

Abstract
Expression of the pro-inflammatory and pro-angiogenic chemokine interleukin-8 (IL-8), which is regulated at the transcriptional level by NFκB, is constitutively increased in the androgen independent metastatic prostate cancer and correlates with poor prognosis. Inhibition of NFκB-dependent transcription was used as an anti-cancer strategy for the development of the first clinically approved 26S proteasome inhibitor, bortezomib (BZ). Even though BZ has shown remarkable anti-tumor activity in hematological malignancies, it has been less effective in prostate cancer and other solid tumors; however, the mechanisms have not been fully understood. Here we report that the proteasome inhibition by BZ unexpectedly increases the IL-8 expression in androgen independent prostate cancer PC3 and DU145 cells, while expression of other NFκB-regulated genes is inhibited or unchanged. The BZ-increased IL-8 expression is associated with increased in vitro p65 NFκB DNA binding activity and p65 recruitment to the endogenous IL-8 promoter. In addition, proteasome inhibition induces a nuclear accumulation of IKKα and inhibition of IKKα enzymatic activity significantly attenuates the BZ-induced p65 recruitment to IL-8 promoter and IL-8 expression, demonstrating that the induced IL-8 expression is mediated, at least partly, by IKKα. Together, these data provide the first evidence for the gene specific increase of IL-8 expression by the proteasome inhibition in prostate cancer cells and suggest that targeting both IKKα and the proteasome may increase the BZ effectiveness in androgen independent prostate cancer treatment.
Keywords: Interleukin-8, IκB kinase, proteasome inhibition, NFκB, prostate cancer, bortezomib
Introduction
Interleukin-8 (IL-8), originally discovered as the neutrophil chemoattractant and inducer of leukocyte-mediated inflammation, contributes to cancer progression through its induction of tumor cell proliferation, survival, and migration (1, 2). In addition, tumor-derived IL-8 activates endothelial cells to promote angiogenesis, induces neutrophil recruitment, and activates neutrophils and the tumor-associated macrophages to release more IL-8, which further amplifies the pro-survival, pro-angiogenic and metastatic effect. IL-8 expression is increased in many types of advanced cancers, including the metastatic androgen independent prostate cancer, and correlates with poor prognosis (2–4).
Prostate cancer is the third most common cause of death from cancer in men. It proceeds from a localized, curable, androgen dependent disease to an invasive, metastatic, and always fatal, androgen independent prostate cancer. One of the critical factors regulating progression to the metastatic androgen refractory prostate cancer is the increased activity of NFκB, which induces synthesis of anti-apoptotic and pro-inflammatory genes, including the IL-8 (5–8). NFκB activity and IL-8 levels are increased in metastatic prostate cancer cells and in patients with hormone refractory prostate cancer, where they promote angiogenesis, tumor growth and metastasis (9–15).
In the canonical NFκB pathway, NFκB p65/p50 dimers are retained in the cytoplasm in an inactive form through their interaction with IκBα. Following cell stimulation with pro-inflammatory signals and other forms of cellular stress, IκBα is phosphorylated through a cascade of inducible protein kinases that involve the enzymes of IκB kinase (IKK) complex, ubiquitinated, and selectively degraded by the 26S proteasome. The proteasomal degradation of IκBα releases NFκB dimers to translocate to the nucleus and stimulate transcription of NFκB-dependent pro-inflammatory and anti-apoptotic genes (16, 17). The inhibition of NFκB-dependent transcription was used as an anti-cancer strategy for the development of the first clinically approved 26S proteasome inhibitor, bortezomib (18–22). Even though bortezomib (BZ, Velcade, PS-341) has shown remarkable anti-tumor activity in multiple myeloma and other hematological malignancies (18–22), it has been less effective in solid tumors (23–30); however, the mechanisms have not been fully understood.
We have previously shown that the proteasome inhibition induces nuclear accumulation of IκBα that has a gene specific effect on the regulation of NFκB-dependent genes, depending on the subunit composition of NFκB dimers (31–33). In this study, we have investigated the mechanism of how the proteasome inhibition by BZ regulates NFκB-dependent transcription in the androgen independent prostate cancer cells. Unexpectedly, we found that the proteasome inhibition significantly increased expression of IL-8, while expression of other NFκB-regulated genes was inhibited or unchanged. The BZ-increased IL-8 expression was associated with increased NFκB p65 DNA binding activity and p65 recruitment to the IL-8 promoter. In addition, the proteasome inhibition induced a nuclear accumulation of IKKα, and suppression of IKKα attenuated the BZ-increased p65 recruitment and IL-8 expression, demonstrating the IKKα requirement for the BZ-increased IL-8 expression in metastatic prostate cancer cells. These data provide the first evidence for the gene specific increase of IL-8 expression by the proteasome inhibition in prostate cancer cells, and suggest that the BZ-increased IL-8 expression may represent one of the underlying mechanisms responsible for the decreased effectiveness of bortezomib in androgen independent prostate cancer treatment.
Materials and Methods
Antibodies and reagents
Purified polyclonal antibodies against human NFκB p65 (sc-372), NFκB p50 (sc-7178), IκBα (sc-371), IKKα (sc -7218), IKKβ (sc-8014), IKKε/i (sc-376114), phosphorylated p65 at S536 (sc-33020), and lamin B (sc-6216) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified polyclonal antibody against lactate dehydrogenase (LDH; 20-LG22) was from Fitzgerald Industries International (Concord, MA, USA), and actin antibody was from Sigma (St Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-mouse and anti-goat secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Bortezomib was from ChemieTek (Indianapolis, IN, USA). The IKK inhibitors Bay-117082 and PS-1145 were purchased from Sigma (St Louis, MO, USA). All other reagents were molecular biology grade and were from Sigma (St Louis, MO).
Cell culture
All cell lines were obtained from American Type Culture Collection (ATCC; Rockville, MD, USA). PC3 cells were cultured in Ham’s F12K medium (ATCC; Rockville, MD, USA) supplemented with 2 mM L-glutamine, 1% penicillin and streptomycin, and 10% fetal bovine serum (FBS; Invitrogen, Grand Island, NY, USA) as described (31). DU145 cells were grown in Eagle’s Minimum Essential medium supplemented with FBS and antibiotics. Hut-78 cells, HeLa cells and U-937 cells were grown in RPMI 1640 medium with FBS and antibiotics as described (31–34). Prior to cell treatment, cells were seeded (106 cells/ml) for 24 hours in 6-well plates and grown at 37°C with 5% CO2. Bortezomib and the IKK inhibitors Bay-117082 and PS-1145 were dissolved in DMSO and stored at −80°C. An equivalent volume of DMSO was used in all experiments as a solvent control. Cell viability was measured by using Trypan Blue exclusion.
Western analysis of cytoplasmic and nuclear extracts
Nuclear (NE) and cytoplasmic extracts (CE) were prepared as described previously (31–33). Contamination of nuclear and cytoplasmic fractions by cytoplasmic and nuclear proteins, respectively, was determined by western analysis using LDH and lamin B as specific markers as described (31). Denatured proteins were separated on 12% (IκBα, p50, p65) or 10% (IKK) denaturing polyacrylamide gels, and immunoblotting analysis was performed as described (31).
