

Abstract
Although calcineurin inhibitors (CNIs) are very useful in preventing allograft rejection, they can mediate a rapid progression of post-transplantation malignancies. The CNI cyclosporine A (CsA) can promote renal tumor growth through activation of the proto-oncogene ras and over-expression of the angiogenic cytokine VEGF; the ras activation also induces over-expression of the cytoprotective enzyme HO-1, which promotes survival of renal cancer cells. Here, we show that the natural product honokiol significantly inhibited CsA-induced and Ras-mediated survival of renal cancer cells through the down-regulations of VEGF and HO-1. Thus, honokiol treatment may help to prevent tumor-promoting effects of CsA in transplant patients.
Keywords: Honokiol, Ras, VEGF, HO-1, Renal tumor
1. INTRODUCTION
The immunosuppressive drugs are essential to prevent allograft rejection in transplant patients. However, the development and progression of cancer is a major problem in patients receiving immunosuppressive therapy; the most common post-transplantation cancers being kidney and skin cancers, lymphoma, and Kaposi’s sarcoma[1-3]. It has been demonstrated that apart from promoting malignancy via impaired functions of the immune system, some of these agents, especially calcineurin inhibitors (CNIs), can directly induce cancer-promoting pathways [4-7].
Classically, the CNI cyclosporine A (CsA) inhibits the phosphatase activity of calcineurin and prevents the nuclear translocation of NFAT, a crucial factor for T cell activation; and these events lead to the suppression of immune system [8]. However, independent of its immunosuppressive functions, CsA can directly promote malignancy through pathways that include production of transforming growth factor (TGF)-β and modulation of DNA repair and apoptosis [4, 9]. Recent studies from our laboratory have demonstrated that CsA treatment can promote an accelerated growth of renal tumors through the over-expression of the angiogenic cytokine vascular endothelial growth factor (VEGF) [6, 10]; in addition, CsA induces a significant activation of the proto-oncogene ras in renal cancer cells [7].
Activated Ras proteins transmit their signals to a cascade of protein kinases that have MAP kinase kinase (MEK) as the substrate, such as MEK kinase, c-Raf-1, and B-Raf, and results in the activation of MAP kinase (MAPK) [11]. Although the Ras mutation is not very common in renal cell carcinoma (RCC), the Raf-ERK pathway is hyperactive [12]. We have recently shown that activation of the Ras-Raf-ERK pathway induces the over-expression of the cytoprotective enzyme heme oxygenase-1 (HO-1), which plays a major role in the survival of renal cancer cells [13, 14]. Interestingly, HO-1 can also regulate angiogenesis [15]. The key role of the Ras/Raf/MEK/ERK pathway in the development and progression of RCC, and the fact that CsA can induce Ras activation, have evoked interest in targeting members of this pathway as a possible therapeutic strategy for CNI-induced renal cancer, which otherwise is highly resistant to conventional chemotherapy [16].
Honokiol, a natural product originally isolated from Magnolia obovata [17], is a promising agent for mediating anti-inflammatory, anti-oxidant, pro-apoptotic and chemopreventive functions [18-20]. It has been demonstrated that honokiol can inhibit the growth of tumors in animal xenograft models and induce tumor cell apoptosis [21-23]. Honokiol-induced apoptosis can be mediated through the activation of caspases, and the induction of pro-apoptotic Bax and Bad [24, 25]. Honokiol may also suppress Ras-mediated activation of phospholipase D, and induce cyclophilin D to promote mitochondrial permeability transition pore-associated cell death [21, 26, 27].
It appears that Ras and cyclophilin D are critical targets of both CsA and honokiol, though in an antagonistic way. While CsA promotes tumor growth through activation of Ras and inhibition of cyclophilin D, honokiol may exert an anti-tumor effect through suppression of Ras pathways and induction of cyclophilin D-mediated mitochondrial transition pores [7, 21, 26]. Thus, a combination therapy using CsA and honokiol might be considered as a potential therapeutic strategy for CNI-induced post-transplantation cancer, which involves Ras activation. Using honokiol in combination with CsA may inhibit the cancer-promoting effects of CNI while retaining its immunosuppressive function, which is essential to prevent allograft rejection.
In the present study, we observe that honokiol can effectively down-regulate CsA-induced activation of the Ras-Raf pathway. It can inhibit CsA-induced over-expression of the pro-angiogenic VEGF; we also demonstrate for the first time that honokiol promotes apoptosis by inhibiting CsA-induced expression of the cytoprotective HO-1, which plays a major role in Ras-mediated survival of renal cancer cells [13, 14].
2. MATERIALS AND METHODS
2.1. Reagents
Honokiol used in this study was synthesized in Dr. J. Arbiser’s laboratory (Emory University School of Medicine, Atlanta, GA) [26]. It was dissolved in DMSO at a concentration of 25 mmol/L, and aliquots were stored at −80°C. Stock solutions were diluted to appropriate concentrations with growth medium immediately before use. CsA (Novartis) was purchased from Children’s Hospital Boston pharmacy and Cobalt protoporphyrin (CoPP) was obtained from Frontier Scientific. Gene-specific small interfering RNAs (siRNA) for H-Ras, Raf-1 and HO-1, along with control siRNA were purchased from Qiagen. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen).
