

Abstract
SopB is a type 3 secreted effector with phosphatase activity that Salmonella employs to manipulate host cellular processes, allowing the bacteria to establish their intracellular niche. One important function of SopB is activation of the pro-survival kinase Akt/protein kinase B in the infected host cell. Here, we examine the mechanism of Akt activation by SopB during Salmonella infection. We show that SopB-mediated Akt activation is only partially sensitive to PI3-kinase inhibitors LY294002 and wortmannin in HeLa cells, suggesting that Class I PI3-kinases play only a minor role in this process. However, depletion of PI(3,4) P2/PI(3–5) P3 by expression of the phosphoinositide 3-phosphatase PTEN inhibits Akt activation during Salmonella invasion. Therefore, production of PI(3,4) P2/PI(3–5) P3 appears to be a necessary event for Akt activation by SopB and suggests that non-canonical kinases mediate production of these phosphoinositides during Salmonella infection. We report that Class II PI3-kinase beta isoform, IPMK and other kinases identified from a kinase screen all contribute to Akt activation during Salmonella infection. In addition, the kinases required for SopB-mediated activation of Akt vary depending on the type of infected host cell. Together, our data suggest that Salmonella has evolved to use a single effector, SopB, to manipulate a remarkably large repertoire of host kinases to activate Akt for the purpose of optimizing bacterial replication in its host.
Introduction
Salmonella enterica serovar Typhimurium ( S . Typhimurium ) is a major cause of food poisoning worldwide and infections can be lethal in young children or immunocompromised hosts [1,2]. These bacteria have the ability to invade host cells and grow intracellularly. To accomplish this, S . Typhimurium uses type 3 secretion systems (T3SS) to translocate effector proteins directly into host cells [3]. The T3SS effectors activate signal transduction pathways leading to actin rearrangements that drive internalization of the bacteria into membrane-bound compartments, known as Salmonella-containing vacuoles (SCVs), within which the bacteria can survive and replicate [4,5]. One T3SS effector, SopB, is known to contribute to S . Typhimurium invasion [6]. SopB also contributes to other important phenotypes associated with infection, including production of proinflammatory cytokines [7], controlling SCV maturation [8], intracellular positioning of the SCV [9], blocking apoptosis [10], intracellular bacterial replication [8] and induction of epithelial-mesenchymal transition [11].
SopB is an important modulator of signal transduction pathways in host cells during infection and, together with two other T3SS effectors –SopE and SopE2- SopB influences members of the small Rho GTPase family [6]. It was demonstrated that SopB activates RhoG via its guanine nucleotide exchange factor SGEF, leading to actin rearrangements that promote bacterial invasion [7]. In addition, SopB can activate the pro-survival kinase Akt, preventing apoptosis in infected cells [10,12]. SopB was found to be solely responsible for Akt activation and subsequent inhibition of caspase-3 activity, protecting infected epithelial cells from apoptosis [10]. Thus, SopB acts as a pro-survival factor, preventing apoptosis and allowing the generation of a replicative niche in infected cells. Yet, the mechanisms by which SopB promotes Akt activation are unclear.
SopB contains a catalytic domain with phosphoinositide phosphatase activity that is essential for all of its associated phenotypes [6,13,14]. It has broad substrate specificity in vitro, but in vivo substrates have been the subject of controversy and the mechanisms by which it acts are unclear. The main substrate of SopB during infection is PI(4,5) P2, leading to the generation of PI(5) P [15,16]. Paradoxically, despite the fact that in vitro SopB can dephosphorylate PI(3,4) P2 and PI(3–5) P3, Mallo et al. showed that SopB is actually required and responsible for the accumulation of these lipids at invasion ruffles [15]. In the canonical pathway of Akt activation, e.g. during growth factor stimulation, Class I PI3-kinase generates PI(3,4) P2 and PI(3–5) P3 in the plasma membrane, where Akt binds and then becomes phosphorylated on Thr308 and Ser473 by PDK1 and mTORC2 complex, respectively [17–19]. However, the equivalent PI-accumulation during Salmonella invasion was found to be resistant to the PI3-kinase inhibitor LY294002 [15], suggesting that in this instance PI(3,4) P2 and PI(3–5) P3 are generated by a Class I PI3-kinase independent mechanism. Indeed, siRNA-mediated depletion of Class I PI3-kinase regulatory subunits p85α and p85β had no inhibitory effect on SopB-mediated Akt activation [20]. Despite this, Steele-Mortimer and colleagues showed that PDK1 and mTORC2 are required for SopB-mediated Akt activation [20]. These authors concluded that SopB activates Akt by a mechanism independent of Class I PI3-kinase that nevertheless incorporates some elements of the canonical pathway. Still, the role of PI(3,4) P2 and PI(3–5) P3 in Akt activation by SopB has not been tested directly.
The mechanisms that regulate Akt activation during Salmonella infection remain unclear. Here, we show that localized PI(3,4) P2 and PI(3–5) P3 production at invasion ruffles is responsible for Akt activation. Furthermore, we show that SopB exploits several different host kinases, depending on the infected cell type, to activate Akt during invasion. Our studies provide important novel insight into bacterial modulation of host kinase activity that can lead to the establishment of an intracellular replicative niche.
Materials and Methods
Plasmids and bacterial strains
A plasmid encoding untagged SopB was previously described [21]. GFP-SopB was previously described [22] and provided by D. Zhou (Purdue University). pEGFP-C1 was from Clontech. PTEN-A4-YFP was described previously [23]. GFP-Akt was a gift from M. Molina (Universidad Complutense de Madrid). Plasmids expressing His-SopB and catalytically inactive His-SopB-C460S [13] were provided by B. Finlay (University of British Columbia). Wild type Salmonella enterica serovar Typhimurium ( S . Typhimurium ) SL1344 [24] and isogenic ΔsopB mutant [12] strain were used in this study.
