

Abstract
The iodocyclization of O-methyloximes of 2-alkyn-1-ones affords 4-iodoisoxazoles, which undergo various palladium-catalyzed reactions to yield 3,4,5-trisubstituted isoxazoles. The palladium-catalyzed processes have been adapted to parallel synthesis utilizing commercially available boronic acid, acetylene, styrene, and amine sublibraries. Accordingly, a diverse 51-member library of 3,4,5-trisubstituted isoxazoles has been generated.
Introduction
Low molecular weight nitrogen-containing heterocycles are securing their place among the most highly recognized pharmacophores.1 Among them, the isoxazole scaffold is of particular interest, since it is known to exhibit a broad range of biological activity.2 Isoxazoles have also had a significant impact as intermediates in the synthesis of various natural products.3 Consequently, isoxazoles are prized as potential drug candidates and biological probes. There have been several reports relating to the synthesis of functionalized isoxazoles by combinatorial techniques,4 although the use of 4-iodoisoxazoles as the key intermediate for library generation is thus far unreported. Many of the reported libraries have been limited to the preparation of disubstituted isoxazoles. Additionally, functionalization at the C4 position of the isoxazole core, with respect to library generation, has previously met with limited success. Until recently, restricted access to 4-haloisoxazoles, by a route involving mild reaction conditions, has prevented them from being an important intermediate for library generation. The halogenation of isoxazoles at the C4 position generally requires the use of high temperatures and harsh acids.5
Previous work in our laboratory has demonstrated that the electrophilic cyclization of Z-2-alkyn-1-one O-methyl oximes (1), using ICl, provides a mild and selective route to 3,5-disubstituted-4-iodoisoxazoles (2) (Scheme 1).6 The requisite 2-alkyn-1-ones (3) required for this methodology can be readily synthesized by established chemistry utilizing commercially available starting materials (Scheme 2).7
Scheme 1.
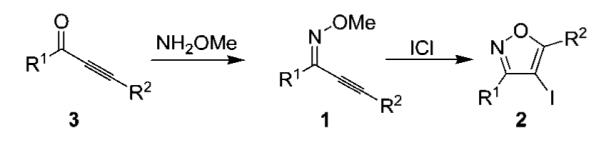
Scheme 2.
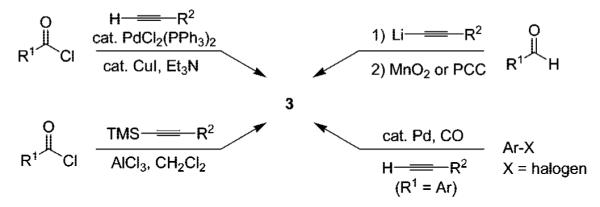
We have previously demonstrated the significance of this methodology by reporting individual examples of Sonogashira,8 Suzuki-Miyaura,9 Heck,10 and carbonylative amidation cross-coupling reactions,11 which provide the corresponding 3,4,5-trisubstituted isoxazoles in good yields, including the highly potent COX-2 inhibitor valdecoxib (4) (Scheme 3).12 With optimized conditions in hand for each of these processes, we wished to adapt these reactions to library generation. Herein, we report the success of this objective.
Scheme 3.
Results and Discussion
Our 4-iodoisoxazole synthesis has previously been demonstrated to tolerate, but not be limited to, alkyl and aryl groups at the R1 or R2 positions of the isoxazole core.6 During our initial study, we found that employing of Z-O-methyloximes was essential to the success of our methodology, since the corresponding E-O-methyloximes proved ineffective in the electrophilic cyclization process. Since bulky groups at the R1 position provided the highest yields of the necessary Z-O-methyloximes required for our 4-iodoisoxazole synthesis, R1 was restricted to aryl groups for library generation. Since R2 substituents had less of an effect on the outcome of our methodology, there was no need to adhere to strict guidelines when choosing R2. Accordingly, we chose a subset of various 4-iodoisoxazoles which could be synthesized from readily available starting materials (Figure 1).
Figure 1.