2.2. Cell Lines
Human renal cancer cell lines (786-0 and Caki-1) were obtained from American Type Culture Collection. 786-0 cells were grown in RPMI-1640, and Caki-1 cells were grown in McCoy’s 5A medium supplemented with 10% fetal bovine serum (GIBCO).
2.3. Plasmid
A 2.6-kb VEGF promoter-luciferase construct in pGL2 basic vector (Promega) containing full-length VEGF promoter sequence (−2361 to +298 bp relative to the transcription start site) (10), and a human HO-1 promoter-luciferase construct (phO4luc) in pSKluc vector containing the human HO-1 promoter sequence (bp −4067 to +70 relative to the transcription start site) (19) were used in transient transfection assays.
2.4. Transfection and Luciferase Assays
786-0 (0.5 × 105) cells were transfected with VEGF or HO-1 promoter-luciferase plasmid using Effectene Transfection Reagent (Qiagen), according to the manufacturer’s protocol. After 18 hours of transfection, the cells were treated with different combinations of CsA and honokiol. The cells were harvested 48 hours after transfection and luciferase activity was quantified using a standard assay kit (Promega) in a luminometer. As an internal control for transfection efficiency, the cells were co-transfected with β-galactosidase gene under control of cytomegalovirus immediate early promoter, and β-galactosidase activity was measured by standard assay system (Promega).
2.5. Western Blot Analysis
Protein samples (15 μg) were run on SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Millipore Corporation). The membranes were incubated with rabbit polyclonal anti-VEGF, mouse monoclonal anti-H-Ras, rabbit polyclonal anti-Raf-1, rabbit polyclonal anti-phospho-RKIP, goat polyclonal anti-RKIP (Santa Cruz Biotechnology), goat polyclonal anti-HO-1 (R&D Systems) or mouse monoclonal anti-β-Actin (Sigma-Aldrich). All primary antibodies were diluted at 0.5 μg/ml. Following incubation with primary antibody, the membranes were incubated with peroxidase-linked secondary antibody (goat anti-mouse IgG/goat anti-rabbit IgG/donkey anti-goat IgG) (0.2 μg/ml). Reactive bands were detected using chemiluminescent substrate (Pierce).
2.6. Preparation of Nuclear Extracts
Cells were treated with different combinations of CsA and honokiol. After 6 hours of treatment, nuclear extracts were prepared from the cells using a Nuclear Extract Kit (Active Motif). Briefly, 0.8 × 105 cells were seeded/well of 6-well plate. Following treatments, cells were collected in phosphate buffered saline (PBS) in presence of phosphatase inhibitors. The cells were resuspended in hypotonic buffer (supplied with the kit) and incubated on ice for 15 min. Cytoplasmic fractions were collected after adding detergent (supplied with the kit) and centrifuging the cells. The nuclear pellets were resuspended in complete lysis buffer, and incubated on ice for 30 min. The nuclear fractions were collected by centrifuging the cells for 10 min.
2.7. DNA Binding ELISA for Activated Nrf2
Binding of activated Nrf2 to specific DNA sequence was quantified by TransAM Nrf2 ELISA kit (Active Motif) following the manufacturer’s protocol. The kit contains strip-well plate to which multiple copies of specific double-stranded oligonucleotide for Nrf2 consensus-binding site anti-oxidant response element (ARE) have been immobilized. When nuclear extract is added to the wells, active Nrf2 contained in the extract binds specifically to the plate-bound oligonucleotide. Nrf2-specific primary antibody is then added followed by subsequent incubation with HRP-conjugated secondary antibody and developing solution. Quantitative analysis is performed by spectrophotometry at 450 nm. The reading at 450 nm is directly proportional to DNA binding activity of Nrf2 and serves as a convenient tool to determine activation of the transcription factor.
2.8. Cell Proliferation Assay
Cell proliferation was measured by MTT Cell Proliferation Assay (ATCC) following the manufacturer’s protocol. Briefly, cells (non-starved) were seeded in 96-well plates (0.025 × 105 cells/well), and treated with different combinations of CsA and honokiol. After 48 hour of drug treatment, 10 μl of MTT reagent was added to each well. Once purple crystals of formazan became clearly visible under microscope, 100 μl of Detergent Reagent was added and the cells were incubated at dark for 4 hour. Absorbance was measured at 570 nm and corrected against blanks, which consisted of culture medium processed in the same way as above in the absence of cells. The reading at 570 nm is directly proportional to cell proliferation (number of viable cells).
2.9. Apoptosis Assay
Cellular apoptosis was measured by Annexin-V and Propidium Iodide (PI) staining using APC Annexin-V Apoptosis Detection Kit (eBioscience) according to the manufacturer’s protocol. Briefly, 0.5 × 105 cells were seeded /well of 6-well plate and treated as described. For the assay, the cells were trypsinized, washed with PBS and 1X binding buffer, and then resuspended in the binding buffer at a concentration of 4-5 × 106 cells/ml. 5 μl APC-conjugated Annexin V was added to 100 μl of the cell suspension and incubated at room temperature for 15 min. The cells were washed and resuspended in 200 μl of binding buffer. 5 μl PI was added to the cells; and the cells were analyzed by flow cytometry on a FACSCalibur.