Proteasome activity assay
Activity of the 20S catalytic proteasomal core unit was measured in whole cell extracts by using the 20S Proteasome Activity Assay Kit (Chemicon, APT280; Temecula, CA, USA) as described by the manufacturer. Briefly, cells were lysed in a lysis buffer (50 mM Hepes, pH 7.5; 5 mM EDTA; 150 mM NaCl; 1% Triton X-100) for 30 minutes on ice, and the lysates were collected by centrifugation (10,000g, 15 min, 4°C). The cell lysates were incubated (2h, 37°C) with a labeled substrate, LLVY-7-amino-4-methylcoumarin, and the cleavage activity was monitored by detection of the free fluorophore 7-amino-4-methylcoumarin using a fluorescence plate reader (Berthold Mithras LB940, Berthold Technologies, USA) at 355/460 nm.
NFκB DNA binding activity assay
NFκB p65 DNA binding activity was measured in nuclear extracts of PC3 cells by using the TransAM NFκB p65 assay kit (Active Motif, 40096; Carlsbad, CA, USA), which measures the amount of p65 NFκB bound to the NFκB consensus GGGACTTTCC oligonucleotide. The assay was performed according to the manufacturer’s instruction. Absorbance was measured at 450 nm using a microplate reader (Biorad 680; Hercules, CA, USA).
Real time PCR
Total RNA was isolated by using RNeasy mini-kit (Qiagen, Valencia, CA, USA). The iScript one-step RT-PCR kit with SYBR Green (BioRad, Hercules, CA, USA) was used as a supermix and 100 ng of RNA was used as template on a Bio-Rad MyIQSingle Color Real-Time PCR Detection System (Hercules, CA, USA). The primers used for quantification of cIAP-1, cIAP-2, Bcl-2, Bcl-3, IL-8 and control actin mRNA were purchased from SA Biosciences (Frederick, MD, USA).
Transfection with siRNA
Human IKKα (sc- 29365), IKKβ (sc-35644), IKKε/i RNA (sc-39056), IκBα (sc-29360) and non-silencing control (sc-37007) small interfering RNAs (siRNAs) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Prior to transfection, PC3 cells were seeded into a 6-well plate and incubated in a humidified 5% CO2 atmosphere at 37°C in antibiotic-free Ham’s F12K medium supplement with 10% FBS for 24 hours to 80% confluence. For each transfection, 60 μmol of either non-silencing siRNA-A control or specific siRNA (Santa Cruz Biotechnology, CA) were used. The cells were transfected for 6 hours in transfection medium with siRNA transfection reagent according to manufacturer’s instructions (Santa Cruz Biotechnology). After transfection, fresh Ham’s F12K medium supplemented with FBS and antibiotics was added and the cells were treated with BZ for 24 hours.
Chromatin immunoprecipitation (ChIP)
ChIP analyses were performed by using the protocol from Upstate Biotechnology Inc., (Billerica, MA, USA). Proteins and DNA were cross-linked by adding formaldehyde to the growth medium to a final concentration of 1% for 15 min at 37 °C and glycine was added at a final concentration of 0.125 M to neutralize formaldehyde. Cells were washed with PBS containing protease inhibitors and collected by centrifugation. Cells were then resuspended in SDS lysis buffer, incubated at 4 °C for 10 min, and sonicated. The lysates were centrifuged at 15,000 g for 10 min at 4 °C, and the supernatant extracts were diluted with ChIP dilution buffer and pre-cleared with Protein A/G Agarose (Santa Cruz Biotechnology) for 30 min at 4 °C. Immunoprecipitation was performed overnight at 4 °C, with p65 or p50 antibodies. Following immunoprecipitation, the samples were incubated with Protein A/G Agarose for 1 h, and the immune complexes were collected by centrifugation (150 g at 4 °C), washed, and eluted with 1% SDS–0.1 M NaHCO3. The cross-linking was reversed by heating with 5 M NaCl at 65 °C for 4 h. Proteins were digested with proteinase K, and the samples were extracted with phenol/chloroform, followed by precipitation with ethanol. The pellets were resuspended in nuclease-free water and subjected to real time PCR. Immunoprecipitated DNA was analyzed by real-time PCR (25 μl reaction mixture) using the iQ SYBR Green Supermix (BioRad, Hercules, CA, USA) and the Bio-Rad MyIQ Single Color Real-Time PCR Detection System as described (34). The occupancy was calculated by using the ChIP-qPCR Human IGX1A Negative Control Assay (GPH100001C(−)01A; SA Biosciences, Frederick, MD, USA) as a negative control and corrected for the efficiency of the primers, which detect specific genomic DNA sequences within ORF-free intergenic regions or “promoter deserts” lacking any known or predicted structural genes. The primers used for real time PCR were the following: cIAP-1: forward, 5′-TGACTGGCAGGCAGAAATGA-3′ and reverse, 5′-TTTGCCCGTTGAATCCGAT-3′; cIAP-2: forward, 5′-TTCAGTAAATGCCGCGAAGAT-3′ and reverse, 5′-TGGTTT-GCATGTGCACTGGT-3′ Bcl-2: forward, 5′-TGCATCTCATGCCAAGGG-3′ and reverse, 5′-CCCCAGAGAAAGAAGAGGAGTT-3′; Bcl-3: forward, 5′-TTGCGGAGAGAAA-CACCTACT-3′ and reverse, 5′-CGCTCTCTCTGCCTCTGTT-3′; and IL-8: forward, 5′-GGGCCATCATCAGTTGCAAATC-3′ and reverse, 5′-GCTTGTGTGCTCTGCTGTCTC-3′. The NFκB promoter sequences of the above genes are shown in Table 1.
Table 1.
NFκB binding sites in the NFκB-regulated promoters
| Gene | NFκB site location | NFκB site sequence |
|---|---|---|
| NFκB consensus oligonucleotide used in ELISA assay | N/A | GGGACTTTCC |
| IL-8 | −82 | GGAATTTCCT |
| Bcl-3 | −293 | GGTGGGGACA |
| cIAP-1 | −1153 | GGAATTCCCC |
| cIAP-2 | −174 | GGAAATCCCC |
| Bcl-2 | −161 | GGGAAACACC |
ELISA
The IL-8 release was measured in cell culture supernatants by commercially available ELISA kit (R&D, Minneapolis, MN, USA) as previously described (32).
Statistical analysis
The results represent at least three independent experiments. Numerical results are presented as means ± SE. Data were analyzed by using an InStat software package (GraphPAD, San Diego, CA, USA). Statistical significance was evaluated by using Mann-Whitney U test with Bonferroni correction for multiple comparisons, and p<0.05 was considered significant.
Results
Proteasome inhibition by BZ increases NFκB p65 nuclear accumulation and DNA binding activity in androgen independent prostate cancer PC3 cells
The androgen independent prostate cancer cells are characterized by high levels of constitutive NFκB p65/p50 DNA binding and proteasome activity (35–38). To investigate the mechanism of how the proteasome inhibition regulates NFκB-dependent transcription in prostate cancer cells, we first analyzed the nuclear-cytoplasmic distribution of NFκB p65 and p50 subunits in PC3 cells cultured 24h in the presence of increasing concentrations of BZ. Unexpectedly, we found that BZ increased the nuclear levels of p65; the increased nuclear accumulation of p65 was most pronounced at 0.1 and 1 μM BZ concentrations (Fig. 1A). In contrast, BZ did not significantly increase the nuclear levels of p50. The highest BZ concentration (10 μM) was associated with a decreased p50 expression, both in the cytoplasm and in the nucleus (Fig. 1A). In addition, BZ induced IκBα translocation from the cytoplasm to the nucleus; this was consistent with our previous studies showing that the proteasome inhibition induces IκBα nuclear translocation and accumulation in prostate cancer and leukemia cells (31, 33). BZ at concentrations up to 10 μM did not have any effect on the actin levels used as a loading control, or lactate dehydrogenase (LDH) and lamin B, which were used as specific cytoplasmic and nuclear markers, respectively.