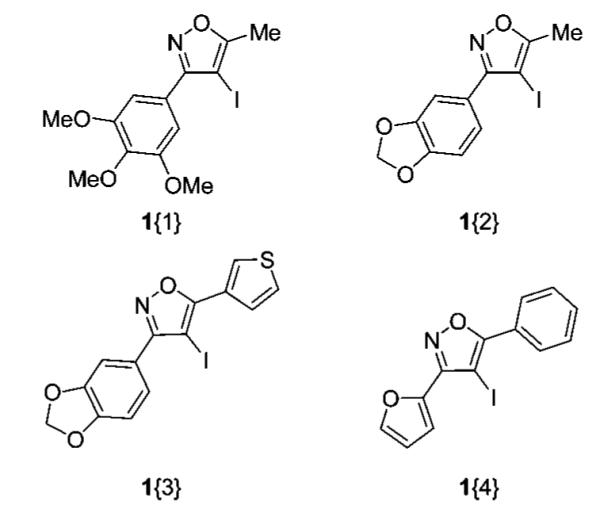
4-Iodoisoxazole sublibrary.
Isoxazoles 1{1–4} were selected because their synthesis is straightforward and the oxygen heteroatoms provide desired polarity in the resulting library.
The alkyne sublibrary, used for Sonogashira cross-coupling, was chosen based on commercially available, heteroatom-containing acetylenes that could provide hydrogen bond donors and/or acceptors (Figure 2).
Figure 2.
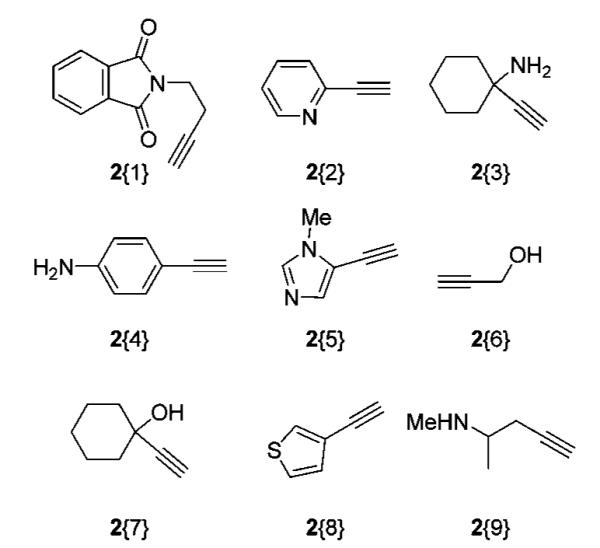
Terminal acetylene sublibrary.
A small styrene sublibrary for Heck cross-coupling was chosen to demonstrate vinylic functionalization. However, only a few functionally diverse styrenes were available from commercial sources (Figure 3). We avoided the use of better Michael acceptors, such as acrylates or acrylonitriles, since the resulting Heck products would also be excellent Michael acceptors and thus undesirable for subsequent pharmaceutical applications.
Figure 3.
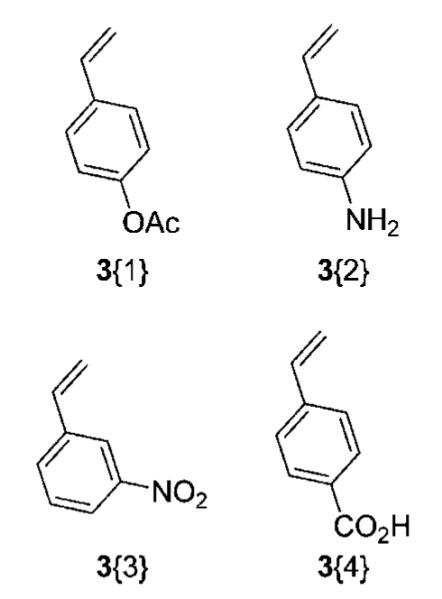
Styrene sublibrary.
Commercially available boronic acids, for Suzuki-Miyaura cross-coupling, were chosen based on their diverse functionality (Figure 4). 5-Indoleboronic acid 4{1} was chosen because the resulting cross-coupling products could generate druglike scaffolds with potentially positive physiochemical properties. Arylboronic acids 4{3} and 4{6} were chosen because the resulting cross-coupling products would contain fluorine atoms. Organofluorine compounds are of considerable interest because of their versatile applications in industry and medicine.13 Other boronic acids within the sublibrary were chosen because they contain heterocycles and polar functionality that would incorporate druglike moieties in the resulting cross-coupling products.