2.10. Statistical Analysis
Statistical significance was determined by Student’s t test. Differences with P < 0.05 were considered statistically significant.
3. RESULTS
3.1. Honokiol Inhibits CsA-induced Transcriptional Activation and Protein Expression of VEGF
VEGF is one of the most important angiogenic cytokines involved in tumor growth [28]. We have previously demonstrated that CsA treatment can induce VEGF transcriptional activation and protein expression in renal cancer cells, and promote cancer progression [6]. Here, we first evaluated if honokiol treatment could inhibit CsA-induced VEGF over-expression in 786-0 and Caki-1 cells. To study promoter activity, cells were transfected with the VEGF promoter-luciferase plasmid and then treated with CsA in absence or presence of honokiol. We observed that CsA–induced VEGF transcriptional activation in both 786-0 and Caki-1 cells was significantly inhibited following honokiol treatment as assessed by luciferase activity (Fig. 1A, and Supplementary Fig. 1). Moreover, CsA-induced VEGF protein expression was markedly inhibited by honokiol as observed by Western blot (Fig. 1B). Thus, our data suggest that honokiol can down-regulate CsA-induced VEGF over-expression.
FIGURE 1. Honokiol inhibits CsA-induced promoter activity and over-expression of VEGF.

(a) 786-O cells were transfected with VEGF promoter-luciferase construct (0.5 μg) and treated with 5 μg/ml CsA, 20 μM honokiol (HNK) or vehicle. The cells were harvested 48 h after transfection and fold change in luciferase activity was calculated as the relative luciferase counts from each group of cells compared to that of vehicle-treated control. Results are representative of three independent experiments. Columns, average of triplicate readings from two different samples; error bars, standard error of the mean (SEM). * p < 0.05. compared with vehicle-treated control, and ** p < 0.05 compared with CsA-treated but HNK-untreated cells. (b) 786-O cells were treated with 5 μg/ml CsA, 20 μM HNK or vehicle for 48 h. The cells were lysed and the expression of VEGF was analyzed by western blot (top). β-actin was used as loading control (bottom). Results are representative of three independent experiments.
3.2. Treatment with Honokiol Inhibits Phosphorylation of Raf Kinase Inhibitory Protein (RKIP)
Raf kinase inhibitory protein (RKIP) acts as an endogenous inhibitor of the Raf-1-MEK pathway [29]. Nonphosphorylated RKIP inhibits Raf and blocks Raf-mediated signaling events [30]. We have previously shown that CsA treatment activates Ras-Raf pathway in renal cancer cells through increased phosphorylation of RKIP [7]. Here, we checked if honokiol could inhibit CsA-induced tumor-promoting pathway through the regulation of RKIP. Renal cancer cells were treated with CsA in absence or presence of honokiol. As shown in Fig. 2, CsA increased the phosphorylation of RKIP; and CsA-induced RKIP phosphorylation was significantly inhibited following honokiol treatment. Thus, we suggest that honokiol may down-regulate CsA-induced Raf/MEK signaling in renal cancer cells through inhibition of RKIP phosphorylation.
FIGURE 2. Honokiol reduces CsA-induced phosphorylation of RKIP.
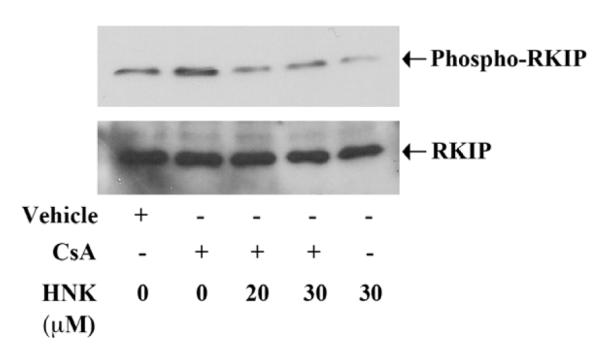
786-O cells were treated with different combinations of 5 μg/ml CsA, 20-30 μM honokiol (HNK) or vehicle alone for 6 h. The cells were lysed and the expression of phospho-RKIP and RKIP was analyzed by Western blot. Data shown are representative of three independent experiments.
3.3. Honokiol Treatment Down-Regulates CsA-induced and Ras-mediated Over-expression of HO-1
We have recently demonstrated that Ras-induced activation of the cytoprotective enzyme HO-1 can play a significant role in accelerating tumor-promoting pathways in renal cancer cells [13, 14]. We have also found (data not shown) that HO-1 can regulate VEGF expression in these cells. Here, we first wished to evaluate if CsA, which has been shown to promote renal cancer progression, could also induce HO-1. As demonstrated in Fig. 3A (top panel), CsA treatment markedly induced the expression of HO-1 in renal cancer cells. However, when we knocked down either H-Ras or Raf-1, CsA-induced HO-1 over-expression was inhibited. These observations suggest that CsA can induce HO-1; and Ras and Raf-1 act as important intermediary signaling molecules in this pathway.
FIGURE 3. CsA-induced and Ras-mediated over-expression of HO-1 is inhibited by honokiol.