Antibodies and Reagents
Rabbit polyclonal antibodies to Ser473-phosphorylated-Akt and pan-Akt were purchased from Cell Signaling. Vps34 antibody was described elsewhere [25] and provided by J. Backer (Albert Einstein College of Medicine). Rabbit polyclonal antibody to S . Typhimurium O antiserum Group B was from Difco. Murine monoclonal anti-GFP antibody was obtained from Invitrogen. Mouse anti-GAPDH antibody was from Milipore. Secondary antibodies used for immunofluorescence study and Phalloidin were Alexa-conjugated and purchased from Molecular Probes. Recombinant human EGF and Wortmannin were purchased from Invitrogen. LY294002 was from Cell Signaling.
Cell Culture and bacterial infections
HeLa and MEFs were grown in high-glucose DMEM (Hyclone) supplemented with 10% FBS (Wisent) at 37°C in 5% CO2. IPMK knockout MEFs were previously described [26]. IPMK knockout embryonic stem cells were previously described [27] and maintained in high-glucose DMEM (Hyclone) supplemented with 12.5% ES cell-qualified FBS (Gibco), 1x Penicillin/Streptomycin (Wisent), 100 µM MEM non-essential amino acids (Wisent), 55 µM 2-mercaptoethanol (Gibco) and Esgro (Chemicon International) at 37°C in 5% CO2. Cells were transfected with Genejuice (Oncogene Research Products) or Amaxa (Lonza) according to manufacturer’s instructions and used 16 hours after transfection.
S . Typhimurium infections were carried out as described previously [28]. Briefly, bacteria were grown overnight shaking at 37°C and then subcultured in LB without antibiotics for 3 hours. Bacterial inoculum was prepared by pelleting 1 mL of the subculture at 10,000 x g for 2 min, washed once and diluted 1:100 in PBS. PI3-kinase inhibitors were added 30 min prior to the infection and maintained throughout the experiment.
siRNA/esiRNA oligonucleotides and transfections
Control siRNA and those directed against Class II PI3-kinases and IPMK were obtained from Sigma. Vps34 siRNA was described elsewhere [15]. esiRNA against all kinases were obtained from the SIDNET facility at the Hospital for Sick Children. Cells grown in 24-well plates were transfected with 200 nM of individual siRNAs or 200 ng of individual esiRNAs using Lipofectamine® RNAiMAX transfection reagent (Life Technologies) and used for experiments 48 hours after transfection. To confirm knockdown, RNA was isolated with an RNeasy Mini kit (Qiagen) and cDNA was transcribed with the iScript cDNA synthesis kit (BioRad). A qPCR reaction was performed using the SSO advanced SYBR green mix (BioRad).
Immunofluorescence and Western Blot analysis
Fixed cells were immunostained as described [29]. To differentiate between intra- and extracellular bacteria, cells were immunostained before permeabilization with saponin. Images were acquired with a Quorum spinning disk microscope with a 63 x oil immersion objective (Leica DMIRE2 inverted fluorescence microscope equipped with a Hamamatsu Back-Thinned EM-CCD camera, spinning disk confocal scan head). Quantification of samples was done with an epifluorescence microscope (DMIRE2; Leica) equipped with a 100x/NA 1.4 oil objective (Plan Apochromat; Leica).
For Western Blot analysis, cells were washed three times with PBS and then lysed with RIPA buffer. Samples were separated on 10% SDS-PAGE gels, transferred to PVDF-membranes and blocked in 5% milk for 1 hour. Primary and Secondary antibodies were incubated in 2% BSA solution. Bands were detected using the ECL detection system (GE Healthcare).
Calculation of the relative phospho-Akt expression level
Intensity of the phospho-Akt protein band on a Western blot was estimated using ImageJ software. In order to account for potential differences in the total amount of proteins from one sample to another, intensity of the phospho-Akt band was divided by that of the total Akt band. This quotient is the relative phospho-Akt expression level. In an experiment, all relative p-Akt expression levels were normalized to that of the control sample.
In vitro dephosphorylation assay and thin layer chromatography
His-SopB and His-SopB-C460S were recombinantly expressed in E. coli BL21 and affinity purified with HisTrap Ni-Sepharose columns according to Manufacturer’s instructions (GE Healthcare). BODIPY® FL PI(4,5) P2 C6 and BODIPY® FL PI(3–5) P3 C6 were from Cedarlane. To make liposomes, the required amount of stock lipids (in chloroform) was dried under N2 and then resuspended in 50mM Tris buffer. Liposomes were incubated with recombinant SopB and then separated on 250 µM silica gel TLC plates (Whatman) with a resolving solution of 2-Propanol, NH4OH and H2O (65:20:15). Lipids were visualized with a Storm 840 chemiluminescence imager system (Molecular Dynamics).
Results
SopB-mediated Akt activation at Salmonella invasion ruffles is PI (3,4) P2/PI (3–5) P3-dependent
SopB is necessary for the activation of Akt during Salmonella invasion, as judged by western blotting with phospho-specific antibodies that recognize the active form of Akt, which is phosphorylated at Serine 473 (pAkt-Ser473) [12]. Cooper et al. previously showed that phosphorylated Akt is recruited to Salmonella ruffles in SopB-dependent manner [20]. Consistent with this earlier finding, we observed accumulation of pAkt-Ser473 at the actin-rich bacterial invasion site, which was visualized by phalloidin staining (Figure 1A). Such accumulation of pAkt-Ser473 at invasion sites was not observed following infection by a bacterial SopB-deficient (ΔsopB) mutant (Figure 1A). We also observed that cells transfected with plasmids expressing SopB showed significant increase in Akt activation compared to control (GFP-transfected) cells (Figure S1). Thus, SopB is both necessary and sufficient for Akt activation.
Figure 1. SopB-mediated Akt activation at Salmonella invasion ruffles is PI(3,4) P2/PI(3–5) P3-dependent.