Figure 1. Proteasome inhibition by BZ induces nuclear accumulation of p65 NFκB in prostate cancer PC3 cells.

(A) Western blotting of cytoplasmic (CE) and nuclear extracts (NE) prepared from PC3 cells treated with increasing concentrations of BZ for 24 hours, and analyzed by using p65, p50 and IκBα antibodies. The presence of cytoplasmic proteins in nuclear fraction was evaluated by re-probing the membrane with lactate dehydrogenase (LDH) antibody. Nuclear contamination in the cytoplasmic fraction was assessed by using lamin B specific antibody. To confirm equal protein loading, the membranes were stripped and re-probed with actin antibody. Each lane corresponds to approximately 5×104 cells. (B) The proteasome activity was measured in whole cell extracts prepared from PC3 cells treated with increasing BZ concentrations for 24 hours. The activity is expressed as relative fluorescence units (RFU) of BZ-treated cells compared to untreated (UT) cells. (C) Cell viability and total cell count of PC3 cells incubated 24 hours with increasing BZ concentrations were measured by using Trypan Blue exclusion. The data are expressed as the percentage compared to untreated (UT) cells. The values represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) inhibition compared to control untreated cells.
To confirm that the 0.1 μM BZ concentration, which induced the maximal nuclear accumulation of p65 (Fig. 1A) also inhibits proteasome activity in PC3 cells, we measured activity of the 26S proteasome in whole cell extracts prepared from cells treated 24h with increasing BZ concentrations. As shown in Fig. 1B, 0.1 μM BZ, which approximately corresponds to the clinically used BZ concentrations in cancer patients (39), inhibited about 90% of the original proteasome activity. In contrast, the 0.1 μM BZ concentration reduced the PC3 cell viability and total cell count only by about 5% and 15%, respectively (Fig. 1C), which is consistent with the relative resistance of metastatic prostate cancer cells to BZ (26).
To determine whether the increased nuclear p65 levels resulted in an increased p65 NFκB DNA binding activity, we analyzed the in vitro p65 DNA binding activity in nuclear extracts prepared from PC3 cells incubated 24 hours with increasing concentrations of BZ. As shown in Fig. 2A, BZ significantly increased the p65 DNA binding activity measured by TransAM assay, which measures the amount of p65 NFκB bound to the NFκB consensus GGGACTTTCC oligonucleotide. Cells treated with 0.1 and 1 μM BZ exhibited three times higher p65 DNA binding activity compared to untreated cells. Fig. 2B demonstrates specificity of p65 DNA binding for the NFκB binding site, since the mutated oligonucleotide did not exhibit any p65 binding. Even though the increased p65 DNA binding activity induced by proteasome inhibition was surprising, since the proteasome inhibition suppresses NFκB activity in most tumor cells (19–21), it correlated well with the BZ-increased p65 nuclear levels in PC3 cells (Fig. 1A).
Figure 2. Proteasome inhibition by BZ increases p65 NFκB DNA binding activity in PC3 cells.

(A) NFκB p65 DNA binding activity was measured in nuclear extract prepared from PC3 cells treated with increasing concentrations of BZ for 24 hours. (B) Specificity analysis of the constitutive p65 NFκB DNA binding activity in PC3 cells, measured in nuclear extracts of untreated (UT) cells in the absence and presence of mutant (mut) or wild type (WT) oligonucleotides. The values represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) inhibition compared to control untreated (UT) cells.
Proteasome inhibition by BZ significantly increases IL-8 expression in metastatic prostate cancer cells while it decreases or does not affect expression of other NFκB-dependent genes
To determine whether the increased p65 nuclear levels and DNA binding activity correlate with the expression of NFκB-dependent genes, we analyzed mRNA levels of the regulatory gene belonging to the IκB family, Bcl-3, the anti-apoptotic genes Bcl-2, cIAP-1 and cIAP-2, and IL-8 in PC3 cells treated with increasing concentrations of BZ. As shown in Fig. 3A, expression of Bcl-3, cIAP-1, and cIAP-2 was suppressed, and Bcl-2 was unchanged. This is in an agreement with previous studies demonstrating that the proteasome inhibition suppresses most NFκB-dependent genes, while it does not affect Bcl-2 expression (31, 33). Remarkably however, proteasome inhibition significantly increased the IL-8 expression and protein release in PC3 cells (Figs. 3B–D). Compared to untreated PC3 cells, in cells incubated 24h with 0.1 and 1 μM BZ, the IL-8 mRNA levels increased almost 30- and 50-fold, respectively (Fig. 3B). However, it is important to note that even in the absence of BZ, PC3 cells expressed a considerable amount of IL-8 mRNA, which is consistent with the IL-8 release from untreated cells (Fig. 3D). To better demonstrate the level of IL-8 mRNA expression in PC3 cells, we compared IL-8 mRNA levels in different untreated cell lines: HeLa cells, used as a reference, two metastatic prostate cancer cell lines PC3 and DU145 cells, and two leukemia cell lines, U937 and Hut-78 cells (Fig. 3E). Compared to HeLa cells, untreated PC3 and DU145 cells expressed significantly more IL-8 mRNA; the highly metastatic prostate cancer PC3 cells had approximately 25-fold higher IL-8 mRNA levels than HeLa cells, and the moderately metastatic prostate cancer DU145 cells had about 3-times more IL-8 mRNA (Fig. 3E). In contrast, untreated leukemia U937 and Hut-78 cells had very low IL-8 mRNA levels. However, the control Bcl-2 mRNA levels were comparable in all cell lines tested, and cIAP-1 mRNA levels were comparable between PC3 and Hut-78 cells, and DU145 and HeLa cells (Fig. 3E). These results demonstrate that the metastatic prostate cancer cells express significantly more IL-8 than other cancer and leukemia cells, and that the IL- 8 mRNA levels in prostate cancer cells correlate with their metastatic potential, as was previously reported (3, 4).
Figure 3. Proteasome inhibition by BZ increases IL-8 expression in PC3 cells, while it inhibits expression of other NFκB-dependent genes.

(A) Real time RT-PCR analysis of mRNA levels of Bcl-2, Bcl-3, cIAP-1 and cIAP-2 measured in PC3 cells treated with increasingBZ concentrations for 24 hours. (B) Real time RT-PCR of IL-8 mRNA levels in PC3 cells treated 24 hours with increasing BZ concentrations. (C) Real time RT-PCR of IL-8 mRNA levels in PC3 cells treated with 1 μM BZ for 0, 6, 24 and 48 hours. (D) IL-8 protein release measured by ELISA in cell culture supernatants from PC3 cells treated 24 hours with increasing BZ concentrations. The values represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) change compared to control untreated (UT) cells. (E) Real time RT-PCR analysis of mRNA levels of IL-8, Bcl-2 and cIAP-1 in untreated HeLa, PC3, DU145, Hut-78 and U937 cells. The data are expressed as percentage of mRNA compared to HeLa cells, which was considered 100%; the values represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) change compared to HeLa cells.
