

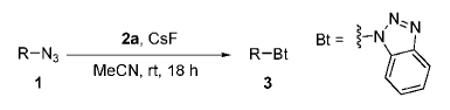
Abstract
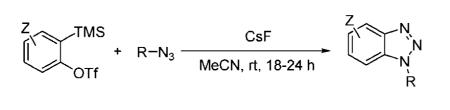
A variety of substituted benzotriazoles have been prepared by the [3 + 2] cycloaddition of azides to benzynes. The reaction scope is quite general, affording a rapid and easy entry to substituted, functionalized benzotriazoles under mild conditions.
Recent years have seen rapid development of the Cucatalyzed [3 + 2] cycloaddition1 reaction between terminal alkynes and azides, commonly referred to as “click chemistry”.2 Such chemistry has found wide applications not only in synthetic organic chemistry3 but also in dendrimer and polymer chemistry,4 the material sciences,5 bioconjugation chemistry,6 and the pharmaceutical sciences.7
Although there have been several reports on the annulation of arynes by azides,8 efforts to modernize this reaction are certainly necessary. Early examples utilizing potentially explosive diazotized anthranilic acid as the benzyne precursor8a–c suffer from the use of a potentially explosive reagent and dangerous reaction conditions, and more recent examples utilizing o-(trimethylsilyl)aryliodonium salts8e,f suffer from the limited availability and difficulties in preparation of these reagents. Nowadays, arynes are more readily and conveniently generated in situ by the fluoride-promoted ortho-elimination of commercially available or easily prepared o-(trimethylsilyl)aryl triflates.9 Arynes generated in this way retain their high reactivity toward nucleophilic additions and annulations.10
With our recent success in the development of benzyne annulation chemistry,11 particularly the [3 + 2] cycloaddition reaction between benzynes and diazo compounds,11a,12 we envisioned the [3 + 2] annulation of benzynes by azides as a very promising extension of the current click chemsitry. Such chemistry should not only expand the scope and utility of the present click chemistry but also potentially provide a rapid entry to substituted, functionalized benzotriazoles, which are known to possess important biological activity13 and exhibit utility as synthetic auxiliaries.14 Herein, we report our preliminary results on the [3 + 2] annulation reaction of benzynes and azides, new benzyne click chemistry.
We started our investigation using commercially available benzyl azide (1a) and o-(trimethylsilyl)phenyl triflate (2a) under a variety of different reaction conditions (Table 1). TBAF and CsF were chosen as fluoride sources, and the reaction was examined in several dipolar aprotic solvents. While most reaction conditions gave low yields (entries 1–4), the reaction carried out in acetonitrile using CsF as the fluoride source afforded a superior yield of 76% (entry 5). These optimized conditions are identical to the optimal conditions observed previously by us for the synthesis of indazoles by the cycloaddition of diazo compounds to arynes.11a We have thus chosen these conditions as our general procedure for all subsequent work.15
Table 1.
Reaction Optimizationa
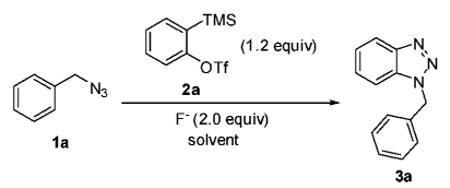
| entry | fluoride source | solvent | T (°C) | time (h) | yieldb (%) |
|---|---|---|---|---|---|
| 1 | TBAF | THF | 0 to rt | 3 | 37 |
| 2 | TBAF | MeCN | 0 to rt | 3 | 45 |
| 3 | TBAF | DCM | 0 to rt | 5 | 34 |
| 4 | CsF | THF | rt | 18 | 37 |
| 5 | CsF | MeCN | rt | 18 | 76 |
All reactions were carried out on a 0.3 mmol scale in 0.1 M concentration.
Isolated yield.