(a) Caki-1 cells were transfected with H-Ras siRNA (25 nM), Raf-1 siRNA (25 nM) or control siRNA. After 48 h of siRNA transfection, the cells were treated with 5 μg/ml CsA or vehicle for 24 h. The cells were lysed and the expression of HO-1, H-Ras, Raf-1 and β-actin was analyzed by Western blot. Data shown are representative of three independent experiments. (b) 786-O cells were transfected with HO-1 promoter-luciferase construct (0.5 μg), and then treated with 5 μg/ml CsA, 20 μM honokiol (HNK) or vehicle. The cells were harvested 48 h after transfection and fold change in luciferase activity was calculated as the relative luciferase counts from each group of cells compared to that of vehicle-treated control. Results are representative of three independent experiments. Columns, average of triplicate readings from two different samples; error bars, standard error of the mean (SEM). * p < 0.05. compared with vehicle-treated control, and ** p < 0.05 compared with CsA-treated but HNK-untreated cells. (c) Caki-1 cells were treated with different combinations of 1-5 μg/ml CsA, 10-20 μM honokiol (HNK) or vehicle for 24 h. The cells were lysed and the expression of HO-1 and β-actin was analyzed by Western blot. Data shown are representative of three independent experiments. (d) Caki-1 cells were treated with different combinations of 5 μg/ml CsA, 20 μM HNK or vehicle for 6 h. Nuclear fraction was isolated from the cells and DNA binding activity of Nrf2 present in the nuclear extract was analyzed using TransAM Nrf2 ELISA kit as described in the “Materials and Methods” section. Nrf2 consensus binding site oligo was used to compete Nrf2 binding in order to monitor specificity of the assay. Results are representative of three different experiments. Columns, average of triplicate readings from two different samples; error bars, SEM. * p < 0.05 compared with vehicle treated control, and ** p < 0.05 compared with CsA-treated but HNK-untreated cells.
We next wanted to determine if honokiol could down-regulate CsA-induced over-expression of HO-1. To assess HO-1 transcriptional activation, we transfected both 786-0 and Caki-1 cells with the HO-1 promoter-luciferase plasmid and then treated with CsA in absence or presence of honokiol, and then luciferase assay was performed. To check the protein expression, 786-0 and Caki-1 cells were treated with different combinations of CsA and honokiol, and then Western blot analysis was performed. We observed that CsA-induced HO-1 transcriptional activation as well as protein over-expression in both the cell types was significantly inhibited by honokiol treatment (Fig. 3B & 3C, and Supplementary Fig. 2A & 2B).
Nrf2 is one of the most important transcription factors for HO-1 expression and binds to ARE, located at the enhancer sites (E1 and E2) of the HO-1 promoter [31, 32]. We have recently demonstrated that the Ras-Raf pathway activates Nrf2 to promote the over-expression of HO-1 in renal cancer cells [13]. Here, we checked if CsA treatment in renal cancer cells could induce the binding of Nrf2 to the ARE site, and how honokiol may modulate this binding. Using Nrf2 DNA binding ELISA kit, we observed that CsA markedly increased the binding of activated Nrf2 to specific DNA sequence containing the ARE site of the HO-1 promoter; and the honokiol treatment significantly attenuated CsA-induced binding of Nrf2 to the specific sequence (Fig. 3D). Binding of Nrf2 to the DNA was confirmed by competition assay using Nrf2 binding site oligo. Together, our observations suggest that honokiol can play an important role in preventing CsA-induced and Ras-mediated over-expression of HO-1, probably through the regulation of the transcription factor Nrf2.
3.4. Honokiol Inhibits CsA-induced Proliferation of Renal Cancer Cells
In our previous studies, we have demonstrated that CsA treatment as well as HO-1 over-expression can promote increased proliferation of renal cancer cells [7, 14]. Here, we sought to determine the effect of honokiol on CsA-induced proliferation of 786-0 and Caki-1 cells. The cell were treated with CsA in absence or presence of honokiol, and subjected to cell proliferation assay. As shown in Fig. 4A and 4B, CsA increased the proliferation of renal cancer cells compared with controls; and the treatment with honokiol significantly decreased CsA-induced cell proliferation in both the cell types. Thus, we suggest that honokiol can prevent CsA-induced renal tumor progression through the inhibition of cancer cell proliferation.
FIGURE 4. Honokiol attenuates CsA-induced proliferation of renal cancer cells.
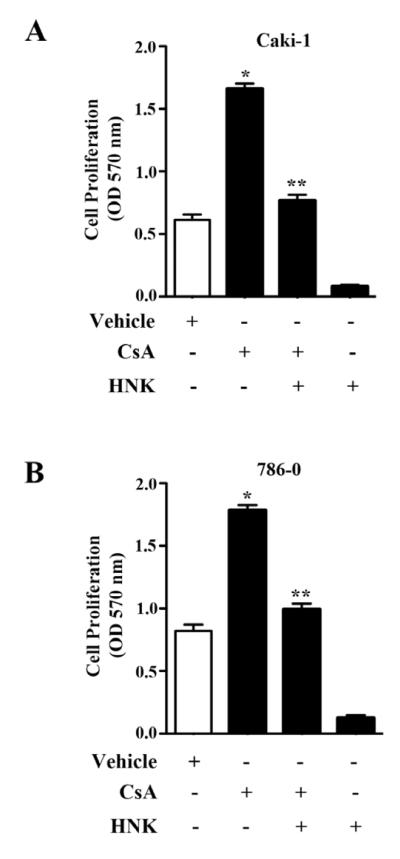
(a) Caki-1 and (b) 786-O cells were treated with different combinations of 5 μg/ml CsA, 20 μM honokiol (HNK) or vehicle alone for 48 h. Cell proliferation was measured by MTT assay. Results are representative of three independent experiments. Columns, average of triplicate readings from three different samples; error bars, SEM. * p < 0.05 compared with vehicle treated control, and ** p < 0.05 compared with CsA-treated but HNK-untreated cells..
