regulation of energy balance (Hervey, 1959). This hypothesis was supported by subsequent observations of a suppression of food intake and loss of body fat in parabiotic partners of genetically obese Zucker rats (Harris, Hervey, Hervey, & Tobin, 1987) and rats overeating due to electrical stimulation of the LH (Parameswaran, Steffens, Hervey, & de Ruiter, 1977). A series of studies with rats made obese by overfeeding further defined the metabolic phenotype of partners of obese rats and excluded several hormones and metabolites as potential circulating satiety signals (Harris & Martin, 1984) (Harris & Martin, 1986). The overfed rats appeared to produce a factor that selectively inhibited adipose tissue lipogenesis, but did not have a significant effect on food intake. Observations that obese ob/ob mice did not produce the hypothesized satiety signal led Freidman’s group to identify leptin as the protein that was missing in these mice (Zhang, et al., 1994). Subsequent studies indicate that leptin fulfills many of the functions ascribed to the parabiotic factor. A more detailed investigation of the energy balance status of ob/ob mice and their parabiotic partners (Harris, 1999) also raises the possibility that leptin induces release of one or more circulating factors that inhibit growth of adipose tissue, but promote accretion of lean tissue.
Highlights.
Early parabiosis studies show that obese rodents produce a circulating satiety factor
Studies with overfed parabiotic rats characterized metabolic responses to the factor
Leptin fulfills many of the functions ascribed to the parabiotic factor
It also is possible that leptin-inducible proteins influence lean and fat mass
Footnotes
This is the third paper in the Part-Issue entitled Harvey, Harris and the Parabiotic Search for Lipostatic Signals; Guest Editors: Gerard P. Smith and David A. Booth.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahima RS, Bjorbaek C, Osei S, Flier JS. Regulation of neuronal and glial proteins by leptin: implications for brain development. Endocrinology. 1999;140:2755–2762. doi: 10.1210/endo.140.6.6774. [DOI] [PubMed] [Google Scholar]
- Armitage G, Harris RB, Hervey GR, Tobin G. The relationship between energy expenditure and environmental temperature in congenitally obese and non-obese Zucker rats. J Physiol. 1984;350:197–207. doi: 10.1113/jphysiol.1984.sp015196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage G, Hervey GR, Tobin G. Energy expenditure of rats tube-fed at different energy levels [proceedings] J Physiol. 1979;290:17P–18P. [PMC free article] [PubMed] [Google Scholar]
- Baskin DG, Blevins JE, Schwartz MW. How the brain regulates food intake and body weight: the role of leptin. J Pediatr Endocrinol Metab. 2001;14(Suppl 6):1417–1429. [PubMed] [Google Scholar]
- Becker EE, Grinker JA. Meal patterns in the genetically obese Zucker rat. Physiol Behav. 1977;18:685–692. doi: 10.1016/0031-9384(77)90067-1. [DOI] [PubMed] [Google Scholar]
- Binhammer RT, Epstein S, Whitehouse A. Development of parabiosis intoxication in rat parabionts. Anat Rec. 1963;145:503–511. doi: 10.1002/ar.1091450403. [DOI] [PubMed] [Google Scholar]
- Bogacka I, Roane DS, Xi X, Zhou J, Li B, Ryan DH, Martin RJ. Expression levels of genes likely involved in glucose-sensing in the obese Zucker rat brain. Nutr Neurosci. 2004;7:67–74. doi: 10.1080/10284150410001710401. [DOI] [PubMed] [Google Scholar]
- Bray GA, York DA. Studies on food intake of genetically obese rats. Am J Physiol. 1972;223:176–179. doi: 10.1152/ajplegacy.1972.223.1.176. [DOI] [PubMed] [Google Scholar]
- Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, Su K, Cheng B, Li X, Harvey-White J, Schwartz GJ, Kunos G, Rossetti L. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med. 2008;14:667–675. doi: 10.1038/nm1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- Chua SC, Jr, White DW, Wu-Peng XS, Liu SM, Okada N, Kershaw EE, Chung WK, Power-Kehoe L, Chua M, Tartaglia LA, Leibel RL. Phenotype of fatty due to Gln269Pro mutation in the leptin receptor (Lepr) Diabetes. 1996;45:1141–1143. doi: 10.2337/diab.45.8.1141. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- Coleman DL. A historical perspective on leptin. Nat Med. 2010;16:1097–1099. doi: 10.1038/nm1010-1097. [DOI] [PubMed] [Google Scholar]
- Coleman DL, Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol. 1969;217:1298–1304. doi: 10.1152/ajplegacy.1969.217.5.1298. [DOI] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Jr, Lowell BB, Elmquist JK. The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Cruce JA, Greenwood MR, Johnson PR, Quartermain D. Genetic versus hypothalamic obesity: studies of intake and dietary manipulations in rats. J Comp Physiol Psychol. 1974;87:295–301. doi: 10.1037/h0036862. [DOI] [PubMed] [Google Scholar]
- Dube MG, Xu B, Kalra PS, Sninsky CA, Kalra SP. Disruption in neuropeptide Y and leptin signaling in obese ventromedial hypothalamic-lesioned rats. Brain Res. 1999;816:38–46. doi: 10.1016/s0006-8993(98)00985-8. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003;964:107–115. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- Finerty JC, Panos TC. Parabiosis intoxication. Proc Soc Exp Biol Med. 1951;76:833–835. doi: 10.3181/00379727-76-18647. [DOI] [PubMed] [Google Scholar]
- Fruhbeck G, Aguado M, Gomez-Ambrosi J, Martinez JA. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem Biophys Res Commun. 1998;250:99–102. doi: 10.1006/bbrc.1998.9277. [DOI] [PubMed] [Google Scholar]
- Gibney B, Chamoto K, Lee GS, Simpson DC, Miele L, Tsuda A, Konerding MA, Wagers A, Mentzer SJ. Cross-circulation and cell distribution kinetics in parabiotic mice. J Cell Physiol. 2012;227:821–828. doi: 10.1002/jcp.22796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbole V, York DA, Bloxham DP. Developmental changes in the fatty (fafa) rat: evidence for defective thermogenesis preceding the hyperlipogenesis and hyperinsulinaemia. Diabetologia. 1978;15:41–44. doi: 10.1007/BF01219327. [DOI] [PubMed] [Google Scholar]
- Greenwood MR, Quartermain D, Johnson PR, Cruce JA, Hirsch J. Food motivated behavior in genetically obese and hypothalamic-hyperphagic rats and mice. Physiol Behav. 1974;13:687–692. doi: 10.1016/0031-9384(74)90241-8. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Harris RB. Loss of body fat in lean parabiotic partners of ob/ob mice. Am J Physiol. 1997;272:R1809–1815. doi: 10.1152/ajpregu.1997.272.6.R1809. [DOI] [PubMed] [Google Scholar]
- Harris RB. Parabiosis between db/db and ob/ob or db/+ mice. Endocrinology. 1999;140:138–145. doi: 10.1210/endo.140.1.6449. [DOI] [PubMed] [Google Scholar]
- Harris RB, Bowen HM, Mitchell TD. Leptin resistance in mice is determined by gender and duration of exposure to high-fat diet. Physiol Behav. 2003;78:543–555. doi: 10.1016/s0031-9384(03)00035-0. [DOI] [PubMed] [Google Scholar]
- Harris RB, Bruch RC, Martin RJ. In vitro evidence for an inhibitor of lipogenesis in serum from overfed obese rats. Am J Physiol. 1989;257:R326–336. doi: 10.1152/ajpregu.1989.257.2.R326. [DOI] [PubMed] [Google Scholar]
- Harris RB, Hervey E, Hervey GR, Tobin G. Body composition of lean and obese Zucker rats in parabiosis. Intl J Obesity. 1987;11:275–283. [PubMed] [Google Scholar]
- Harris RB, Hervey GR, Tobin G. Food intake in relation to dietary energy and nitrogen concentrations in ‘lean’ and ‘fatty’ Zucker rats. Proc Nutr Soc. 1979;38:126A. [PubMed] [Google Scholar]