(A) HeLa cells were infected with wild type or ΔsopB mutant S . Typhimurium and fixed at 30 min p.i. Cells were examined by epifluorescence microscopy after co-staining for activated Akt with a phospho-specific (Ser473) antibody, actin with fluorescently-labeled phalloidin and bacteria with DAPI. Insets are enlarged from boxed areas. (B) HeLa cells were transiently transfected with GFP-PH-Akt for 16 h prior to Salmonella infection. Cells were infected and fixed as in A. Cells were co-stained for actin with fluorescently-labeled phalloidin and bacteria with a polyclonal antibody to S . Typhimurium . Insets are enlarged from boxed areas. (C) HeLa cells were transiently transfected with GFP or PTEN-A4-YFP for 16 h prior to Salmonella infection. Cells were infected with wild type S . Typhimurium and fixed as in A. Cells were co-stained for actin with fluorescently-labeled phalloidin, Akt with a phospho-specific (Ser473) antibody and bacteria with a polyclonal antibody to S . Typhimurium . Insets are enlarged from boxed areas. Size bars, 10 µm. (D) The percentage of phospho-Akt+ Salmonella invasion ruffles from C was quantified (n ≥ 50). Averages ± SD for three separate experiments are shown. Asterisk indicates that the percent value is significantly different from the control (P < 0.001) as determined by one-way ANOVA analysis. (E) HeLa cells were transiently transfected with GFP-Akt and PTEN-A4-YFP or GFP for 16 h prior to infection. Cells were uninfected, infected as in A, or incubated with 100 ng/mL EGF for 5 min. Where indicated, cells were treated with 100 µM LY294002 for 30 min prior to infection. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. GFP antibodies were used to ensure equal Akt transfection.
We previously showed that SopB leads to the formation of PI(3,4) P2 and PI(3–5) P3 at bacterial invasion sites [15]. Visualization of PI(3,4) P2/PI(3–5) P3 is accomplished by expression of GFP-PH-Akt, a fluorescent probe for these phosphoinositides. Wild type bacteria led to recruitment of GFP-PH-Akt to invasion ruffles, while a bacterial ΔsopB mutant did not, consistent with our previous studies [15] (Figure 1B). Since formation of PI(3,4) P2 and PI(3–5) P3 at membranes is required for canonical Akt activation, we tested if these phosphoinositides are required for SopB-dependent Akt activation by inhibiting their accumulation at invasion sites. This was accomplished by expression of PTEN, a PI-3- phosphatase that specifically dephosphorylates PI(3,4) P2 and PI(3–5) P3. Expression of YFP-tagged PTEN-A4 (with 4 alanine substitutions S380A, T382A, T383A, and S385A) has been successfully used to block accumulation of these PIs [23]. Mutations in this construct are at the regulatory phosphorylation sites at the C-terminus of PTEN, making the resultant protein more active by targeting it to the plasma membrane [23]. After transfection of this modified PTEN construct, SopB-mediated Akt activation was almost completely abolished at invasion sites, as determined by immunofluorescence (Figure 1C, D). A similar decrease in Akt activation can be seen by immunoblotting (Figure 1E). Together, these results suggest that PI(3,4) P2/PI(3–5) P3 production is required for Akt activation by SopB.
SopB-Mediated Akt Activation in HeLa Cells Is Only Partially Sensitive to PI3-Kinase Inhibitors LY294002 and Wortmannin
PI3-kinases, which phosphorylate PIs at the 3-position of the inositol ring, can be divided into three main classes depending on their substrate specificity [30]. We tested all 3 classes of PI3-kinases for their ability to influence SopB-mediated Akt activation. Class I PI3-kinases can be effectively inhibited by the PI3-kinase inhibitors LY294002 and Wortmannin. HeLa cells were treated with varying concentrations of LY294002 (Figure 2) or Wortmannin (Figure S2), then infected with wild type Salmonella . Akt activation was measured in cell lysates with anti-pAkt-Ser473 antibodies. Addition of low concentrations of LY294002 did not significantly impair SopB-mediated Akt activation during infection. However, higher concentrations, e.g. 50-100 µM, LY294002 significantly reduced the Akt activation by approximately 25% compared to the control. Wortmannin showed a very similar impact on Akt activation. In contrast, Akt activation by EGF was abolished by low concentrations of either inhibitor. This indicates that Class I PI3-kinases, which are normally inhibited by LY294002 or Wortmannin, play a minor role in the SopB-mediated Akt activation in HeLa cells.
Figure 2. SopB-mediated Akt activation in HeLa cells is only partially sensitive to PI3-Kinase inhibitor LY294002.

(A) HeLa cells were treated with different concentrations of LY294002 (1 µM to 100 µM) for 30 min. Cells were then infected with wild type S . Typhimurium for 30 min or incubated with 100 ng/mL EGF for 5 min. As controls, cells were either uninfected or infected with ΔsopB mutant S . Typhimurium for 30 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (B) Western blot results from A were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD induced by wild type S . Typhimurium or EGF for three separate experiments, calculated as outlined in the Materials and Methods. Asterisks indicate that the percent value is significantly different from the control (P < 0.05).
Class II PI3-Kinase beta isoform (PI3K-C2β) contributes to SopB-mediated Akt activation
Class II PI3-kinases are less sensitive to PI3-kinase inhibitors [31] and Class II-mediated Akt activation has been shown to be required for glucose-stimulated insulin secretion [32]. Therefore, we targeted expression of all Class II PI3-kinase isoforms using siRNA and analyzed Akt activation by Salmonella . Knockdown of PI3K-C2β led to a significant decrease in SopB-dependent Akt activation, while that of other Class II PI3-kinase isoforms was without effect (Figure 3). Though silencing PI3K-C2β reduced Akt phosphorylation, this kinase seemed to only have a partial role in Akt activation in HeLa cells, indicating that other kinases are also involved.
Figure 3. Class II PI3-Kinase beta isoform (PI3K-C2β) plays a role in SopB-mediated Akt activation.