Figure 4.
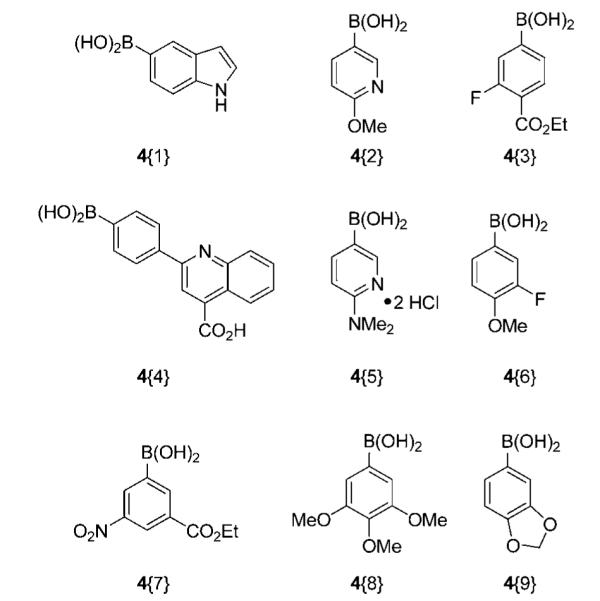
Boronic acid sublibrary.
Primary amines were chosen as a sublibrary for palladium-catalyzed amide formation, because the resulting nitrogen-containing products would enable us to make compounds with potentially attractive druglike features, including molecular weight, solubility, and molecular polarizability. Amines 5{1–11} were chosen to represent compounds found in natural products and derivatives of substances known to possess biological activity (Figure 5).
Figure 5.
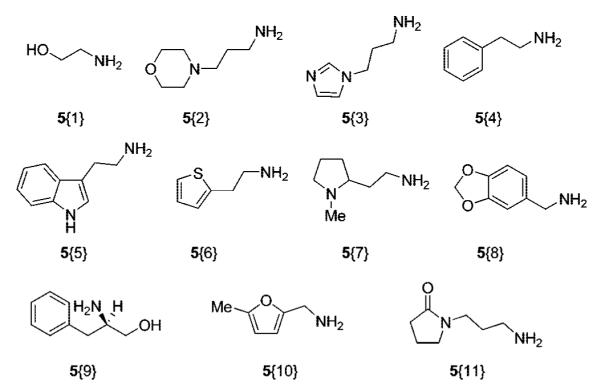
Primary amine sublibrary.
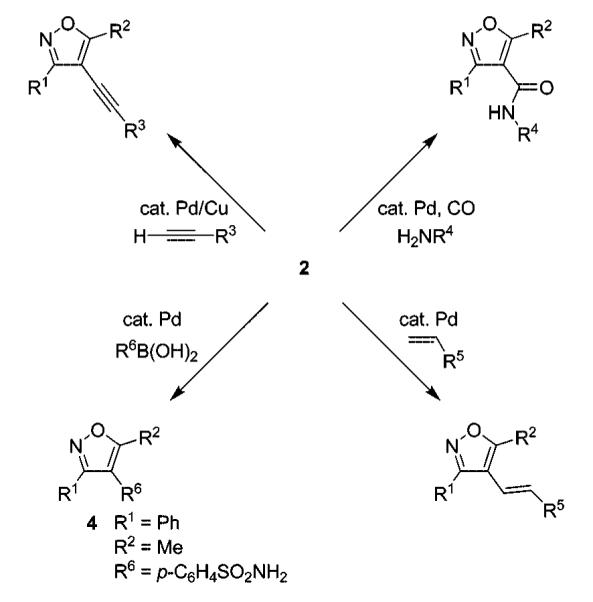
To ease the transition from our isoxazole methodology to library generation, we wished to preserve the protocols we had originally employed for the cross-coupling of 4-iodoisoxazoles with terminal acetylenes, styrenes, boronic acids, and amines.6b With these preliminary findings, we proceeded to prepare a diverse library of 3,4,5-trisubstituted isoxazoles as outlined in Scheme 3. The crude products were analyzed by LC/MS, followed by purification by preparative HPLC.