3.5. Honokiol Treatment Prevents CsA-induced Down-regulation of Tumor Cell Apoptosis
CsA is known to inhibit cellular apoptosis [9, 33, 34]. Here, we first studied the effect of CsA treatment on apoptosis of renal cancer cells, and the role of Ras in the process. The cells were transfected with control siRNA or H-Ras siRNA, and then treated with either CsA or vehicle alone. Cells were stained with Annexin-V and propidium iodide and analyzed by flow cytometry to check the apoptotic index. As shown in Fig. 5A, CsA treatment decreased apoptosis in control siRNA-transfected cells compared with vehicle-treated controls; the percentage of apoptotic cells (early + late) decreased from 1.948% (0.678% + 1.27%) to 0.686% (0.562% + 0.124%). However, the knock-down of H-Ras markedly increased cellular apoptosis in CsA-treated cells; the percentage of apoptotic cells increased from 0.686% (control siRNA-transfected and CsA-treated cells) to 1.738% (0.718% + 1.02%) (H-Ras siRNA-transfected and CsA-treated cells). Thus, our findings suggest that CsA treatment inhibits renal cancer cell apoptosis through the induction of Ras.
FIGURE 5. Honokiol inhibits CsA-induced and Ras-mediated survival of renal cancer cells.

(a) Caki-1 cells were transfected with 50 nM H-Ras siRNA or control siRNA, and treated with 10 μg/ml CsA or vehicle for 72 h. (b) Caki-1 cells were treated with different combinations of 10 μg/ml CsA, 20 μM honokiol (HNK) or vehicle alone for 72 h. In (a) and (b), apoptotic index of the cells was determined by Annexin-V (APC) and propidium iodide staining. Data shown are representative of three independent experiments.
As our earlier experiments suggested that honokiol can inhibit Ras-mediated signaling events, we next tested its effect on renal cancer cell apoptosis following CsA treatment. As shown in Fig. 5B, CsA treatment decreased cellular apoptosis in Caki-1 cells compared with vehicle-treated control cells; the percentage of apoptotic cells (early + late) decreased from 2.255% (0.625% + 1.63%) to 0.624% (0.519% + 0.105%). Interestingly, honokiol reverted CsA-induced down-regulation of cellular apoptosis; percentage of apoptotic cells increased from 0.624% (CsA-treated cells) to 2.093% (0.543% + 1.55%) (CsA + honokiol- treated cells). We also checked (data not shown) the apoptotic effect of honokiol in 786-0 cells. However, the effect of honokiol to induce apoptosis in CsA-treated cells was much stronger in Caki-1 compared with 786-0 cells. Together, our observations suggest that honokiol may restrict CsA-induced survival of renal cancer cells through the induction of apoptosis.
3.6. Honokiol Promotes Apoptosis of Renal Cancer Cells through Inhibition of HO-1
We examined if honkiol-induced renal cancer cell apoptosis is mediated through the regulation of HO-1. As the apoptotic effect of honokiol was much stronger in Caki-1 cells, we performed these experiments in this cell type. The cells were treated with different combinations of honokiol and CoPP (HO-1 inducer), and then subjected to apoptosis assay as described earlier. As shown in Fig. 6A, honokiol treatment increased apoptosis (early + late) of renal cancer cells compared with vehicle-treated control; the percentage of total apoptotic cells increased from 2.95% (1.08% + 1.87%) to 7.54% (2.63% + 4.91%). However, following HO-1 induction by CoPP treatment, honokiol-induced cellular apoptosis was markedly decreased; the percentage of total apoptotic cells decreased from 7.54% to 2.618% (0.518% + 2.1%). The induction of HO-1 by CoPP treatment was confirmed by Western blot (bottom panel).
FIGURE 6. HO-1 modulates honokiol-induced apoptosis of renal cancer cells.

(a) Caki-1 cells were treated with different combinations of 20 μM CoPP, 20 μM honokiol (HNK) or vehicle for 72 h. (b) Caki-1 cells were transfected with 50 nM HO-1 siRNA or control siRNA, and treated with 20 μM HNK or vehicle for 72 h. In A and B (top panel), apoptotic index of the cells was determined by Annexin-V (APC) and propidium iodide staining. The over-expression of HO-1 in CoPP-treated cells and the knockdown of HO-1 in siRNA-transfected cells was confirmed by Western blot (a and b, bottom panel). Data shown are representative of three independent experiments.