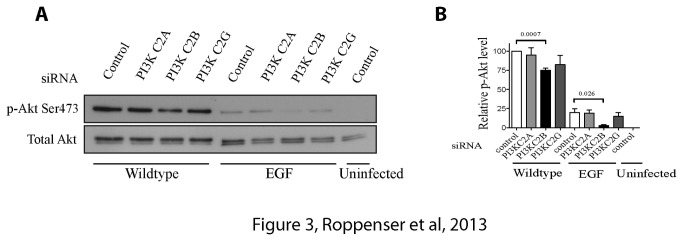
(A) HeLa cells were treated with control, PI3K-C2A, PI3K-C2B or PI3K-C2G siRNA for 48 h. Cells were then infected with wild type S . Typhimurium for 30 min. As controls, cells were uninfected or incubated with 100 ng/mL EGF for 5 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (B) Western blot results from A were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD induced by wild type S . Typhimurium or EGF for three separate experiments, calculated as outlined in the Materials and Methods. The p-values from one-way ANOVA analysis are shown.
Class III PI3-Kinase Vps34 does not play a role in SopB-mediated Akt activation
Class III PI3-kinase Vps34 mediates phosphorylation of PI, generating PI3P. Previous studies suggested that phosphorylation of PI3P can lead to production of PI(3,4) P2 and PI(3–5) P3 [33]. Therefore, we tested the role of Vps34 in Salmonella -induced Akt activation, targeting its expression with siRNA. Knockdown of Vps34 did not impair SopB-dependent activation of Akt, indicating that the Class III PI3-kinase does not contribute to SopB-mediated Akt activation (Figure 4).
Figure 4. Class III PI3-Kinase does not contribute to SopB-mediated Akt activation.
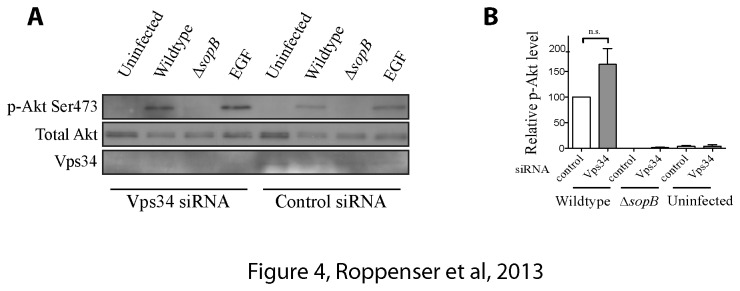
(A) HeLa cells were treated with control or Vps34 siRNA for 48 h. Cells were infected with wild type or ΔSopB mutant S . Typhimurium for 30 min. As controls, cells were uninfected or incubated with 100 ng/mL EGF for 5 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. Anti-Vps34 antibody was used to verify the knockdown of Vps34 in cells that were treated with siRNA. (B) Western blot results from A were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD for three separate experiments, calculated as outlined in the Materials and Methods. Statistical significance was assessed by one-way ANOVA analysis.
SopB does not directly generate PI (3–5) P3 by phosphotransfer
Since the canonical PI3-kinases cannot fully account for Akt activation during Salmonella invasion, we considered other possible mechanisms by which SopB could lead to the production of PI(3,4) P2 and PI(3–5) P3. Because some bacterial proteins can catalyze phosphotransfer reactions between proteins [34], we considered the possibility that SopB might be mediating phosphotransfer reactions between phosphoinositides. Such a catalytic activity for SopB has not been formally tested in vitro. To determine if SopB has unrecognized catalytic activity and is by itself sufficient to induce PI(3–5) P3 formation through a phosphotransfer reaction, we performed an in vitro assay with recombinant SopB and fluorescently tagged PIs. In our assays, SopB induced a robust dephosphorylation of PI(4,5) P2 and PI(3–5) P3, generating phosphatidylinositol monophosphates (Figure S3). However, formation of PI(3–5) P3 by SopB was not detected in our assay. Our studies therefore indicate that SopB does not directly mediate production of PI(3–5) P3 during infection, but rather modulates unknown host factors to generate this phosphoinositide by a non-canonical pathway.
A kinase screen reveals that IPMK plays a role in SopB-mediated Akt activation in HeLa cells
In search for the non-canonical host kinase(s) involved in SopB-mediated Akt activation, we performed an esiRNA screen of 778 mammalian kinases. The results of our screen are shown in Table 1. Many of the kinases identified have been previously implicated in regulating Akt activity (highlighted in yellow; see references), supporting the validity of our screen. The top ‘hit’ in our screen was inositol polyphosphate multikinase (IPMK). SopB-mediated Akt activation was impaired by as much as 91% when IPMK expression was targeted by esiRNA (Table 1). We also targeted expression of IPMK using siRNA for comparison. siRNA-mediated silencing of IPMK expression similarly impaired Akt activation, providing additional evidence for its role in HeLa cells (Figure 5A).
Table 1. Kinases that play a role in Akt activation during Salmonella infection.