To determine whether proteasome inhibition increases the IL-8 expression also in the moderately metastatic, androgen-independent prostate cancer DU145 cells, we analyzed IL-8 mRNA expression and protein release in DU145 cells treated 24h with increasing BZ concentrations. As shown in Fig. 4, both IL-8 mRNA levels (Fig. 4A) and protein release (Fig. 4B) were significantly increased in BZ-treated DU145 cells; 0.1 μM BZ increased IL-8 mRNA expression approximately 20-fold compared to untreated cells, and protein release about 5-fold. These data demonstrate that the increased IL-8 expression induced by proteasome inhibition is not unique to the highly metastatic PC3 cells, but is induced in other metastatic prostate cancer cells as well.
Figure 4. BZ increases IL-8 expression in androgen-independent prostate cancer DU145 ells.

(A) Real time RT-PCR of IL-8 mRNA levels in DU145 cells treated 24 hours with increasing BZ concentrations. (B) IL-8 protein release measured by ELISA in cell culture supernatants from DU145 cells treated 24 hours with increasing BZ concentrations. The values represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) change compared to control untreated (UT) cells.
Proteasome inhibition increases p65 but not p50 recruitment to the endogenous IL-8 promoter
Since proteasome inhibition increased the in vitro p65 DNA binding activity (Fig. 2), we analyzed whether it also increases p65 recruitment to the endogenous IL-8 promoter. Cells were incubated 24h with 0, 0.1, or 1 μM BZ, cross-linked with formaldehyde, lysed, and chromatin was sheared by sonication. NFκB p65 and p50 recruitment to IL-8, cIAP-1, cIAP-2, Bcl-2 and Bcl-3 promoters was analyzed by chromatin immunoprecipitation (ChIP) using p65 and p50 antibodies and quantified by real time PCR. The NFκB binding sites of the above genes are shown in Table 1. Fig. 5A illustrates the proximal NFκB binding site in human IL-8 promoter that was shown to be required for the IL-8 expression (40–42).
Figure 5. Proteasome inhibition by BZ increases p65 but not p50 recruitment to the endogenous IL-8 promoter in PC3 cells.

(A) Schematic illustration of the proximal NFκB binding site in human IL-8 promoter and the ChIP primers used in the assay. Recruitment of NFκB p65 (B) and p50 (C) subunits to NFκB-dependent promoters of IL-8, Bcl-2, Bcl-3, cIAP-1 and cIAP-2 genes in PC3 cells treated 24 hours with 0, 0.1 and 1 μM BZ was analyzed by ChIP and quantified by real time PCR. The data are presented as the change in occupancy over the human IGX1A (SA Biosciences) sequence control and represent the mean +/−SE of four experiments. Asterisks denote a statistically significant (p<0.05) change compared to control untreated cells.
The proteasome inhibition by BZ significantly increased p65 recruitment to IL-8 promoter (Fig. 5B). In PC3 cells treated 24h with 0.1 and 1 μM BZ, p65 occupancy at the IL-8 promoter increased approximately 6- and 9-fold compared to untreated cells, respectively. In contrast to p65, p50 recruitment to IL-8 promoter was not changed (Fig. 5C). The proteasome inhibition also increased p65 recruitment to cIAP-1 and cIAP-2 promoters even though the p65 occupancy at these promoters was significantly lower than on the IL-8 promoter (Fig. 5B). However, in contrast to IL-8, BZ increased p50 recruitment to Bcl-3, cIAP-1 and cIAP-2 promoters (Fig. 5C). Interestingly, the high p50 occupancy on Bcl-3 and cIAP-2 promoters (Fig. 5C) was associated with the maximum inhibition by BZ (Fig. 3A).
The BZ-increased IL-8 expression in PC3 cells is mediated by IKKα
Based on previous studies that indicated that the constitutively increased NFκB activity in metastatic prostate cancer cells is mediated by the increased IKK activity (9, 11; 43–46), we hypothesized that the proteasome inhibition increases IL-8 expression by an IKK-dependent mechanism. To this end, we first analyzed whether the proteasome inhibition increased the intracellular levels of IKKα, IKKβ or IKKε. PC3 cells were treated 24h with 0, 0.1 or 1 μM BZ, and the cytoplasmic and nuclear extracts were analyzed by western blotting followed by densitometry. As shown in Figs. 6A and B, IKKα and IKKβ were localized both in the cytoplasm and in the nucleus, and BZ further increased their nuclear accumulation. The increased nuclear accumulation of IKKα and IKKβ in response to proteasome inhibition is likely caused by inhibited proteasomal degradation of IKKα and IKKβ, since BZ did not increase the IKKα or IKKβ mRNA levels in PC3 cells (data not shown). This is supported by a previous study that indicated that IKKα and IKKβ are subject to the proteasomal degradation (47). In contrast to IKKα and IKKβ, IKKε was localized mainly in the nucleus in PC3 cells, and its levels did not change after the proteasome inhibition (Figs. 6A, B).
Figure 6. The BZ-increased IL-8 expression in PC3 cells is mediated by IKKα. (A).

Western analysis of CE and NE prepared from PC3 cells treated with 0, 0.1, and 1 μM BZ for 24 hours, and analyzed by using IKKα, IKKβ, IKKε, and control actin, LDH and lamin B antibodies. For IKKα, IKKβ, and the control proteins, each lane corresponds to approximately 5x104 cells. For IKKε, each lane corresponds to approximately 5x105 cells. (B) Densitometric evaluation of IKKα, IKKβ and IKKε levels in nuclear extracts of BZ-treated PC3 cells. The nuclear IKKα, IKKβ and IKKε bands were scanned and their densities were normalized to the densities of actin used as a loading control. The values for NE of untreated cells were arbitrarily set to 1, and the other values are presented relative to these values. The data represent the means of three experiments +/-SE. (C) Real time RT-PCR analysis of IL-8 mRNA levels in PC3 cells pre-treated 12 hours with control DMSO or the IKK inhibitors Bay-117082 (5 μM) and PS-1145 (20 μM) before 24 hour incubation with 0, 0.1, or 1 μM BZ. (D) Real time RT-PCR of IL-8 mRNA levels in DU145 cells pre-treated 12 hours with DMSO, Bay-117082 (5 μM) or PS-1145 (20 μM) before 24 hour incubation with 0, 0.1, or 1 μM BZ. (E) Real time RT-PCR analysis of IL-8 mRNA levels in PC3 cells transfected with control, IKKα, IKKβ IKKε or IκBα siRNA and then incubated 24 hours with 0 or 0.1 μM BZ. The values represent the mean +/−SE of four experiments. Asterisks denote a statistically significant (p<0.05) inhibition compared to cells pre-treated with control DMSO or transfected with control non-specific (NS) siRNA.