We next tested different benzyne precursors in this reaction (Table 2). As can be seen, the reaction shows good compatibility with a range of different benzyne precursors. Thus, benzyne precursors 2b and 2c gave comparable yields of the desired benzotriazole products (entries 1 and 2). However, the electron-poor benzyne precursor 2d gave only a 56% yield (entry 3). An unsymmetrical benzyne precursor 2e afforded a single regioisomer in a 78% yield (entry 4), which is consistent with our previous results.11a
Table 2.
Reaction with Different Benzyne Precursorsa
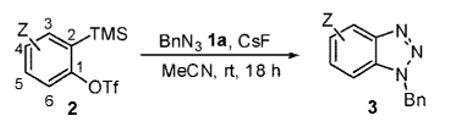
| entry | benzyne precursor |
Z | product | yield (%)b |
|---|---|---|---|---|
| 1 | 2b | 4,5-Me2 |
|
71 |
| 2 | 2c | 4,5- (OMe)2 |
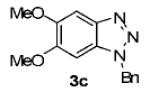
|
71 |
| 3 | 2d | 4,5-F2 |
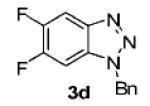
|
56 |
| 4 | 2e | 3-OMe |
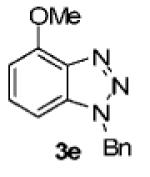
|
78c |
All reactions were carried out on a 0.3 mmol scale with 1.2 equiv of benzyne precursor and 2.0 equiv of CsF.
Isolated yield.
The product was assigned by a 2D-NOESY experiment; see the Supporting Information for details.
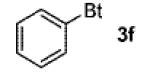
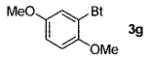
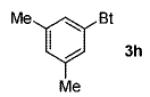
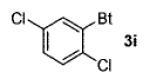
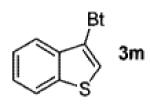
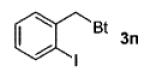
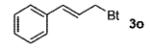
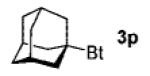
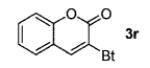
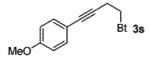
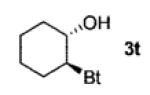
A wide range of azides have also been screened (Table 3). Among them, aryl and heteroaryl azides are generally good substrates, affording the desired benzotriazole products in 83–90% yields (entries 1–8). The substrate scope includes electron-rich (entries 2 and 3), electron-poor (entries 4–7), sterically hindered (entries 2, 4, and 7), and heterocyclic (entry 8) aryl azides. All of these substrates gave clean reactions under mild conditions and tolerated functional groups, such as ester, ether, cyano, and halogen groups. Alkyl azides are also good substrates. Other than benzyl azide (1a), functionalized benzylic (entry 9) and allylic (entry 10) azides also have afforded excellent yields of the desired products. Sterically demanding adamantyl azide (1l) (entry 11) reacted smoothly to afford a 78% yield as well. Azides with functional groups can be easily transformed into the corresponding benzotriazoles. Thus, ethyl azidoacetate (1m) reacted cleanly to give a quantitative yield of the corresponding benzotriazole (entry 12). Coumarin-derived azide 1n also afforded the desired product in a moderate 51% yield (entry 13). An alkyne moiety is well tolerated under the reaction conditions, as seen in the smooth reaction of alkyne 1o with benzyne, affording the alkynyl benzotriazole 3s (entry 14). A free hydroxyl group is tolerated, although an additional, unidentified side product was observed (entry 15). Although the reaction tolerates alkenes quite well (see entries 10 and 13), the vinylic azide 1q was not a suitable substrate in this annulation process (entry 16). After the reaction was complete, a complex mixture was obtained. After isolation, purification, and identification, the product was found in only a 20% yield, and we were unable to identify the rest of the products. Trimethylsilyl azide (1r) was also examined in this reaction (entry 17). Surprisingly, the reaction did not stop at the [3 + 2] cycloaddition stage but underwent further desilylation, followed by phenylation with another equivalent of 2a to afford 3f as the final product. The same reaction afforded unidentified products when MeOH was used as a cosolvent.16 The current limitation on the scope of the azide substrate is that azides bearing electron-withdrawing groups directly attached to the azide moiety do not work in this annulation. Thus, the reaction of sulfonyl azide 1s did not give any annulation product.17
Table 3.