| Gene symbol | % Activation compared to control | References | Gene symbol | % Activation compared to control | References |
|---|---|---|---|---|---|
| IPMK | 8.47 | [26] | PRKCN | 39.76 | - |
| CHKA | 11.09 | [40] | PDXK | 39.76 | - |
| HCK | 12.03 | [41] | PRKCD | 44.74 | - |
| NADK | 13.15 | - | PRKAR1A | 45.86 | - |
| MAST2 | 15.18 | - | ULK2 | 47.05 | - |
| MINK | 16.77 | - | PRKR | 48.05 | - |
| SSTK | 18.97 | - | DAPK2 | 56.70 | - |
| PNKP | 19.11 | - | FASTK | 60.99 | - |
| MAP3K2 | 20.76 | - | ADCK4 | 62.56 | - |
| MAP3K9 | 21.09 | - | PAK1 | 62.88 | [44] |
| CDC2 | 23.06 | - | ULK3 | 63.49 | - |
| DGUOK | 23.32 | - | MARK3 | 64.04 | - |
| ACK1 | 25.33 | [42] | LCK | 64.21 | [45] |
| SNRK | 27.46 | - | ILK | 67.01 | [46] |
| ERK8 | 28.08 | - | PIK3C2A | 70.06 | - |
| AGK | 28.09 | - | SRC | 71.09 | [47] |
| CDKL5 | 28.25 | - | PANK1 | 72.28 | - |
| PFKFB1 | 29.10 | - | BLNK | 73.06 | - |
| LYK5 | 30.26 | - | STK11 | 73.36 | - |
| MIDORI | 30.80 | - | PRKCH | 74.43 | - |
| AK1 | 32.42 | - | CDC2L2 | 75.12 | - |
| HK3 | 36.07 | - | AURKC | 79.06 | - |
| BLK | 36.77 | - | PRKCQ | 79.72 | [48] |
| PLK1 | 38.36 | [43] | MGC8407 | 80.59 | - |
| PRKACG | 39.20 | - | IKBKE | 83.02 | [49] |
| CKB | 39.33 | - | PIP5K1A | 89.48 | - |
HeLa cells were treated with esiRNAs to mammalian kinases (778 were tested individually) for 48 h. Cells were then infected with wild type S . Typhimurium for 30 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. GAPDH antibodies were used to ensure equal protein loading. Relative phospho-Akt levels were determined using ImageJ software. Activation of Akt shown in the table was normalized to that of control esiRNA-treated sample. Among the listed 52 most highly ranked kinases, of which their knockdowns resulted in the most significant decrease in Akt activation, those with indicated references have been previously implicated in the positive regulation of Akt.
Figure 5. IPMK plays a role in SopB-mediated Akt activation.

(A) HeLa cells were treated with control or IPMK siRNA for 48 h. Cells were infected with wild type S . Typhimurium for 30 min. As a control, cells were uninfected. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (B) Wild type (ES10) or IPMK knockout (ES2 and ES5) embryonic stem cells were infected with wild type or ΔSopB mutant S . Typhimurium for 30 min. As a control, cells were uninfected. Where indicated, cells were treated with 100 µM LY294002 for 30 min. prior to infection. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (C) Western blot results of ES10 and ES5 from B were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD for three separate experiments, calculated as outlined in the Materials and Methods. The p-values from one-way ANOVA analysis are shown.
To further explore the role of IPMK in SopB-mediated Akt activation, we used two different cell types lacking IPMK. In IPMK knockout embryonic stem (ES) cells [27], we observed a significant decrease in Akt activation upon Salmonella infection, compared to wild type control cells (Figure 5B and C). We also examined mouse embryonic fibroblast cells (MEFs) lacking IPMK [35]. In contrast to ES cells, we did not see a decrease in Akt activation in IPMK-/- MEFs when infected with wild type Salmonella compared to WT MEFs (Figure S4). However, when we treated the ES and MEF cells with the PI3-kinase inhibitor LY294002, we saw a significant decrease in SopB-mediated Akt activation, suggesting that Class I PI3-kinase may play a dominant role in these cell types unlike in HeLa cells. Together, our studies demonstrate that IPMK contributes to Akt activation in HeLa cells, but has a minor or negligible role in other tested cell types.
Discussion
Recently, we showed that PI(3,4) P2 and PI(3–5) P3 are generated at invasion ruffles in a SopB-dependent manner [15]. In our studies of HeLa cells, formation of these phosphoinositides and the phosphorylation of Akt were found to be sensitive to PI3-kinase inhibitors LY294002 and wortmannin at only high concentrations, suggesting that kinases other than Class I PI3-kinase are also involved. In contrast, Cooper et al. previously showed that SopB-mediated Akt activation is resistant to 100 nM wortmannin, but completely inhibited by 50 µM LY294002 [20]. However, we believe that such differences in sensitivity may be cell-type dependent, since we saw a significant decrease in SopB-mediated Akt activation when Salmonella -infected embryonic stem cells or mouse embryonic fibroblasts were treated with LY294002. Our findings and that of Cooper et al. suggest that the mechanisms of Akt activation by Salmonella vary depending on the cell types used for infection.
When Class II PI3-kinase C2β was knocked down in HeLa cells, we observed that SopB-mediated Akt activation was significantly decreased, although this kinase seemed to only play a minor role. No further reduction in Akt activation was noted when such silenced cells were additionally treated with LY294002. We concluded that other kinases must be activated downstream of SopB to induce PI(3,4) P2/PI(3–5) P3 formation at membrane ruffles. After performing a screen of ~780 host kinases for their potential role in SopB-mediated Akt activation, we identified about 90 kinases that influenced Akt activation after Salmonella invasion. This remarkably large repertoire of kinases that contribute to SopB-mediated Akt activation is not surprising, given that SopB is known to be able to manipulate many host kinase phosphorylation events in infected cells [36]. In fact, Rogers et al. have proposed that approximately half of all Salmonella -induced phosphorylation events in the host cell require the SopB effector [36]. Manipulation of the phosphorylation status of host kinases may be necessary for SopB-mediated PI(3,4) P2 and/or PI(3–5) P3 synthesis. It is noteworthy that a complete screen for kinases required for Akt activation has not been previously published. Therefore, the results of our kinase screen are likely to open up new avenues of study into the regulation of Akt.
The kinase that showed the greatest effect on SopB-mediated Akt activation in HeLa cells was IPMK. IPMK is reported to have PI3-kinase activity and it was shown to phosphorylate PI(4,5) P2 to generate PI(3–5) P3 [37]. Also, IPMK was shown to activate Akt [26]. However, as measured by RT-PCR, IPMK knockdown efficiency was not consistent in HeLa cells, which led to variable extent of reduction in phospho-Akt expression. Thus, we decided to try 2 different knockout cell lines lacking IPMK. To our surprise, we did not see a large decrease in SopB-induced Akt activation in these cells. However, we noted a significant effect of the PI3-kinase inhibitor LY294002 on SopB-induced Akt activation in embryonic stem cells and mouse embryonic fibroblasts, suggesting that Class I PI3-kinases play a more prominent role in these cells. This suggests that SopB can recruit and/or activate various kinases in different cell types to induce PI(3,4) P2/PI(3–5) P3 formation at invasion ruffles.