The results of the library synthesis are summarized in Tables 1–3. The crude products were subjected to preparative HPLC, and purities in the range of 87–100% were achieved after purification. Most of the reactions proceeded well, with the exception of the Heck reaction of 4-vinylbenzoic acid. In the Suzuki–Miyaura reaction of boronic acids 4{5} and 4{7}, poor or no yield of the desired product was observed. Palladium-catalyzed amide formation using carbon monoxide and primary amines proceeded smoothly and provided the corresponding amides in satisfactory yields with high purities. Out of a total of 54 palladium-catalyzed processes attempted, only three failed completely.
Table 1.
Library Data for Compounds 6{1–13}
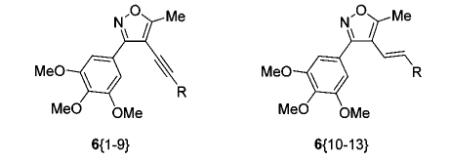
| compound | R | yielda (%) | purityb (%) |
|---|---|---|---|
| 6{1} | 2-ethylphthalimide | 40 | 94 |
| 6{2} | 2-pyridinyl | 30 | 97 |
| 6{3} | 1-amino-1-cyclohexyl | 42 | 95 |
| 6{4} | 4-H2NC6H4 | 62 | 99 |
| 6{5} | 1-methyl-1H-imidazo-5-yl | 83 | 95 |
| 6{6} | CH2OH | 67 | 97 |
| 6{7} | 1-hydroxy-1-cyclohexyl | 59 | 99 |
| 6{8} | 3-thiophenyl | 75 | 97 |
| 6{9} | 2-(N-methyl)propyl | 50 | 97 |
| 6{10} | 4-AcOC6H4 | 60 | 94 |
| 6{11} | 4-H2NC6H4 | 67 | 93 |
| 6{12} | 3-O2NC6H4 | 73 | 95 |
| 6{13} | 4-HO2CC6H4 | 8 | 87 |
Isolated yield after preparative HPLC.
UV purities determined at 214 nm after preparative HPLC.
Table 3.
Library Data for Compounds 6{44-54}
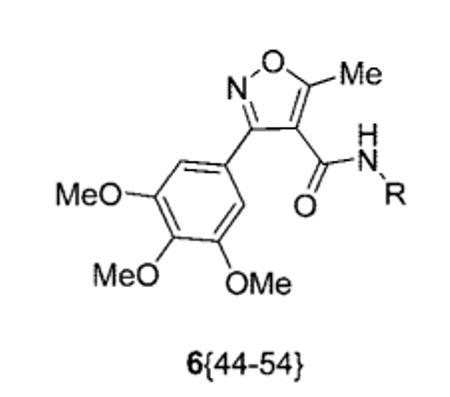
|
| |||
|---|---|---|---|
| compound | R | yielda(%) | purityb(%) |
|
| |||
| 6{44} | (CH2)2OH | 24 | 99 |
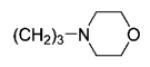
| 6{45} |
|
44 | 99 |
| 6{46} |
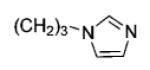
|
50 | 100 |
| 6{47} | (CH2)2Ph | 58 | 96 |
| 6{48} |
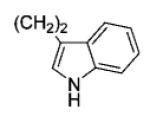
|
39 | 97 |
| 6{49} |
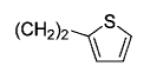
|
79 | 99 |
| 6{50} |
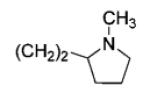
|
65 | 96 |
| 6{51} |
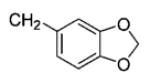
|
40 | 99 |
| 6{52} |
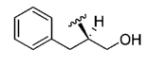
|
27 | 99 |
| 6{53} |
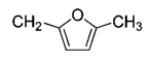
|
80 | 99 |
| 6{54} |
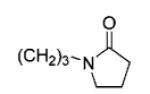
|
69 | 99 |
Isolated yield after preparative HPLC.
UV purities determined at 214 nm after preparative HPLC.