- Harris RB, Kasser TR, Martin RJ. Dynamics of recovery of body composition after overfeeding, food restriction or starvation of mature female rats. J Nutr. 1986;116:2536–2546. doi: 10.1093/jn/116.12.2536. [DOI] [PubMed] [Google Scholar]
- Harris RB, Kelso EW, Flatt WP, Bartness TJ, Grill HJ. Energy expenditure and body composition of chronically maintained decerebrate rats in the fed and fasted condition. Endocrinology. 2006;147:1365–1376. doi: 10.1210/en.2005-1156. [DOI] [PubMed] [Google Scholar]
- Harris RB, Martin RJ. Specific depletion of body fat in parabiotic partners of tube-fed obese rats. Am J Physiol. 1984;247:R380–386. doi: 10.1152/ajpregu.1984.247.2.R380. [DOI] [PubMed] [Google Scholar]
- Harris RB, Martin RJ. Metabolic response to a specific lipid-depleting factor in parabiotic rats. Am J Physiol. 1986;250:R276–286. doi: 10.1152/ajpregu.1986.250.2.R276. [DOI] [PubMed] [Google Scholar]
- Harris RB, Martin RJ. Site of action of putative lipostatic factor: food intake and peripheral pentose shunt activity. Am J Physiol. 1990;259:R45–52. doi: 10.1152/ajpregu.1990.259.1.R45. [DOI] [PubMed] [Google Scholar]
- Harris RB, Martin RJ, Bruch RC. Dissociation between food intake, diet composition, and metabolism in parabiotic partners of obese rats. Am J Physiol. 1995;268:R874–883. doi: 10.1152/ajpregu.1995.268.4.R874. [DOI] [PubMed] [Google Scholar]
- Harris RB, Mitchell TD, Yan X, Simpson JS, Redmann SM., Jr Metabolic responses to leptin in obese db/db mice are strain dependent. Am J Physiol Regul Integr Comp Physiol. 2001;281:R115–132. doi: 10.1152/ajpregu.2001.281.1.R115. [DOI] [PubMed] [Google Scholar]
- Harris RB, Ramsay TG, Smith SR, Bruch RC. Early and late stimulation of ob mRNA expression in meal-fed and overfed rats. J Clin Invest. 1996;97:2020–2026. doi: 10.1172/JCI118637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RB, Zhou J, Redmann SM, Jr, Smagin GN, Smith SR, Rodgers E, Zachwieja JJ. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139:8–19. doi: 10.1210/endo.139.1.5675. [DOI] [PubMed] [Google Scholar]
- Harris RB, Zhou J, Weigle DS, Kuijper JL. Recombinant leptin exchanges between parabiosed mice but does not reach equilibrium. Am J Physiol. 1997;272:R1800–1808. doi: 10.1152/ajpregu.1997.272.6.R1800. [DOI] [PubMed] [Google Scholar]
- Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J Physiol, London. 1959;145:336–352. doi: 10.1113/jphysiol.1959.sp006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff RL, Trautman R, Van Dyke DC. Nature of exchanges in parabiotic rats. Am J Physiol. 1950;161:56–74. doi: 10.1152/ajplegacy.1950.161.1.56. [DOI] [PubMed] [Google Scholar]
- Kasser TR, Harris RB, Martin RJ. Level of satiety: fatty acid and glucose metabolism in three brain sites associated with feeding. Am J Physiol. 1985a;248:R447–452. doi: 10.1152/ajpregu.1985.248.4.R447. [DOI] [PubMed] [Google Scholar]
- Kasser TR, Harris RB, Martin RJ. Level of satiety: GABA and pentose shunt activities in three brain sites associated with feeding. Am J Physiol. 1985b;248:R453–458. doi: 10.1152/ajpregu.1985.248.4.R453. [DOI] [PubMed] [Google Scholar]
- Kasser TR, Harris RB, Martin RJ. Site of action of putative lipostatic factor: hypothalamic metabolism of parabiotic rats. Am J Physiol. 1989;257:R224–228. doi: 10.1152/ajpregu.1989.257.1.R224. [DOI] [PubMed] [Google Scholar]
- Kowalski TJ, Liu SM, Leibel RL, Chua SC., Jr Transgenic complementation of leptin-receptor deficiency. I. Rescue of the obesity/diabetes phenotype of LEPR-null mice expressing a LEPR-B transgene. Diabetes. 2001;50:425–435. doi: 10.2337/diabetes.50.2.425. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Opland DM, Louis GW, Leinninger GM, Patterson CM, Rhodes CJ, Munzberg H, Myers MG., Jr Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci. 2010;30:5713–5723. doi: 10.1523/JNEUROSCI.1001-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade CJ, Ashwell M, Sowter C. Is genetically transmitted obesity due to an adipose tissue defect? Proc R Soc Lond B Biol Sci. 1979;205:395–410. doi: 10.1098/rspb.1979.0073. [DOI] [PubMed] [Google Scholar]