Reaction Scope with Different Azidesa
| entry | R (compound) |
product | yield (%)b |
|---|---|---|---|
| 1 | Ph (1b) |
|
87 |
| 2 | 2,5-(MeO)2C6H3 (1e) |
|
88 |
| 3 | 3,5-Me2C6H3 |
|
85 |
| 4 | 2,5-Cl2C6H3(1e) |
|
87 |
| 5 | 4-BrC6H4 (1f) |
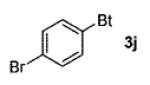
|
83 |
| 6cd | 4-EtO2CC6H4 (1g) |
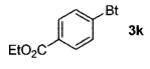
|
90 |
| 7 | 4-(NC)-2IC6H3 (1h) |
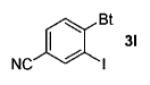
|
86 |
| 8 |
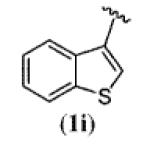
|
|
85 |
| 9 | 2-IC6H4CH2 (1j) |
|
100 |
| 10 | cinnamyl (1k) |
|
91 |
| 11e | 1-adamantyl(1l) |
|
78 |
| 12 | EtO2CCH2 (1m) |
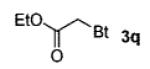
|
100 |
| 13 |
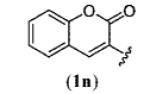
|
|
51 |
| 14 |
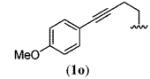
|
|
93 |
| 15e |
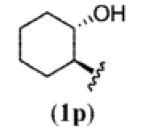
|
|
68 |
| 16e |
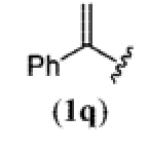
|
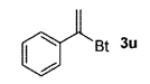
|
20 |
| 17f | TMS (1r) |
3f | 58 |
| 18 | 4-AcNHc6H4SO2 (1s) |
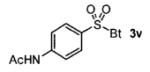
|
0 |
All reactions were carried out on a 0.3 mmol scale with 1.2 equiv of 2a and 2.0 equiv of CsF.
Isolated yield.
The reaction was allowed to run for 24 h.
A trace of unreacted starting azide remained even after 24 h.
Other unidentified products were present.
The reaction was carried out using 2.4 equiv of 2a and 5.0 equiv of CsF.
In conclusion, we have developed a facile, efficient, and general method for the synthesis of substituted, functionalized benzotriazoles by the 1,3-dipolar cycloaddition of benzynes with azides under very mild reaction conditions. The reaction has good substrate scope and tolerates a board range of functional groups. It provides a useful new route to benzotriazoles in much the same manner as present “click” chemistry affords triazoles. We believe that this methodology should find broad applications in synthetic organic chemistry, as well as the combinatorial, pharmaceutical, and polymer sciences.
Supplementary Material
Acknowledgment
We are grateful to the National Institutes of Health (GM079593 and GM070620) and the Kansas University NIH Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support. We also thank Mr. Donald C. Rogness at Iowa State University for his help in the preparation of the benzyne precursors.
Footnotes
Supporting Information Available: Preparation of azide starting materials, experimental details, and characterization of the final products, including full 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Huisgen R. HelV. Chim. Acta. 1967;50:2421–2439. [Google Scholar]
- (2).For comprehensive reviews, see: Moses JE, Moorhouse AD. Chem. Soc. ReV. 2007;36:1249–1262. doi: 10.1039/b613014n. Gil MV, Arevalo MJ, Lopez O. Synthesis. 2007:1589–1620.