To determine whether IKK activity is required for the BZ-induced IL-8 expression in metastatic prostate cancer cells, we analyzed IL-8 expression in PC3 and DU145 cells treated with IKK inhibitors, Bay-117082 and PS-1145. Cells were pre-treated 12h either with 5 μM Bay-117082, a broad-spectrum IKK inhibitor (48, 49), or 20 μM PS-1145, IKKβ specific inhibitor (43), before 24h incubation with 0, 0.1 or 1 μM BZ. Both in PC3 cells (Fig. 6C) and in DU145 cells (Fig. 6D), the IKK inhibition by Bay-117082 resulted in a significantly reduced IL-8 expression in BZ-treated cells, while PS-1145 did not have any effect. These data indicated that the increased IL-8 expression induced by proteasome inhibition in metastatic prostate cancer cells is mediated by IKK, but not IKKβ.
To confirm the above data and determine which IKK isoform is responsible for the BZ-increased IL-8 expression, we used IKK suppression by siRNAs. PC3 cells were transfected with IKKα, IKKβ, IKKε or control non-silencing siRNA before 24h treatment with 0 or 0.1 μM BZ. In addition, to determine whether the BZ-induced nuclear IκBα regulates IL-8 expression in PC3 cells, cells were transfected also with IκBα siRNA. As shown in Fig. 6E, transfection using IKKα siRNA significantly suppressed the BZ-increased IL-8 expression. In contrast, transfection with IKKβ siRNAs did not have any effect on the BZ-increased IL-8 expression compared to cells transfected with control siRNA. Even though transfection using IKKε siRNA somewhat decreased the BZ-induced IL-8 expression in PC3 cells, it was not statistically significant. Transfection using IκBα specific siRNA did not have any effect on the BZ-increased IL-8 expression, suggesting that the nuclear IκBα induced by proteasome inhibition does not regulate IL-8 expression in prostate cancer cells. Together, these results demonstrated that the IL-8 expression induced by the proteasome inhibition in prostate cancer cells is mediated, at least partly, by IKKα.
IKKα mediates the BZ-increased p65 recruitment to IL-8 promoter in PC3 cells
To investigate the function of IKKα in the BZ-induced IL-8 expression, we analyzed whether the enzymatic IKKα activity is required for the BZ-increased p65 recruitment to IL-8 promoter by ChIP. As illustrated in Fig. 7A, 12h pre-incubation of PC3 cells with 5 μM Bay-117082 significantly attenuated the BZ-induced p65 recruitment to IL-8 promoter. Considering that IKKβ is not involved in the IL-8 regulation (Fig. 6), and that suppression of IKKε has only an insignificant effect on the IL-8 expression in PC3 cells (Fig. 6E), these results strongly indicated that the IKKα enzymatic activity is required for the BZ-increased p65 recruitment to IL-8 promoter.
Figure 7. IKKα activity is required for the BZ-increased p65 recruitment to IL-8 promoter in PC3 cells.

(A) p65 recruitment to IL-8 promoter was analyzed by ChIP in PC3 cells pre-incubated 12 hours with control DMSO or 5 μM Bay-117082, and then treated 24 hours with 0, 0.1 or 1 μM BZ. The results were quantified by real time PCR and presented as the change in occupancy over the human IGX1A (SA Biosciences) sequence control. The data represent the mean +/−SE of four experiments; asterisks denote a statistically significant (p<0.05) change compared to control cells pre-treated with DMSO. (B) Western analysis of CE and NE prepared from PC3 cells treated with increasing concentrations of BZ for 24 hours, and analyzed by using p65, S536P-p65 and actin antibodies. Each lane corresponds to approximately 5x104 cells. (C) Densitometric evaluation of p65 and S536P-p65 levels in nuclear extracts of BZ-treated PC3 cells. The nuclear p65 and S536P-p65 bands were scanned and their densities were normalized to actin. The values for NE of untreated cells were arbitrarily set to 1, and the other values are presented relative to these values. The data represent the means of three experiments +/−SE.
Since previous studies showed that IKKα can phosphorylate p65 at S536, resulting in its increased transcriptional activity (50, 51), we investigated whether BZ increases S536 p65 phosphorylation and recruitment to IL-8 promoter. Western analysis using S536P-p65 specific antibody demonstrated that similarly to p65, S536P-p65 is localized mainly in the nucleus of PC3 cells (Fig. 7B). However in contrast to p65, BZ decreased the nuclear levels of S536P-p65 (Figs. 7B, C), suggesting that the proteasome inhibition reduces p65 phosphorylation at S536. Importantly, we did not detect any S536P-p65 recruitment to the IL-8 promoter in untreated or BZ-treated PC3 cells (data not shown), indicating that the S536P-p65 is not recruited to IL-8 promoter in prostate cancer cells. Together, these results show that the proteasome inhibition induces IL-8 expression in prostate cancer cells through the increased p65 recruitment that is facilitated by IKKα independently of p65 phosphorylation at S536.
Discussion
The present data show that the proteasome inhibition by bortezomib unexpectedly increases expression of the pro-angiogenic and pro-inflammatory chemokine IL-8 in androgen independent metastatic prostate cancer PC3 and DU145 cells, while expression of other NFκB-dependent genes is inhibited or unchanged. The increased IL-8 expression is associated with increased p65 nuclear accumulation and recruitment to the IL-8 promoter. Importantly, the proteasome inhibition also increases nuclear accumulation of IKKα, and suppression of IKKα protein levels and enzymatic activity significantly decreases the BZ-induced p65 recruitment and IL-8 expression. These data provide the first evidence for the gene specific increase of IL-8 expression by the proteasome inhibition in prostate cancer cells, and indicate that the BZ-increased IL-8 expression is mediated by the IKKα dependent enhanced p65 recruitment to the IL-8 promoter.
Bortezomib is the first clinically approved proteasome inhibitor that has been very effective in the treatment of multiple myeloma and other hematological malignancies (18–21). One of the main mechanisms of BZ function is the suppressed proteasomal degradation of IκBα in the cytoplasm, resulting in the inhibition of inducible NFκB activity and expression of NFκB-dependent pro-inflammatory and anti-apoptotic genes (19–21). NFκB activity is constitutively increased in metastatic prostate cancer cells through the increased activation of IKK, resulting in the increased cell survival and resistance to chemotherapy (9–12). BZ has so far failed to exhibit a significant clinical activity in prostate cancer patients (23–30); however, the mechanisms underlying the prostate cancer resistance to BZ are largely unknown.
We have found that BZ unexpectedly increases the nuclear levels of p65 in prostate cancer cells; the highest p65 nuclear accumulation was achieved by 0.1 μM BZ (Fig. 1A), which approximately corresponds to the clinically used BZ concentrations (39). Since BZ did not have any effect on p65 mRNA levels in PC3 cells (data not shown), it seems likely that BZ prevents the proteasomal degradation of nuclear p65 in prostate cancer cells. This is supported by previous studies that demonstrated that p65 is a target of the proteasomal degradation both in canonical and atypical pathways of NFκB activation (52–54). The increased p65 nuclear accumulation was associated with increased in vitro p65 DNA binding activity (Fig. 2A) and with the increased p65 recruitment to promoters of NFκB-regulated genes, especially to the IL-8 promoter (Fig. 5B).