- Meyer RK, Biddulph C, Finerty JC. Pituitary-gonad interaction in immature female parabiotic rats. Endocrinology. 1946;39:23–31. doi: 10.1210/endo-39-1-23. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Munzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakic B, Nakic Z, Silobrcic V. Graft versus host reaction in parabiotic disease. Nature. 1960;186:322–323. doi: 10.1038/186322a0. [DOI] [PubMed] [Google Scholar]
- Nishizawa Y, Bray GA. Evidence for a circulating ergostatic factor: studies on parabiotic rats. Am J Physiol. 1980;239:R344–351. doi: 10.1152/ajpregu.1980.239.3.R344. [DOI] [PubMed] [Google Scholar]
- Parameswaran SV, Steffens AB, Hervey GR, de Ruiter L. Involvement of a humoral factor in regulation of body weight in parabiotic rats. Am J Physiol. 1977;232:R150–157. doi: 10.1152/ajpregu.1977.232.5.R150. [DOI] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Pullar JD, Webster AJ. Heat loss and energy retention during growth in congenitally obese and lean rats. Br J Nutr. 1974;31:377–392. doi: 10.1079/bjn19740046. [DOI] [PubMed] [Google Scholar]
- Radcliffe JD, Webster AJ. Regulation of food intake during growth in fatty and lean female Zucker rats given diets of different protein content. Br J Nutr. 1976;36:457–469. doi: 10.1079/bjn19760100. [DOI] [PubMed] [Google Scholar]
- Ross BD. Techniques for investigation of tissue metabolism. In: Kornberg H, Metcalfe JC, Northcote DE, editors. Techniques of Metabolic Research. New York: Elsevier-North Holland; 1979. pp. 1–22. [Google Scholar]
- Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, James WP. Thermoregulation and non-shivering thermogenesis in the genetically obese (ob/ob) mouse. Pflugers Arch. 1978;373:189–193. doi: 10.1007/BF00584859. [DOI] [PubMed] [Google Scholar]
- Truett GE, Bahary N, Friedman JM, Leibel RL. Rat obesity gene fatty (fa) maps to chromosome 5: evidence for homology with the mouse gene diabetes (db) Proc Natl Acad Sci U S A. 1991;88:7806–7809. doi: 10.1073/pnas.88.17.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, Elmquist J, Lowell BB, Barsh GS, de Luca C, Myers MG, Jr, Schwartz GJ, Chua SC., Jr Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke DC, Simpson ME, Li CH, Evans HM. Survival in the circulation of the growth and adrenocorticotrophic hormones as evidenced by parabiosis. Am J Physiol. 1950;163:297–309. doi: 10.1152/ajplegacy.1950.163.2.297. [DOI] [PubMed] [Google Scholar]
- Wagoner B, Hausman DB, Harris RB. Direct and indirect effects of leptin on preadipocyte proliferation and differentiation. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1557–1564. doi: 10.1152/ajpregu.00860.2005. [DOI] [PubMed] [Google Scholar]
- Wang MY, Lee Y, Unger RH. Novel form of lipolysis induced by leptin. J Biol Chem. 1999;274:17541–17544. doi: 10.1074/jbc.274.25.17541. [DOI] [PubMed] [Google Scholar]
- Woods SC, Porte D, Jr, Bobbioni E, Ionescu E, Sauter JF, Rohner-Jeanrenaud F, Jeanrenaud B. Insulin: its relationship to the central nervous system and to the control of food intake and body weight. Am J Clin Nutr. 1985;42:1063–1071. doi: 10.1093/ajcn/42.5.1063. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zucker LM. Some effects of caloric restriction and deprivation on the obese hyperlipemic rat. J Nutr. 1967;91:247–254. doi: 10.1093/jn/91.2.247. [DOI] [PubMed] [Google Scholar]
- Zucker LM, Zucker TF. Fatty, a new mutation in the rat. J Heredity. 1961;52:275–278. [Google Scholar]