- (3) (a).Tilliet M, Lundgren S, Moberg C, Levacher V. Adv. Synth. Catal. 2007;349:2079–2084. [Google Scholar]; (b) Yan Z-Y, Niu Y-N, Wei H-L, Wu L-Y, Zhao Y-B, Liang Y-M. Tetrahedron: Asymmetry. 2007;17:3288–3293. [Google Scholar]; (c) Luo S, Xu H, Mi X, Li J, Zheng X, Cheng J-P. J. Org. Chem. 2006;71:9244–9247. doi: 10.1021/jo061657r. [DOI] [PubMed] [Google Scholar]
- (4) (a).Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JMJ, Sharpless KB, Fokin VV. Angew. Chem., Int. Ed. 2004;43:3928–3932. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]; (b) Lee JW, Kim JH, Kim B-K, Kim JH, Shin WS, Jin S-H. Tetrahedron. 2006;62:9193–9200. [Google Scholar]
- (5) (a).Nandivada H, Jiang X, Lahann J. Adv. Mater. 2007;19:2197–2208. [Google Scholar]; (b) Lutz J-F. Angew. Chem., Int. Ed. 2007;46:1018–1025. doi: 10.1002/anie.200604050. [DOI] [PubMed] [Google Scholar]
- (6) (a).Rozkiewicz DI, Gierlich J, Burley GA, Gutsmiedl K, Carell T, Ravoo BJ, Reinhoudt DN. ChemBioChem. 2007;8:1997–2002. doi: 10.1002/cbic.200700402. [DOI] [PubMed] [Google Scholar]; (b) Lutz J-F, Boerner HG, Weichenhan K. Macromolecules. 2006;39:6376–6383. [Google Scholar]
- (7) (a).Sharpless KB, Manetsch R. Expert Opin. Drug Discovery. 2006;1:525–538. doi: 10.1517/17460441.1.6.525. [DOI] [PubMed] [Google Scholar]; (b) Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (c) Whiting M, Muldoon J, Lin Y-C, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Angew. Chem., Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- (8) (a).Huisgen R, Knorr R. Naturwissenschaften. 1962;48:716. [Google Scholar]; (b) Reynolds GA. J. Org. Chem. 1964;29:3733–3734. [Google Scholar]; (c) Huisgen R, Knorr R, Moebius L, Szeimies G. Chem. Ber. 1965;98:4014–4021. [Google Scholar]; (d) Mitchell G, Rees CW. J. Chem. Soc., Perkin Trans. 1. 1987:403–412. [Google Scholar]; (e) Kitamura T, Fukatsu N, Fujiwara Y. J. Org. Chem. 1998;63:8579–8581. [Google Scholar]; (f) Kitamura T, Todaka M, Shin-machi I, Fujiwara Y. Heterocycl. Commun. 1998;4:205–208. [Google Scholar]
- (9).Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211–1214. [Google Scholar]
- (10).For recent reports, see: Yoshida H, Mimura Y, Ohshita J, Kunai A. Chem. Commun. 2007:2405–2407. doi: 10.1039/b701581j. Xie C, Zhang Y. Org. Lett. 2007;9:781–784. doi: 10.1021/ol063017g. Yoshida H, Fukushima H, Ohshita J, Kunai A. J. Am. Chem. Soc. 2006;128:11040–11041. doi: 10.1021/ja064157o. Liu Z, Larock RC. J. Org. Chem. 2006;71:3198–3209. doi: 10.1021/jo0602221. Liu Z, Larock RC. J. Am. Chem. Soc. 2005;127:13112–13113. doi: 10.1021/ja054079p. Yoshida H, Watanabe M, Ohshita J, Kunai A. Chem. Commun. 2005:3292–3294. doi: 10.1039/b505392g. Tambar UK, Stoltz BM. J. Am. Chem. Soc. 2005;127:5340–5341. doi: 10.1021/ja050859m. Yoshida H, Watanabe M, Fukushima H, Ohshita J, Kunai A. Org. Lett. 2004;6:4049–4051. doi: 10.1021/ol048298b.