We next checked the effect of HO-1 knock-down on honokiol-induced apoptosis of renal cancer cells. As shown in Fig. 6B, honokiol treatment increased cellular apoptosis (early + late) in control siRNA-transfected renal cancer cells compared with vehicle-treated controls; the percentage of total apoptotic cells increased from 3.02% (1.03% + 1.99%) to 6.16% (2.01% + 4.15%). The knock-down of HO-1 markedly elevated cellular apoptosis in honokiol-treated cells; the percentage of total apoptotic cells increased from 6.16% to 18.02% (3.52% + 14.5%). The knock-down of HO-1 was confirmed by Western blot (bottom panel). Thus, our observations suggest that the down-regulation of HO-1 is one of the critical events for the regulation of renal cancer cell apoptosis following honokiol treatment.
4. DISCUSSION
Honokiol, a small molecular weight polyphenol, is a promising agent for its anti-tumor and anti-carcinogenic properties. In this study, we show that the natural product honokiol can effectively inhibit CsA-induced over-expression of the angiogenic cytokine VEGF and the cytoprotective enzyme HO-1 in renal cancer cells. It reduces CsA-induced cancer cell proliferation and inhibits H-Ras- and HO-1-mediated cell survival. Our findings clearly suggest the clinical significance of honokiol treatment in preventing CsA-induced post-transplantation cancer progression that depends on the activation of Ras, and over-expression of VEGF and HO-1.
The effect of CsA on post-transplantation therapies can be best described as a double-edged sword; while its immunosuppressive function is indispensable for preventing transplant rejection, its pro-tumorigenic property leads to the development and progression of post-transplantation malignancies [1-3]. As discussed earlier, CsA can induce cancer progression by directly promoting tumorigenic pathways [4-7]. Recent studies from our laboratory show that CsA promotes renal tumor growth through activation of the proto-oncogene ras and over-expression of VEGF [6, 7]. Thus, using inhibitors of these pathways in combination with CsA can be an ideal approach to attenuate the pro-tumorigenic effects of CsA without compromising its essential immunosuppressive functions. It has recently been suggested that honokiol, which has a great potential to inhibit tumor growth, may also down-regulate the Ras pathway [21, 26, 35]. A combination treatment with CsA and honokiol may be considered as a therapeutic strategy for preventing the development of post-transplantation renal cancer. This is supported by the fact that honokiol exhibits not only anti-cancer effects, but also immunosuppressive and anti-inflammatory properties [36]. Thus, along with blocking the cancer-promoting effects of CsA, honokiol might actually reduce the dose of CsA needed to prevent transplant rejection. Here, we show that honokiol can effectively inhibit CsA-induced transcriptional activation and over-expression of VEGF in renal cancer cells. It is known that renal tumors are highly vascular with markedly elevated expression of pro-angiogenic VEGF and VEGF receptor [37]. The inhibition of CsA-induced VEGF expression by honokiol indicates possible potency of the molecule to attenuate CsA-induced angiogenesis and vascularization that plays a crucial role in development of post-transplantation renal cancer.
The Ras-Raf-ERK pathway can regulate the proliferation, differentiation, survival of cancer cells, and thus may serve as potential therapeutic target [38]. RKIP inhibits the Raf/MEK/ERK pathway by disrupting their interaction and blocking downstream signaling [29, 30]. It has been shown that phosphorylation of RKIP by protein kinase C (PKC) releases the molecule from Raf, thus rescuing the Raf/MEK/ERK pathway from inhibitory effects of RKIP [39]. It would be of interest to know whether RKIP is phosphorylated in cancers arising in patients treated with CsA. Our data shows a significant decrease in CsA-induced RKIP phosphorylation and renal cancer cell proliferation upon honokiol treatment. Considering the central role of PKC-mediated VEGF activation in CsA-induced post-transplantation cancer progression [6], it may be possible that honokiol downregulates proliferation of renal cancer cells through the inhibition of PKC-mediated regulation of the Raf/MEK/ERK signaling axis.
Honokiol may be used as an important therapeutic molecule to inhibit tumor angiogenesis and promote apoptosis [21, 26, 35]. Our present study for the first time shows that honokiol can promote apoptosis of renal cancer cells through the regulation of HO-1. Classically, HO-1 is a cytoprotective enzyme under oxidative and cellular stresses [40]; however, HO-1 is often over-expressed in cancer cells to promote tumor growth and progression [41, 42]. We have found (data not shown) that HO-1 can also induce the expression of angiogenic VEGF in renal cancer cells. Recent studies from our laboratory show that HO-1 is induced in renal cancer cells through the activation of the Ras-Raf-ERK signaling pathway involving the transcription factor Nrf2 [13]; and HO-1 can promote cell survival through the inhibition of chemotherapeutic drug-mediated apoptosis and autophagy [14]. Our present study shows a marked up-regulation of HO-1 by CsA via the Ras-Raf pathway; and CsA-induced HO-1 expression is attenuated by honokiol treatment. Honokiol also suppressed CsA-induced activation of Nrf2, which is crucial for Ras-mediated induction of HO-1. Moreover, honokiol markedly inhibited CsA-induced survival of renal cancer cells; and this effect was also mediated through the modulation of HO-1, as honokiol-induced apoptosis was significantly blocked by HO-1. However, we have observed (data not shown) no significant change in renal cancer cell autophagy following CsA and honokiol treatment. Together, honokiol can effectively inhibit CsA-induced and Ras-medited survival of renal cancer cells by down-regulating HO-1 and inducing apoptosis.