How SopB is producing these PIs and exploiting the host kinases needs to be further elucidated. The main substrate of SopB is PI(4,5) P2, generating the rare lipid PI(5) P. Since SopB does not seem to have enzymatic activity other than PI-phosphatase activity, the mechanism behind SopB-mediated Akt activation might be via PI(5) P. It is conceivable that PI(5) P recruits distinct lipid kinases to Salmonella invasion ruffles to synthesize PI(3,4) P2 and/or PI(3–5) P3 and/or kinases that directly activate Akt independently of the lipids. In addition, PI(5) P may have an inhibitory effect on protein phosphatases that deactivate Akt, such as PP2A, or on lipid phosphatases that dephosphorylate PI(3–5) P3, like SHIP2.
Shigella flexneri utilizes a type 3 secreted effector IpgD, a SopB homologue, for infection. Pendaries et al. recently suggested that PI(5) P production by IpgD is necessary for activation of Class I PI3-kinases, thereby activating Akt, in HeLa cells [38]. Although both IpgD and SopB lead to the accumulation of PI(5) P, whether or not SopB-mediated Akt activation is similar to that of IpgD is unclear, because the former is only partially sensitive to PI3-kinase inhibitors in the HeLa cells. It has been shown in vitro that these two homologues have different substrate specificities [14,39]. Hence, a slight difference in the underlying mechanisms of these two bacterial effectors may be possible.
The advantage of bacterial exploitation of several host kinases instead of depending on a single kinase is obvious. Since Akt has been previously shown to play an important role in the Salmonella life cycle in the infected host cell, it would be beneficial for the pathogen to manipulate multiple kinases to ensure its activation. Additionally, our data suggests that Salmonella may have evolved to use different repertoires of kinases for Akt activation in different types of host cells. This may be intended to adapt to variable expression levels of the kinases in different cell types.
In summary, our data indicate that Salmonella can exploit different host kinases during invasion to optimize replication and survival in host cells. SopB generates PI(3,4) P2/PI(3–5) P3 at invasion sites via a specific group of kinases, which depends on the type of infected host cell. This PI-formation at membrane ruffles is responsible for activating Akt and inhibiting apoptosis in infected cells, as well as modulating SCV biogenesis. However, the precise mechanism of how SopB manipulates different kinases for Akt activation remains incompletely understood and needs further investigation.
Supporting Information
SopB expression is sufficient to activate Akt. HeLa cells were transiently transfected with plasmids encoding untagged SopB, GFP-SopB or GFP. As a control, cells were stimulated with 100 ng/mL EGF for 5 min. Akt activation of cells was assessed 8 h after transfection by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading.
(TIF)
SopB-mediated Akt activation in HeLa cells is only partially sensitive to PI3-Kinase inhibitor Wortmannin. HeLa cells were treated with different concentrations of Wortmannin (10 nM to 1000 nM) for 30 min. Cells were then infected with wild type S . Typhimurium for 30 min or incubated with 100 ng/mL EGF for 5 min. As controls, cells were either uninfected or infected with ΔSopB mutant S . Typhimurium for 30 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading.
(TIF)
SopB is not sufficient to produce formation of PI(3–5) P3in vitro. Reaction mixtures of FL-PIP2 or FL-PIP3 and His-tagged recombinant SopB or catalytically inactive SopB (C460S) were separated on TLC. As a control, pure lipids were directly separated on TLC and liposomes without recombinant SopB were also examined.
(TIF)
IPMK does not contribute to SopB-mediated Akt activation in mouse embryonic fibroblasts (MEF). (A) Wild type or IPMK knockout mouse embryonic fibroblasts were infected with wild type or ΔsopB mutant S . Typhimurium for 30 min. As controls, cells were uninfected or treated with 100 ng/mL EGF for 5 min. Where indicated, cells were treated with 100 µM LY294002 for 30 min prior to infection. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (B) Western blot results from A were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD for three separate experiments, calculated as outlined in the Materials and Methods. The p-values from one-way ANOVA analysis are shown.
(TIF)
Acknowledgments
We thank members of the Brumell lab for critical reading of the manuscript. We also thank Doug Chris Fladd for his assistance with esiRNA and Mike Woodside and Paul Paroutis for their support with confocal microscopy.