- (11) (a).Liu Z, Shi F, Martinez PDG, Raminelli C, Larock RC. J. Org. Chem. 2008;73:219–226. doi: 10.1021/jo702062n. [DOI] [PubMed] [Google Scholar]; (b) Zhao J, Larock RC. J. Org. Chem. 2007;72:583–588. doi: 10.1021/jo0620718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Z, Larock RC. Tetrahedron. 2007;63:347–355. doi: 10.1016/j.tet.2006.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Z, Larock RC. J. Org. Chem. 2007;72:223–232. doi: 10.1021/jo0619534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For related indazole work, see: Jin T, Yamamoto Y. Angew. Chem., Int. Ed. 2007;46:3323–3325. doi: 10.1002/anie.200700101.
- (13) (a).Kopańska K, Najda A, Źeebrowska J, Chomicz L, Piekarczyk J, Myjak P, Bretner M. Bioorg. Med. Chem. 2004;12:2617–2624. doi: 10.1016/j.bmc.2004.03.022. [DOI] [PubMed] [Google Scholar]; (b) He F-Q, Liu X-H, Wang B-L, Li Z-M. J. Chem. Res. 2006:809–811. [Google Scholar]; (c) Caliendo G, Greco G, Grieco P, Novellino E, Perissutti E, Santagada V, Barbarulo D, Esposito E, De Blasi A. Eur. J. Med. Chem. 1996;31:207–213. [Google Scholar]; (d) Caliendo G, Di Carlo R, Greco G, Meli R, Novellino E, Perissutti E, Santagada V. Eur. J. Med. Chem. 1995;30:77–84. [Google Scholar]; (e) Wynne GM, Wren SP, Johnson PD, Price PD, De Moor O, Nugent G, Dorgan CR, Tinsley JM, Storer R, Mulvaney A, Pye R. Appl. WO 2007091107 PCT Int. 2007
- (14) (a).Katritzky AR, Lan X, Yang JZ, Denisko OV. Chem. Rev. 1998;98:409–548. doi: 10.1021/cr941170v. [DOI] [PubMed] [Google Scholar]; (b) Katritzky AR, Rogovoy BV. Chem. Eur. J. 2003;9:4586–4593. doi: 10.1002/chem.200304990. [DOI] [PubMed] [Google Scholar]; (c) Katritzky AR, Manju K, Singh SK, Meher NK. Tetrahedron. 2005;61:2555–2581. [Google Scholar]; (d) Katritzky AR, Lan X. Chem. Soc. Rev. 1994;23:363–373. [Google Scholar]; (e) Katritzky AR, Yang Z, Cundy DJ. Aldrichim. Acta. 1994;27:31–38. [Google Scholar]
- (15).General Procedure. To a solution of benzyne precursor (0.35 mmol) and azide (0.30 mmol) in 3 mL of dry MeCN was added CsF (0.60 mmol). The reaction vial was sealed, and the reaction mixture was stirred at room temperature for 18–24 h before being poured into saturated aqueous NaHCO3. The resulting mixture was extracted with EtOAc or DCM, and the combined organic layers were dried over MgSO4 and evaporated. The residue was purified by silica gel chromatography.
- (16).In a similar reaction between benzyne 2a and TMS diazomethane to form indazole, MeOH was needed as a co-solvent; see ref 11a.
- (17).We have also unsuccessfully carried out a reaction with an acyl azide (2-iodobenzoyl azide). However, we observed severe spontaneous decomposition of this azide simply upon standing. Thus, not surprisingly, under our reaction conditions, the reaction between this azide and 2a afforded a complex mixture. Other acyl azides will be investigated, and results will be published in due course.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.