Because of our interest in synthesizing heterocycles for use in high-throughput screening projects, an in silico evaluation of library members was carried out to determine their agreement with Lipinski’s “rule of five” and Veber’s rules. 15 Molecular weight, clog P, number of hydrogen bond donors and acceptors, and the number of rotatable bonds were either specified or calculated for each of the library members using the SYBYL16 program (Table 4). Most of the isoxazole library members were highly Lipinski compliant. In fact, 96% of the library members are entirely compliant with Lipinski rules and only compound 6{31} had multiple violations, including molecular weight and clog P.
Table 4.
In silico Parameters for Gauging Oral Availability/Druglikeness
| mean | st dev | range | |
|---|---|---|---|
| clog P | 3.13 | 1.40 | −0.01–6.87 |
| mol. weight | 380.8 | 46.4 | 292.3–518.5 |
| H-bond acceptors | 6.00 | 1.13 | 3–9 |
| H-bond donors | 0.92 | 0.73 | 0–2 |
| rotatable bonds | 5.14 | 1.67 | 2–8 |
In conclusion, the preparation and subsequent palladium-catalyzed reactions of 4-iodoisoxazoles with various cross-coupling partners has allowed for simple construction of a 51-member library of 3,4,5-trisubstituted isoxazoles. The average yield of the library was 49% and the average purity after preparative HPLC was 96%.
Supplementary Material
Table 2.
Library Data for Compounds 6{14–43}
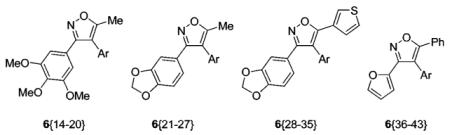
| compound | Ar | yielda(%) | purityb(%) |
|---|---|---|---|
| 6{14} | 5-indolyl | 24 | 92 |
| 6{15} | 6-methoxy-3-pyridinyl | 29 | 95 |
| 6{16} | 4-EtO2C-3-FC6H4 | 53 | 96 |
| 6{17} |
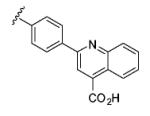
|
5 | 87 |
| 6{18} | 6-(N,N-dimethyl)-3-pyridinyl | 22 | 94 |
| 6{19} | 4-MeO-3-FC6H4 | 58 | 97 |
| 6{20} | 3-MeO2-C-5-O2NC6H4 | 44 | 97 |
| 6{21} | 5-indolyl | 51 | 92 |
| 6{22} | 6-methoxy-3-pyridinyl | 26 | 95 |
| 6{23] | 4-EtO2C-3-FC6H4 | 26 | 97 |
| 6{24} |
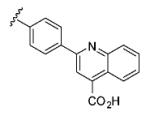
|
0 | |
| 6{25] | 6-(N,N-dimethyl)-3-pyridinyl | 7 | 100 |
| 6{26} | 4-MeO-3-FC6H4 | 46 | 88 |
| 6{27} | 3-MeO2C-5-O2NC6H4 | 0 | |
| 6{28} | 5-indolyl | 79 | 97 |
| 6{29} | 6-methoxy-3-pyridinyl | 81 | 95 |
| 5{30} | 4-EtO2C-3-FC6H4 | 73 | 99 |
| 6{31} |
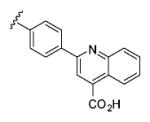
|
57 | 94 |
| 6{32} | 6-(N,N-dimethyl)-3-pyridinyl | 85 | 96 |
| 6{33} | 4-MeO-3-FC6H4 | 73 | 99 |
| 6{34} | 3,4,5-(MeO)3C6H4 | 73 | 99 |
| 6{35} | 5-indolyl | 75 | 96 |
| 6{36} | 6-methoxy-3-pyridinyl | 16 | 91 |
| 6{37} | 4-EtO2C-3-FC6H4 | 64 | 98 |
| 6{38} |
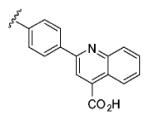
|
7 | 100 |
| 6{39} | 6-(N,N-dimethyl)-3-pyridinyl | 20 | 92 |
| 6{40} | 4-MeO-3-FC6H4 | 60 | 99 |
| 6{41} | 3-EtO2C-5-O2NC6H4 | 0 | |
| 6{42} | 3,4,5-(MeO)3C6H4 | 53 | 99 |
| 6{43} | 3,4-(OCH2CH2O)C6H3 | 44 | 99 |
Isolated yield after preparative HPLC.