In contrast to p65, proteasome inhibition did not increase the nuclear levels of p50 NFκB. On the contrary, PC3 cells treated with 10 μM BZ exhibited somewhat decreased p50 levels both in the cytoplasm and in the nucleus. One possible mechanism that may be responsible for the decreased cellular levels of p50 in BZ-treated cells, is the previously described p50 cleavage by the proteasome regulated, calcium-dependent protease calpain (55, 56). However, even though BZ did not increase the nuclear levels of p50 NFκB in PC3 cells, it increased the p50 recruitment to Bcl-3 and cIAP-2 promoters. Interestingly, the increased occupancy of p50 at Bcl-3 and cIAP-2 promoters was associated with the highest gene suppression by BZ, suggesting that the p50 promoter binding inhibits transcription of these genes in prostate cancer cells. The suppressory role of p50 promoter binding in regulating Bcl-3 and cIAP-2 expression in prostate cancer cells is supported by earlier studies demonstrating that p50 homodimers inhibit transcription of a subset of NFκB-regulated genes (57, 58). In contrast to p65, p50 recruitment to IL-8 promoter was not increased by BZ (Fig. 5C), indicating that in prostate cancer cells, the IL-8 promoter is regulated predominantly by p65 homodimers.
In addition to p65, BZ induced the nuclear accumulation of IκBα in PC3 cells (Fig. 1A). This was consistent with our previous studies that showed that proteasome inhibition suppresses NFκB activity by an additional mechanism that consists of inducing the translocation of IκBα from the cytoplasm to the nucleus, resulting in the gene specific inhibition of NFκB-dependent transcription (31, 33). However, the BZ-increased IL-8 expression in PC3 cells is not regulated by IκBα since suppression of IκBα levels by siRNA did not have any effect on IL-8 expression (Fig. 6E). Together, these results indicate that in prostate cancer cells, the IL-8 promoter is regulated by p65, but not by p50 NFκB or IκBα. This is consistent with our previous study demonstrating that in human leukocytes, the IL-8 promoter is regulated by p65/65 homodimers, independently of the nuclear IκBα (32). In addition, these data suggest that the proteasome inhibition induces IL-8 expression in prostate cancer cells by increasing p65 nuclear accumulation and recruitment to the IL-8 promoter; when proteasome is inhibited, p65 is persistently bound to IL-8 promoter, resulting in the increased transcriptional activity.
Previous studies have shown that proteasome inhibitors block the inducible NFκB activity in prostate cancer cells (44, 59, 60). Moreover, the proteasome inhibitor MG132 reduced the in vitro binding of p65/50 heterodimers to NFκB consensus oligonucleotides measured by electrophoretic mobility shift assay (EMSA) in nuclear extracts from unstimulated PC3 cells (11, 31). However, the in vitro binding of transcription factors to consensus oligonucleotides measured by EMSA does not necessarily predict the in vivo binding to endogenous promoters. Our data indicate that the proteasome inhibition by BZ has a promoter-specific effect on NFκB-dependent transcription. While most genes are inhibited, or not affected, the IL-8 expression is greatly increased, and this is associated with the increased p65 recruitment. Even though the BZ-increased p65 promoter recruitment and IL-8 expression in prostate cancer cells are surprising, they are supported by recent studies that showed that BZ increases the constitutive NFκB activity in endometrial carcinoma cells (61) and multiple myeloma cells (62). In addition, previous studies indicated that BZ induces the proteasome-independent NFκB activation in intestinal epithelial cancer cells, lung cancer cells and bone marrow stromal cells (53, 55, 63).
BZ increased the nuclear accumulation of IKKα and IKKβ in PC3 cells (Figs. 6A, B), without increasing the IKKα/βmRNA levels (data not shown). Thus, it appears that the nuclear IKKα and IKKβ in prostate cancer cells are subject to proteasomal degradation, as was previously suggested (47). However, only IKKα seems to be required for the BZ-induced p65 recruitment and IL-8 expression in metastatic prostate cancer cells, since siRNA suppression of IKKα but not IKKβ, decreased the BZ-induced IL-8 expression (Fig. 6E). A previous study has demonstrated that the nuclear levels of IKKα correlate with prostate cancer progression (64). While no activated IKKα was detected in nuclear fractions of normal human prostate or benign prostate hyperplasia, stage 4 tumors exhibited the highest nuclear levels and activity of IKKα (64). Furthermore, a recent study showed that IKKα regulates expression of androgen receptor in prostate cancer cells (65).
The enzymatic activity of IKKα is required for the BZ-induced p65 recruitment and IL-8 expression in prostate cancer cells, since inhibition of IKK activity significantly attenuates the BZ-induced p65 recruitment (Fig. 7A) and IL-8 mRNA levels in PC3 and DU145 cells (Figs. 6C, D). However, our data indicate that IKKα does not phosphorylate the nuclear p65 at S536 in prostate cancer cells and that S536P-p65 is not recruited to the IL-8 promoter. Recent studies have shown that the kinase-dependent nuclear functions of IKKα include the transcriptional activation by histone H3 phosphorylation, phosphorylation of CREB-binding protein (CBP) resulting in p65 transcriptional activation, and regulation of the metastatic suppressor Maspin (64–70). In addition, the nuclear IKKα can remove the inhibitory HDAC3 from certain promoters, allowing increased p65 recruitment and transcriptional activity (51, 71).
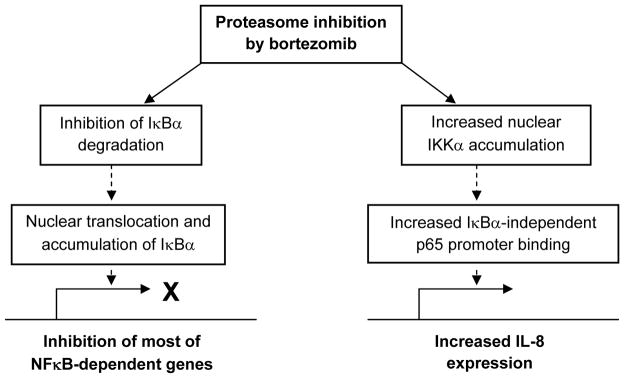
IL-8 contributes to prostate cancer progression through its induction of tumor cell proliferation, survival, and angiogenesis. In androgen independent prostate cancer cells, IL-8 expression enhances tumorigenicity and metastasis. In this study, we show that proteasome inhibition by BZ greatly increases IL-8 expression in androgen independent prostate cancer PC3 and DU145 cells. Interestingly however, in both cell types, the level of IL-8 induction at mRNA level was considerably higher than on the protein release level (Figs. 3, 4), indicating that in addition to inducing the IL-8 mRNA expression, proteasome inhibition has an inhibitory effect on protein(s) controlling mRNA processing, translation or IL-8 release from cells. On the transcriptional level, proteasome inhibition seems to have two opposite effects on the NFκB-dependent genes. One mechanism of action consists of the inhibition of NFκB activity through the inhibition of cytoplasmic IκBα degradation and induction of the nuclear translocation and accumulation of IκBα, resulting in suppression of most of the NFκB-dependent genes. The other, opposite, mechanism consists of the increased expression of IL-8, and perhaps other genes regulated by p65 and IKKα (Fig. 8). Future studies should determine the exact mechanism of IKKα involvement in the enhanced p65 recruitment and IL-8 expression, as well as the mechanisms responsible for the IL-8 post-transcriptional regulation by proteasome inhibition in prostate cancer cells. Understanding the mechanisms of how the proteasome inhibition and IKKα regulate IL-8 expression and secretion could lead to the development of new combination therapies targeting both IKKα and proteasome in androgen independent prostate cancer and other solid tumors characterized by excessive IL-8 release.