In summary, honokiol can inhibit CsA-induced survival of renal cancer cells by targeting the Ras pathway, and suppressing the induction of VEGF and HO-1. As renal cancer is a significant problem in CNI-treated transplant patients, our study clearly suggests the effectiveness of a combination therapy using CsA and honokiol as a strategy to reduce the occurrence of post-transplantation renal cancer. Addition of honokiol to CNI treatment can attenuate Ras-induced cancer-promoting pathways of CsA, without affecting its required immunosuppressive functions.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grants R01 CA131145 (to S. P.) and R01 AR047901 (to J. L. A.); and a Fellowship Grant from American Society of Transplantation (to P. B.).
Footnotes
CONFLICT OF INTEREST STATEMENT: Emory University has filed for a patent on honokiol, and Dr. Arbiser is listed as the inventor. Other authors do not have any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Kasiske BL, Snyder JJ, Gilbertson DT, Wang C. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905–913. doi: 10.1111/j.1600-6143.2004.00450.x. [DOI] [PubMed] [Google Scholar]
- [2].Bustami RT, Ojo AO, Wolfe RA, Merion RM, Bennett WM, McDiarmid SV, Leichtman AB, Held PJ, Port FK. Immunosuppression and the risk of post-transplant malignancy among cadaveric first kidney transplant recipients. Am J Transplant. 2004;4:87–93. doi: 10.1046/j.1600-6135.2003.00274.x. [DOI] [PubMed] [Google Scholar]
- [3].Wimmer CD, Rentsch M, Crispin A, Illner WD, Arbogast H, Graeb C, Jauch KW, Guba M. The janus face of immunosuppression - de novo malignancy after renal transplantation: the experience of the Transplantation Center Munich. Kidney Int. 2007;71:1271–1278. doi: 10.1038/sj.ki.5002154. [DOI] [PubMed] [Google Scholar]
- [4].Hojo M, Morimoto T, Maluccio M, Asano T, Morimoto K, Lagman M, Shimbo T, Suthanthiran M. Cyclosporine induces cancer progression by a cell-autonomous mechanism. Nature. 1999;397:530–534. doi: 10.1038/17401. [DOI] [PubMed] [Google Scholar]
- [5].Guba M, Graeb C, Jauch KW, Geissler EK. Pro- and anti-cancer effects of immunosuppressive agents used in organ transplantation. Transplantation. 2004;77:1777–1782. doi: 10.1097/01.tp.0000120181.89206.54. [DOI] [PubMed] [Google Scholar]
- [6].Basu A, Contreras AG, Datta D, Flynn E, Zeng L, Cohen HT, Briscoe DM, Pal S. Overexpression of vascular endothelial growth factor and the development of post-transplantation cancer. Cancer Res. 2008;68:5689–5698. doi: 10.1158/0008-5472.CAN-07-6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Datta D, Contreras AG, Basu A, Dormond O, Flynn E, Briscoe DM, Pal S. Calcineurin inhibitors activate the proto-oncogene Ras and promote protumorigenic signals in renal cancer cells. Cancer Res. 2009;69:8902–8909. doi: 10.1158/0008-5472.CAN-09-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu J, Farmer JD, Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- [9].Yarosh DB, Pena AV, Nay SL, Canning MT, Brown DA. Calcineurin inhibitors decrease DNA repair and apoptosis in human keratinocytes following ultraviolet B irradiation. J Invest Dermatol. 2005;125:1020–1025. doi: 10.1111/j.0022-202X.2005.23858.x. [DOI] [PubMed] [Google Scholar]
- [10].Basu A, Banerjee P, Contreras AG, Flynn E, Pal S. Calcineurin inhibitor-induced and Ras-mediated overexpression of VEGF in renal cancer cells involves mTOR through the regulation of PRAS40. PLoS One. 2011;6:e23919. doi: 10.1371/journal.pone.0023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crespo P, Xu N, Simonds WF, Gutkind JS. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- [12].Oka H, Chatani Y, Hoshino R, Ogawa O, Kakehi Y, Terachi T, Okada Y, Kawaichi M, Kohno M, Yoshida O. Constitutive activation of mitogen-activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res. 1995;55:4182–4187. [PubMed] [Google Scholar]
- [13].Banerjee P, Basu A, Datta D, Gasser M, Waaga-Gasser AM, Pal S. The heme oxygenase-1 protein is overexpressed in human renal cancer cells following activation of the Ras-Raf-ERK pathway and mediates anti-apoptotic signal. J Biol Chem. 2011;286:33580–33590. doi: 10.1074/jbc.M111.248401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Banerjee P, Basu A, Wegiel B, Otterbein LE, Mizumura K, Gasser M, Waaga-Gasser AM, Choi AM, Pal S. Heme Oxygenase-1 Promotes Survival of Renal Cancer Cells through Modulation of Apoptosis- and Autophagy-regulating Molecules. J Biol Chem. 2012;287:32113–32123. doi: 10.1074/jbc.M112.393140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Costa LJ, Drabkin HA. Renal cell carcinoma: new developments in molecular biology and potential for targeted therapies. Oncologist. 2007;12:1404–1415. doi: 10.1634/theoncologist.12-12-1404. [DOI] [PubMed] [Google Scholar]