UV purities determined at 214 nm after preparative HPLC.
Acknowledgment
We would like to thank Frank Schoenen for his useful discussions and David Hill at the University of Kansas NIH Center of Excellence in Chemical Methodologies and Library Development for obtaining the library NMR spectra. We thank the National Institute of General Medical Sciences (R01 GM070620 and R01 GM079593) and the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (P50 GM069663) for support of this research; Johnson Matthey, Inc. and Kawaken Fine Chemicals Co., Ltd. for donations of palladium catalysts; and Frontier Scientific and Synthonix for donations of boronic acids.
Footnotes
Supporting Information Available. Experimental details and characterization of a representative 20 library members, including full 1H and 13C NMR spectra and conditions for the high throughput liquid chromatography purification. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1)(a).Loughlin WA. Aust. J. Chem. 1998;51:875–893. [Google Scholar]; (b) Balkenhohl F, von dem Bussche-Hünnefeld C, Lansky A, Zechel C. Angew. Chem., Int. Ed. Engl. 1996;35:2288–2337. [Google Scholar]; (c) Nefzi A, Ostresh JM, Houghten RA. Chem. Rev. 1997;97:449–472. doi: 10.1021/cr960010b. [DOI] [PubMed] [Google Scholar]
- (2)(a).For a brief review, see: Carlsen L, Döpp D, Döpp H, Duus F, Hartmann H, Lang-Fugmann S, Schulze B, Smalley RK, Wakefield BJ. In: Houben-Weyl, Methods in Organic Chemistry. Schaumann E, editor. Vol. E8a. Georg Thieme Verlag; Stuttgart, Germany: 1992. pp. 45–204. Rowley M, Broughton HB, Collins I, Baker R, Emms F, Marwood R, Patel S, Ragan CI. J. Med. Chem. 1996;39:1943–1945. doi: 10.1021/jm960072u. Frolund B, Jorgensen AT, Tagmose L, Stensbol TB, Vestergaard HT, Engblom C, Kristiansen U, Sanchez C, Krogsgaard-Larsen P, Liljefors T. J. Med. Chem. 2002;45:2454–2468. doi: 10.1021/jm020027o. Daidone G, Raffa D, Maggio B, Plescia F, Cutuli VMC, Mangano NG, Caruso A. Arch. Pharm. Pharm. Med. Chem. 1999;332:50–54. doi: 10.1002/(sici)1521-4184(19993)332:2<50::aid-ardp50>3.0.co;2-s. Tomita K, Takahi Y, Ishizuka R, Kamamura S, Nakagawa M, Ando M, Nakanishi T, Nakamura T, Udaira H. Ann. Sankyo Res. Lab. 1973;1:25.; Chem. Abstr. 1974;80:120808. Talley JJ. Prog. Med. Chem. 1999;13:201–234. doi: 10.1016/s0079-6468(08)70048-1. Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, Perkins WE, Rogers RS, Shaffer AF, Zhang YY, Zweifel BS, Seibert K. J. Med. Chem. 2000;43:775–777. doi: 10.1021/jm990577v. Giovannoni MP, Vergelli C, Ghelardini C, Galeotti N, Bartolini A, Kal Piaz V. J. Med. Chem. 2003;46:1055–1059. doi: 10.1021/jm021057u. Li W-T, Hwang D-R, Chen C-P, Shen C-W, Huang C-L, Chen T-W, Lin C-H, Chang Y-L, Chang Y-Y, Lo Y-K, Tseng H-Y, Lin C-C, Song J-S, Chen H-C, Chen S-J, Wu S-H, Chen C-T. J. Med. Chem. 2003;46:1706–1715. doi: 10.1021/jm020471r.