Funding Statement
BR was supported by the Austrian Science Fund. HK was supported by a summer studentship from the Canadian Association for Gastroenterology and Crohn’s and Colitis Foundation of Canada. This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP#93634) (to JHB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pang T, Bhutta ZA, Finlay BB, Altwegg M (1995) Typhoid fever and other salmonellosis: a continuing challenge. Trends Microbiol 3: 253-255. doi:10.1016/S0966-842X(00)88937-4. PubMed: 7551636. [DOI] [PubMed] [Google Scholar]
- 2. Tsolis RM, Kingsley RA, Townsend SM, Ficht TA, Adams LG et al. (1999) Of mice, calves, and men. Comparison of the mouse typhoid model with other Salmonella infections. Adv Exp Med Biol 473: 261-274. doi:10.1007/978-1-4615-4143-1_28. PubMed: 10659367. [PubMed] [Google Scholar]
- 3. Hueck CJ (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62: 379-433. PubMed: 9618447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bakowski MA, Braun V, Brumell JH (2008) Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 9: 2022-2031. doi:10.1111/j.1600-0854.2008.00827.x. PubMed: 18778407. [DOI] [PubMed] [Google Scholar]
- 5. Galan JE, Zhou D (2000) Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc Natl Acad Sci U S A 97: 8754-8761. doi:10.1073/pnas.97.16.8754. PubMed: 10922031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou D, Chen LM, Hernandez L, Shears SB, Galán JE (2001) A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol 39: 248-259. doi:10.1046/j.1365-2958.2001.02230.x. PubMed: 11136447. [DOI] [PubMed] [Google Scholar]
- 7. Patel JC, Galán JE (2006) Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol 175: 453-463. doi:10.1083/jcb.200605144. PubMed: 17074883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hernandez LD, Hueffer K, Wenk MR, Galán JE (2004) Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304: 1805-1807. doi:10.1126/science.1098188. PubMed: 15205533. [DOI] [PubMed] [Google Scholar]
- 9. Wasylnka JA, Bakowski MA, Szeto J, Ohlson MB, Trimble WS et al. (2008) Role for myosin II in regulating positioning of Salmonella-containing vacuoles and intracellular replication. Infect Immun 76: 2722-2735. doi:10.1128/IAI.00152-08. PubMed: 18411289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Knodler LA, Finlay BB, Steele-Mortimer O (2005) The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem 280: 9058-9064. PubMed: 15642738. [DOI] [PubMed] [Google Scholar]
- 11. Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS et al. (2012) Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 12: 645-656. doi:10.1016/j.chom.2012.10.009. PubMed: 23159054. [DOI] [PubMed] [Google Scholar]
- 12. Steele-Mortimer O, Knodler LA, Marcus SL, Scheid MP, Goh B et al. (2000) Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J Biol Chem 275: 37718-37724. doi:10.1074/jbc.M008187200. PubMed: 10978351. [DOI] [PubMed] [Google Scholar]
- 13. Marcus SL, Wenk MR, Steele-Mortimer O, Finlay BB (2001) A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett 494: 201-207. doi:10.1016/S0014-5793(01)02356-0. PubMed: 11311241. [DOI] [PubMed] [Google Scholar]
- 14. Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW (1998) SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci U S A 95: 14057-14059. doi:10.1073/pnas.95.24.14057. PubMed: 9826652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mallo GV, Espina M, Smith AC, Terebiznik MR, Alemán A et al. (2008) SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol 182: 741-752. doi:10.1083/jcb.200804131. PubMed: 18725540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM et al. (2002) Elimination of host cell PtdIns(4,5) P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 4: 766-773. [DOI] [PubMed] [Google Scholar]
- 17. Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S et al. (1998) Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17: 313-325. doi:10.1038/sj.onc.1201947. PubMed: 9690513. [DOI] [PubMed] [Google Scholar]
- 18. Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ et al. (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272: 31515-31524. doi:10.1074/jbc.272.50.31515. PubMed: 9395488. [DOI] [PubMed] [Google Scholar]
- 19. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 307: 1098-1101. doi:10.1126/science.1106148. PubMed: 15718470. [DOI] [PubMed] [Google Scholar]
- 20. Cooper KG, Winfree S, Malik-Kale P, Jolly C, Ireland R et al. (2011) Activation of Akt by the bacterial inositol phosphatase, SopB, is wortmannin insensitive. PLOS ONE 6: e22260. doi:10.1371/journal.pone.0022260. PubMed: 21779406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marcus SL, Knodler LA, Finlay BB (2002) Salmonella enterica serovar Typhimurium effector SigD/SopB is membrane-associated and ubiquitinated inside host cells. Cell Microbiol 4: 435-446. doi:10.1046/j.1462-5822.2002.00202.x. PubMed: 12102689. [DOI] [PubMed] [Google Scholar]
- 22. Dai S, Zhang Y, Weimbs T, Yaffe MB, Zhou D (2007) Bacteria-generated PtdIns(3)P recruits VAMP8 to facilitate phagocytosis. Traffic 8: 1365-1374. doi:10.1111/j.1600-0854.2007.00613.x. PubMed: 17645435. [DOI] [PubMed] [Google Scholar]
- 23. Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F et al. (2009) A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci U S A 106: 480-485. doi:10.1073/pnas.0811212106. PubMed: 19114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoiseth SK, Stocker BA (1981) Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291: 238-239. doi:10.1038/291238a0. PubMed: 7015147. [DOI] [PubMed] [Google Scholar]
- 25. Siddhanta U, McIlroy J, Shah A, Zhang Y, Backer JM (1998) Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3'-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol 143: 1647-1659. doi:10.1083/jcb.143.6.1647. PubMed: 9852157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maag D, Maxwell MJ, Hardesty DA, Boucher KL, Choudhari N et al. (2011) Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc Natl Acad Sci U S A 108: 1391-1396. doi:10.1073/pnas.1017831108. PubMed: 21220345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frederick JP, Mattiske D, Wofford JA, Megosh LC, Drake LY et al. (2005) An essential role for an inositol polyphosphate multikinase, Ipk2, in mouse embryogenesis and second messenger production. Proc Natl Acad Sci U S A 102: 8454-8459. doi:10.1073/pnas.0503706102. PubMed: 15939867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steele-Mortimer O, Méresse S, Gorvel JP, Toh BH, Finlay BB (1999) Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell Microbiol 1: 33-49. doi:10.1046/j.1462-5822.1999.00003.x. PubMed: 11207539. [DOI] [PubMed] [Google Scholar]