- [17].Fujita M, Itokawa H, Sashida Y. Studies on the components of Magnolia obovata Thunb. 3. Occurrence of magnolol and honokiol in M. obovata and other allied plants. Yakugaku Zasshi. 1973;93:429–434. doi: 10.1248/yakushi1947.93.4_429. [DOI] [PubMed] [Google Scholar]
- [18].Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. The anti-inflammatory effect of honokiol on neutrophils: mechanisms in the inhibition of reactive oxygen species production. Eur J Pharmacol. 2003;475:19–27. doi: 10.1016/s0014-2999(03)02121-6. [DOI] [PubMed] [Google Scholar]
- [19].Hibasami H, Achiwa Y, Katsuzaki H, Imai K, Yoshioka K, Nakanishi K, Ishii Y, Hasegawa M, Komiya T. Honokiol induces apoptosis in human lymphoid leukemia Molt 4B cells. Int J Mol Med. 1998;2:671–673. doi: 10.3892/ijmm.2.6.671. [DOI] [PubMed] [Google Scholar]
- [20].Konoshima T, Kozuka M, Tokuda H, Nishino H, Iwashima A, Haruna M, Ito K, Tanabe M. Studies on inhibitors of skin tumor promotion, IX. Neolignans from Magnolia officinalis. J Nat Prod. 1991;54:816–822. doi: 10.1021/np50075a010. [DOI] [PubMed] [Google Scholar]
- [21].Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11:1139–1148. doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- [23].Hahm ER, Arlotti JA, Marynowski SW, Singh SV. Honokiol, a constituent of oriental medicinal herb magnolia officinalis, inhibits growth of PC-3 xenografts in vivo in association with apoptosis induction. Clin Cancer Res. 2008;14:1248–1257. doi: 10.1158/1078-0432.CCR-07-1926. [DOI] [PubMed] [Google Scholar]
- [24].Battle TE, Arbiser J, Frank DA. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 2005;106:690–697. doi: 10.1182/blood-2004-11-4273. [DOI] [PubMed] [Google Scholar]
- [25].Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, Podar K, Le Gouill S, Chauhan D, Tamura K, Arbiser J, Anderson KC. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106:1794–1800. doi: 10.1182/blood-2005-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, Arbiser JL. Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin Cancer Res. 2008;14:4267–4274. doi: 10.1158/1078-0432.CCR-08-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, Luo J, Hu X. Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 2007;67:4894–4903. doi: 10.1158/0008-5472.CAN-06-3818. [DOI] [PubMed] [Google Scholar]
- [28].Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- [29].Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- [30].Hagan S, Garcia R, Dhillon A, Kolch W. Raf kinase inhibitor protein regulation of raf and MAPK signaling. Methods Enzymol. 2006;407:248–259. doi: 10.1016/S0076-6879(05)07021-7. [DOI] [PubMed] [Google Scholar]
- [31].Ryter SW, Choi AM. Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal. 2002;4:625–632. doi: 10.1089/15230860260220120. [DOI] [PubMed] [Google Scholar]
- [32].Kivela AM, Kansanen E, Jyrkkanen HK, Nurmi T, Yla-Herttuala S, Levonen AL. Enterolactone induces heme oxygenase-1 expression through nuclear factor-E2-related factor 2 activation in endothelial cells. J Nutr. 2008;138:1263–1268. doi: 10.1093/jn/138.7.1263. [DOI] [PubMed] [Google Scholar]
- [33].Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation. 1998;98:1153–1157. doi: 10.1161/01.cir.98.12.1153. [DOI] [PubMed] [Google Scholar]
- [34].Pritchard DE, Singh J, Carlisle DL, Patierno SR. Cyclosporin A inhibits chromium(VI)-induced apoptosis and mitochondrial cytochrome c release and restores clonogenic survival in CHO cells. Carcinogenesis. 2000;21:2027–2033. doi: 10.1093/carcin/21.11.2027. [DOI] [PubMed] [Google Scholar]
- [35].Yu C, Zhang Q, Zhang HY, Zhang X, Huo X, Cheng E, Wang DH, Arbiser JL, Spechler SJ, Souza RF. Targeting the intrinsic inflammatory pathway: honokiol exerts proapoptotic effects through STAT3 inhibition in transformed Barrett’s cells. Am J Physiol Gastrointest Liver Physiol. 2012;303:G561–569. doi: 10.1152/ajpgi.00033.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Munroe ME, Arbiser JL, Bishop GA. Honokiol, a natural plant product, inhibits inflammatory signals and alleviates inflammatory arthritis. J Immunol. 2007;179:753–763. doi: 10.4049/jimmunol.179.2.753. [DOI] [PubMed] [Google Scholar]
- [37].Finley DS, Pantuck AJ, Belldegrun AS. Tumor biology and prognostic factors in renal cell carcinoma. Oncologist. 2011;16(Suppl 2):4–13. doi: 10.1634/theoncologist.2011-S2-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, Kolch W. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- [40].Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- [41].Alaoui-Jamali MA, Bismar TA, Gupta A, Szarek WA, Su J, Song W, Xu Y, Xu B, Liu G, Vlahakis JZ, Roman G, Jiao J, Schipper HM. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009;69:8017–8024. doi: 10.1158/0008-5472.CAN-09-0419. [DOI] [PubMed] [Google Scholar]
- [42].Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal. 2007;9:2099–2117. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.