- (3)(a).Baraldi PG, Basco A, Benetti S, Pollini GP, Simoni D. Synthesis. 1987:857–869. [Google Scholar]; (b) Kozikowski AP. Acc. Chem. Res. 1984;17:410–416. [Google Scholar]
- (4)(a).Shankar BB, Yang DY, Girton S, Ganguly AK. Tetrahedron Lett. 1998;39:2447–2448. [Google Scholar]; (b) Shang YJ, Wang YG. Tetrahedron Lett. 2002;43:2247–2249. [Google Scholar]; (c) Cereda E, Ezhaya A, Quai M, Barbaglia W. Tetrahedron Lett. 2001;42:4951–4953. [Google Scholar]; (d) Park KH, Kurth MJ. J. Org. Chem. 1999;64:9297–9300. [Google Scholar]; (e) Park KH, Kurth MJ. Tetrahedron Lett. 1999;40:5841–5844. [Google Scholar]; (f) Kantorowski EJ, Kurth MJ. J. Org. Chem. 1997;62:6797–6803. [Google Scholar]; (g) Batra S, Srinivasan T, Rastogi SK, Kundu B, Patra A, Bhaduri AP, Dixit M. Bioorg. Med. Chem. Lett. 2002;12:1905–1908. doi: 10.1016/s0960-894x(02)00333-5. [DOI] [PubMed] [Google Scholar]
- (5)(a).Claisen L. Ber. Dtsch. Chem. Ges. 1891;24:130–138. [Google Scholar]; (b) Quilico A, Justoni R. Chem. Abstr. 1938;32:7455. [Google Scholar]; (c) Day RA, Blake JA, Stephens CE. Synthesis. 2003:1586–1590. [Google Scholar]
- (6)(a).Waldo JP, Larock RC. Org. Lett. 2005;23:5203–5205. doi: 10.1021/ol052027z. [DOI] [PubMed] [Google Scholar]; (b) Waldo JP, Larock RC. J. Org. Chem. 2007;72:9643–9647. doi: 10.1021/jo701942e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7)(a).Tohda Y, Sonogashira K, Hagihara N. Synthesis. 1977:777–778. [Google Scholar]; (b) Kobayashi T, Tanaka M. J. Chem. Soc., Chem. Commun. 1981:333–334. [Google Scholar]; (c) Mohamed Ahmed MS, Mori A. Org. Lett. 2003;5:3057–3060. doi: 10.1021/ol035007a. [DOI] [PubMed] [Google Scholar]; (d) Lin C-F, Lu W-D, Wang I-W, Wu M-J. Synlett. 2003:2057–2061. [Google Scholar]; (e) Birkofer L, Ritter A, Uhlenbrauck H. Chem. Ber. 1963;96:3280–3288. [Google Scholar]
- (8)(a).Sonogashira K. Chapter 5. In: Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim, Germany: 1998. pp. 203–229. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467–4470. [Google Scholar]
- (9)(a).Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]; (b) Suzuki A. J. Organomet. Chem. 1999;576:147–168. and references therein. [Google Scholar]
- (10)(a).Alonso F, Beletskaya IP, Yus M. Tetrahedron. 2005;61:11771–11835. [Google Scholar]; (b) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (c) Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1. Wiley-Interscience; New York: 2002. pp. 1223–1254. and references therein. [Google Scholar]; (d) Tsuji J. Palladium Reagents and Catalysts. John Wiley & Sons; Chichester, England: 2004. pp. 135–175. [Google Scholar]
- (11).Schoenberg A, Heck RF. J. Org. Chem. 1974;39:3327–3321. [Google Scholar]
- (12)(a).Talley JJ. Chem. Abstr. 1999;130:110269.; 5,859,257 U.S. Patent. 1999; G. D. Searle & Company. Talley JJ. Prog. Med. Chem. 1999;36:201–234. doi: 10.1016/s0079-6468(08)70048-1. Talley JJ, Brown DL, Carter JS, Graneto MJ, Koboldt CM, Masferrer JL, Perkins WE, Rogers RS, Shaffer AF, Zhang YY, Zweifel BS, Seibert K. J. Med. Chem. 2000;43:775–777. doi: 10.1021/jm990577v.
- (13)(a).Hudlicky M, Pavlath AE. Chemistry of Organic Fluorine Compounds II. A Critical Review. American Chemical Society; Washington, DC: 1995. pp. 888–912. ACS Monograph 187. [Google Scholar]; (b) Hiyama T, editor. Organofluorine Compounds. Chemistry and Applications. Springer-Verlag; New York: 2000. [Google Scholar]
- (14)(a).Lipinski CA, Lombardo F, Dominay BW, Feeney PJ. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]; (b) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Del. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- (15).Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- (16).SYBYL. version 8.0 The Tripos Associate; St. Louis, MO: 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.