- 29. Bakowski MA, Cirulis JT, Brown NF, Finlay BB, Brumell JH (2007) SopD acts cooperatively with SopB during Salmonella enterica serovar Typhimurium invasion. Cell Microbiol 9: 2839-2855. doi:10.1111/j.1462-5822.2007.01000.x. PubMed: 17696999. [DOI] [PubMed] [Google Scholar]
- 30. Vanhaesebroeck B, Alessi DR (2000) The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346 3: 561-576. doi:10.1042/0264-6021:3460561. PubMed: 10698680. [PMC free article] [PubMed] [Google Scholar]
- 31. Domin J, Pages F, Volinia S, Rittenhouse SE, Zvelebil MJ et al. (1997) Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem J 326(1): 139-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leibiger IB, Brismar K, Berggren PO (2010) Novel aspects on pancreatic beta-cell signal-transduction. Biochem Biophys Res Commun 396: 111-115. doi:10.1016/j.bbrc.2010.02.174. PubMed: 20494121. [DOI] [PubMed] [Google Scholar]
- 33. Zhang X, Loijens JC, Boronenkov IV, Parker GJ, Norris FA et al. (1997) Phosphatidylinositol-4-phosphate 5-kinase isozymes catalyze the synthesis of 3-phosphate-containing phosphatidylinositol signaling molecules. J Biol Chem 272: 17756-17761. doi:10.1074/jbc.272.28.17756. PubMed: 9211928. [DOI] [PubMed] [Google Scholar]
- 34. Huang YH, Ferrières L, Clarke DJ (2006) The role of the Rcs phosphorelay in Enterobacteriaceae. Res Microbiol 157: 206-212. doi:10.1016/j.resmic.2005.11.005. PubMed: 16427772. [DOI] [PubMed] [Google Scholar]
- 35. Kim S, Kim SF, Maag D, Maxwell MJ, Resnick AC et al. (2011) Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab 13: 215-221. doi:10.1016/j.cmet.2011.01.007. PubMed: 21284988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rogers LD, Brown NF, Fang Y, Pelech S, Foster LJ (2011) Phosphoproteomic analysis of Salmonella-infected cells identifies key kinase regulators and SopB-dependent host phosphorylation events. Sci Signal 4: rs9–: rs9 PubMed: 21934108. [DOI] [PubMed] [Google Scholar]
- 37. Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH et al. (2005) Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci U S A 102: 12783-12788. doi:10.1073/pnas.0506184102. PubMed: 16123124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pendaries C, Tronchère H, Arbibe L, Mounier J, Gozani O et al. (2006) PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J 25: 1024-1034. doi:10.1038/sj.emboj.7601001. PubMed: 16482216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F et al. (2002) Conversion of PtdIns(4,5) P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J 21: 5069-5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chua BT, Gallego-Ortega D, Ramirez de Molina A, Ullrich A, Lacal JC et al. (2009) Regulation of Akt (ser473) phosphorylation by choline kinase in breast carcinoma cells. Mol Cancer 8: 131 [DOI] [PMC free article] [PubMed]
- 41. Suh HS, Kim MO, Lee SC (2005) Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol 174: 2712-2719. PubMed: 15728479. [DOI] [PubMed] [Google Scholar]
- 42. Mahajan K, Coppola D, Challa S, Fang B, Chen YA et al. (2010) Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLOS ONE 5: e9646. doi:10.1371/journal.pone.0009646. PubMed: 20333297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF et al. (2011) Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem 286: 35795-35800. doi:10.1074/jbc.C111.269050. PubMed: 21890624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P et al. (2008) Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol 44: 429-434. doi:10.1016/j.yjmcc.2007.10.016. PubMed: 18054038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cuevas B, Lu Y, Watt S, Kumar R, Zhang J et al. (1999) SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem 274: 27583-27589. doi:10.1074/jbc.274.39.27583. PubMed: 10488096. [DOI] [PubMed] [Google Scholar]
- 46. Koul D, Shen R, Bergh S, Lu Y, de Groot JF et al. (2005) Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol Cancer Ther 4: 1681-1688. doi:10.1158/1535-7163.MCT-05-0258. PubMed: 16275989. [DOI] [PubMed] [Google Scholar]
- 47. Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K et al. (2003) Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem 278: 2118-2123. doi:10.1074/jbc.M210828200. PubMed: 12431978. [DOI] [PubMed] [Google Scholar]
- 48. Ma J, Ding Y, Fang X, Wang R, Sun Z (2012) Protein kinase C-theta inhibits inducible regulatory T cell differentiation via an AKT-Foxo1/3a-dependent pathway. J Immunol 188: 5337-5347. doi:10.4049/jimmunol.1102979. PubMed: 22539794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guo JP, Coppola D, Cheng JQ (2011) IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem 286: 37389-37398. doi:10.1074/jbc.M111.287433. PubMed: 21908616. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SopB expression is sufficient to activate Akt. HeLa cells were transiently transfected with plasmids encoding untagged SopB, GFP-SopB or GFP. As a control, cells were stimulated with 100 ng/mL EGF for 5 min. Akt activation of cells was assessed 8 h after transfection by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading.
(TIF)
SopB-mediated Akt activation in HeLa cells is only partially sensitive to PI3-Kinase inhibitor Wortmannin. HeLa cells were treated with different concentrations of Wortmannin (10 nM to 1000 nM) for 30 min. Cells were then infected with wild type S . Typhimurium for 30 min or incubated with 100 ng/mL EGF for 5 min. As controls, cells were either uninfected or infected with ΔSopB mutant S . Typhimurium for 30 min. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading.
(TIF)
SopB is not sufficient to produce formation of PI(3–5) P3in vitro. Reaction mixtures of FL-PIP2 or FL-PIP3 and His-tagged recombinant SopB or catalytically inactive SopB (C460S) were separated on TLC. As a control, pure lipids were directly separated on TLC and liposomes without recombinant SopB were also examined.
(TIF)
IPMK does not contribute to SopB-mediated Akt activation in mouse embryonic fibroblasts (MEF). (A) Wild type or IPMK knockout mouse embryonic fibroblasts were infected with wild type or ΔsopB mutant S . Typhimurium for 30 min. As controls, cells were uninfected or treated with 100 ng/mL EGF for 5 min. Where indicated, cells were treated with 100 µM LY294002 for 30 min prior to infection. Akt activation was determined by immunoblotting the cell lysates with a phospho-specific anti-Ser473 Akt antibody. Pan-Akt antibodies were used to ensure equal protein loading. (B) Western blot results from A were analyzed by estimating the intensities of protein bands with the ImageJ software. Shown on the graph are the relative and normalized expression levels of phospho-Akt ± SD for three separate experiments, calculated as outlined in the Materials and Methods. The p-values from one-way ANOVA analysis are shown.
(TIF)