Figure 8. Model of the transcriptional regulation of NFκB-dependent genes by proteasome inhibition in androgen independent prostate cancer cells.
Abbreviations used in this paper
- BZ
bortezomib
- ChIP
chromatin immunoprecipitation
- IKK
IκB kinase
- NLS
nuclear localization signal
Footnotes
This work was supported by NIH grants AI085497 and CA173452 to I. Vancurova.
References
- 1.Kunkel SL, Strieter RM, Chensue SW, Basha M, Standiford T, Ham J, Remick DG. Tumor necrosis factor-alpha, interleukin-8 and chemotactic cytokines. Prog Clin Biol Res. 1990;349:433–444. [PubMed] [Google Scholar]
- 2.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 3.Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD, Yano S, BarEli M, Radinsky R, Pettaway CA, Dinney CP. Interleukin-8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res. 2000;6:2104–2119. [PubMed] [Google Scholar]
- 4.Araki S, Omori Y, Lyn D, Singh RK, Meinbach DM, Sandman Y, Lokeshwar VB, Lokeshwar BL. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007;67:6854–6862. doi: 10.1158/0008-5472.CAN-07-1162. [DOI] [PubMed] [Google Scholar]
- 5.Aggarwal BB. NFκB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Bassères DS, Baldwin AS. NFκB and IκB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 7.DiDonato JA, Mercurio F, Karin M. NFκB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. NFκB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Constitutive activation of IκB kinase alpha and NFκB in prostate cancer cells is inhibited by ibuprofen. Oncogene. 1999;18:7389–7394. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- 10.Chen CD, Sawyers CL. NFκB activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol Cell Biol. 2002;22:2862–2870. doi: 10.1128/MCB.22.8.2862-2870.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, Budunova IV. The role of IKK in constitutive activation of NFκB transcription factor in prostate carcinoma cells. J Cell Sci. 2002;115:141–151. doi: 10.1242/jcs.115.1.141. [DOI] [PubMed] [Google Scholar]
- 12.Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, Case TC, Ellwood-Yen K, Sawyers CL, Bhowmick NA, Blackwell TS, Yull FE, Matusik RJ. The NFκB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008;68:6762–6769. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain G, Cronauer MV, Schrader M, Möller P, Marienfeld RB. NFκB signaling in prostate cancer: A promising therapeutic target? World J Urol. 2012;30:303–310. doi: 10.1007/s00345-011-0792-y. [DOI] [PubMed] [Google Scholar]
- 14.Singh RK, Lokeshwar BL. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 2011;71:3268–3277. doi: 10.1158/0008-5472.CAN-10-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh RK, Sudhakar A, Lokeshwar BL. Role of Chemokines and Chemokine Receptors in Prostate Cancer Development and Progression. J Cancer Sci Ther. 2010;2:89–94. doi: 10.4172/1948-5956.1000030. [DOI] [PubMed] [Google Scholar]
- 16.Hayden MS, Ghosh S. Shared principles in NFκB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 17.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246:239–253. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5:2638–2645. [PubMed] [Google Scholar]
- 19.Cusack JC, Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin AS. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic NFκB inhibition. Cancer Res. 2001;61:3535–3540. [PubMed] [Google Scholar]
- 20.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076. [PubMed] [Google Scholar]
- 21.Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi NC, Mitsiades N, Anderson KC. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101:1530–1534. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 22.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome inhibition in the treatment of cancer. Cell Cycle. 2005;4:290–296. [PubMed] [Google Scholar]
- 23.Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther. 2003;2:835–843. [PubMed] [Google Scholar]
- 24.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 25.Papandreou CN, Logothetis CJ. Bortezomib as a potential treatment for prostate cancer. Cancer Res. 2004;64:5036–5043. doi: 10.1158/0008-5472.CAN-03-2707. [DOI] [PubMed] [Google Scholar]
- 26.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11:164–179. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Zhu K, Dunner K, Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29:451–462. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voutsadakis IA, Papandreou CN. The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urol Oncol. 2012;30:752–761. doi: 10.1016/j.urolonc.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Wright JJ. Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy. Clin Cancer Res. 2010;16:4094–4104. doi: 10.1158/1078-0432.CCR-09-2882. [DOI] [PubMed] [Google Scholar]
- 30.Kraft AS, Garrett-Mayer E, Wahlquist AE, Golshayan A, Chen CS, Butler W, Bearden J, Lilly M. Combination therapy of recurrent prostate cancer with the proteasome inhibitor bortezomib plus hormone blockade. Cancer Biol Ther. 2011;12:119–124. doi: 10.4161/cbt.12.2.15723. [DOI] [PubMed] [Google Scholar]
- 31.Vu HY, Juvekar A, Ghosh C, Ramaswami S, Le DH, Vancurova I. Proteasome inhibitors induce apoptosis of prostate cancer cells by inducing nuclear translocation of IκBα. Arch Biochem Biophys. 2008;475:156–163. doi: 10.1016/j.abb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh CC, Ramaswami S, Juvekar A, Vu HY, Galdieri L, Davidson D, Vancurova I. Gene-specific repression of proinflammatory cytokines in stimulated human macrophages by nuclear IκBα. J Immunol. 2010;185:3685–3693. doi: 10.4049/jimmunol.0902230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juvekar A, Manna S, Ramaswami S, Chang TP, Vu HY, Ghosh CC, Celiker MY, Vancurova I. Bortezomib induces nuclear translocation of IκBα resulting in gene-specific suppression of NFκB-dependent transcription and induction of apoptosis in CTCL. Mol Cancer Res. 2011;9:183–194. doi: 10.1158/1541-7786.MCR-10-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramaswami S, Manna S, Juvekar A, Kennedy S, Vancura A, Vancurova I. Chromatin immunoprecipitation analysis of NFκB transcriptional regulation by nuclear IκBα in human macrophages. Methods Mol Biol. 2012;809:121–134. doi: 10.1007/978-1-61779-376-9_8. [DOI] [PubMed] [Google Scholar]
- 35.Ross JS, Kallakury BV, Sheehan CE, Fisher HA, Kaufman RP, Jr, Kaur P, Gray K, Stringer B. Expression of NFκB and IκBα proteins in prostatic adenocarcinomas: correlation of NFκB immunoreactivity with disease recurrence. Clin Cancer Res. 2004;10:2466–2472. doi: 10.1158/1078-0432.ccr-0543-3. [DOI] [PubMed] [Google Scholar]
- 36.Shukla S, MacLennan GT, Fu P, Patel J, Marengo SR, Resnick MI, Gupta S. NFκB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia. 2004;6:390–400. doi: 10.1593/neo.04112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sweeney C, Li L, Shanmugam R, Bhat-Nakshatri P, Jayaprakasan V, Baldridge LA, Gardner T, Smith M, Nakshatri H, Cheng L. NFκB is constitutively activated in prostate cancer in vitro and is overexpressed in prostatic intraepithelial neoplasia and adenocarcinoma of the prostate. Clin Cancer Res. 2004;10:5501–5507. doi: 10.1158/1078-0432.CCR-0571-03. [DOI] [PubMed] [Google Scholar]
- 38.Fradet V, Lessard L, Begin LR, Karakiewicz P, Masson AM, Saad F. NFκB nuclear localization is predictive of biochemical recurrence in patients with positive margin prostate cancer. Clin Cancer Res. 2004;10:8460–8464. doi: 10.1158/1078-0432.CCR-04-0764. [DOI] [PubMed] [Google Scholar]
- 39.Levêque D, Carvalho MC, Maloisel F. Review. Clinical pharmacokinetics of bortezomib. In Vivo. 2007;21:273–278. [PubMed] [Google Scholar]
- 40.Kunsch C, Rosen CA. NFκB subunit-specific regulation of the IL-8 promoter. Mol Cell Biol. 1993;13:6137–6146. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein B, Baldwin AS. Distinct mechanisms for regulation of the IL-8 gene involve synergism and cooperativity between C/EBP and NFκB. Mol Cell Biol. 1993;13:7191–7198. doi: 10.1128/mcb.13.11.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma J, Ren Z, Ma Y, Xu L, Zhao Y, Zheng C, Fang Y, Xue T, Sun B, Xiao W. Targeted knockdown of EGR-1 inhibits IL-8 production and IL-8-mediated invasion of prostate cancer cells through suppressing EGR-1/NFκB synergy. J Biol Chem. 2009;284:34600–34606. doi: 10.1074/jbc.M109.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F, Karseladze A, Budunova I. Effects of IKK inhibitor PS1145 on NFκB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. 2006;25:387–398. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 44.Gasparian AV, Guryanova OA, Chebotaev DV, Shishkin AA, Yemelyanov AY, Budunova IV. Targeting transcription factor NFκB: comparative analysis of proteasome and IKK inhibitors. Cell Cycle. 2009;8:1559–1566. doi: 10.4161/cc.8.10.8415. [DOI] [PubMed] [Google Scholar]
- 45.Péant B, Diallo JS, Dufour F, Le Page C, Delvoye N, Saad F, Mes-Masson AM. Over-expression of IκB kinase-epsilon (IKKε/IKKi) induces secretion of inflammatory cytokines in prostate cancer cell lines. Prostate. 2009;69:706–718. doi: 10.1002/pros.20912. [DOI] [PubMed] [Google Scholar]
- 46.Péant B, Forest V, Trudeau V, Latour M, Mes-Masson AM, Saad F. IκB-Kinase-ε (IKKε/IKKi/IκBKε) expression and localization in prostate cancer tissues. Prostate. 2011;71:1131–1138. doi: 10.1002/pros.21329. [DOI] [PubMed] [Google Scholar]
- 47.Broemer M, Krappmann D, Scheidereit C. Requirement of Hsp90 activity for IκB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NFκB activation. Oncogene. 2004;23:5378–5386. doi: 10.1038/sj.onc.1207705. [DOI] [PubMed] [Google Scholar]
- 48.Lee J, Rhee MH, Kim E, Cho JY. BAY 11-7082 Is a Broad-Spectrum Inhibitor with Anti-Inflammatory Activity against Multiple Targets. Mediators Inflamm. 2012;2012:416036. doi: 10.1155/2012/416036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. The NFκB activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem. 2003;278:919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- 51.Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IκB kinase-alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the NFκB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKα limits macrophage NFκB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 54.Cullen SJ, Ponnappan S, Ponnappan U. Proteasome inhibition up-regulates inflammatory gene transcription induced by an atypical pathway of NFκB activation. Biochem Pharmacol. 2010;79:706–714. doi: 10.1016/j.bcp.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demarchi F, Bertoli C, Greer PA, Schneider C. Ceramide triggers an NFκB-dependent survival pathway through calpain. Cell Death Differ. 2005;12:512–522. doi: 10.1038/sj.cdd.4401592. [DOI] [PubMed] [Google Scholar]
- 56.Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, Sun SY. Proteasome inhibitor PS-341 induces calpain-dependent IκBα degradation. J Biol Chem. 2010;285:16096–16104. doi: 10.1074/jbc.M109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plaksin D, Baeuerle PA, Eisenbach L. KBF1 (p50 NFκB homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J Exp Med. 1993;177:1651–1662. doi: 10.1084/jem.177.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bohuslav J, V, Kravchenko V, Parry GC, Erlich JH, Gerondakis S, Mackman N, Ulevitch RJ. Regulation of an essential innate immune response by the p50 subunit of NFκB. J Clin Invest. 1998;102:1645–1652. doi: 10.1172/JCI3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levine L, Lucci JA, 3rd, Pazdrak B, Cheng JZ, Guo YS, Townsend CM, Jr, Hellmich MR. Bombesin stimulates NFκB activation and expression of proangiogenic factors in prostate cancer cells. Cancer Res. 2003;63:3495–3502. [PubMed] [Google Scholar]
- 60.Patrikidou AP, Vlachostergios J, Voutsadakis IA, Hatzidaki E, Valeri RM, Destouni C, Apostolou E, Papandreou CN. Neuropeptide-inducible upregulation of proteasome activity precedes NFκB activation in androgen-independent prostate cancer cells. Cancer Cell Int. 2012 doi: 10.1186/1475-2867-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dolcet X, Llobet D, Encinas M, Pallares J, Cabero A, Schoenenberger JA, Comella JX, Matias-Guiu X. Proteasome inhibitors induce death but activate NFκB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem. 2006;281:22118–22130. doi: 10.1074/jbc.M601350200. [DOI] [PubMed] [Google Scholar]
- 62.Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Bortezomib induces canonical NFκB activation in multiple myeloma cells. Blood. 2009;114:1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Németh ZH, Wong HR, Odoms K, Deitch EA, Szabó C, Vizi ES, Haskó G. Proteasome inhibitors induce IκB kinase activation, IκBα degradation, and NFκB activation in HT-29 cells. Mol Pharmacol. 2004;65:342–349. doi: 10.1124/mol.65.2.342. [DOI] [PubMed] [Google Scholar]
- 64.Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, Cheresh DA, Karin M. Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nature. 2007;446:690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 65.Jain G, Voogdt C, Tobias A, Spindler KD, Möller P, Cronauer MV, Marienfeld RB. IκB kinases modulate the activity of the androgen receptor in prostate carcinoma cell lines. Neoplasia. 2012;14:178–189. doi: 10.1593/neo.111444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;(357):re13. doi: 10.1126/stke.3572006re13. Epub 2006 Oct 17. [DOI] [PubMed] [Google Scholar]
- 67.Gloire G, Dejardin E, Piette J. Extending the nuclear roles of IκB kinase subunits. Biochem Pharmacol. 2006;72:1081–1089. doi: 10.1016/j.bcp.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 68.Lee DF, Hung MC. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin Cancer Res. 2008;14:5656–5662. doi: 10.1158/1078-0432.CCR-08-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Espinosa L, Bigas A, Mulero MC. Alternative nuclear functions for NFκB family members. Am J Cancer Res. 2011;1:446–459. [PMC free article] [PubMed] [Google Scholar]
- 70.Huang WC, Hung MC. Beyond NFκB activation: nuclear functions of IκB kinase α. J Biomed Sci. 2013 Jan 23;20(1):3. doi: 10.1186/1423-0127-20-3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gloire G, Horion J, El Mjiyad N, Bex F, Chariot A, Dejardin E, Piette J. Promoter-dependent effect of IKKα on NFκB/p65 DNA binding. J Biol Chem. 2007;282:21308–21318. doi: 10.1074/jbc.M610728200. [DOI] [PubMed] [Google Scholar]