

Background: The antigen processing compartments in APCs possess a multivesicular morphology.
Results: APCs lacking multivesicular bodies can effectively process and present antigens to T cells.
Conclusion: Multivesicular body integrity is not required for antigen presentation.
Significance: Understanding the nature of antigen processing compartments is important for understanding mechanisms of T cell activation.
Keywords: Antigen Presentation, Antigen Processing, Endosomes, Major Histocompatibility Complex (MHC), T Cell, Niemann-Pick Disease Type C
Abstract
The antigen processing compartments in antigen-presenting cells (APCs) have well known characteristics of multivesicular bodies (MVBs). However, the importance of MVB integrity to APC function remains unknown. In this study, we have altered the ultrastructure of the MVB by perturbing cholesterol content genetically through the use of a deletion of the lipid transporter Niemann-Pick type C1 (NPC1). Immunofluorescence and electron microscopic analyses reveal that the antigen processing compartments in NPC1−/− dendritic cells (DCs) have an abnormal ultrastructure in that the organelles are enlarged and the intraluminal vesicles are almost completely absent and those remaining are completely disorganized. MHC-II is restricted to the limiting membrane of these enlarged MVBs where it colocalizes with the peptide editor H2-DM. Curiously, proteolytic removal of the chaperone protein Invariant chain from MHC-II, degradation of internalized foreign antigens, and antigenic-peptide binding to nascent MHC-II are normal in NPC1−/− DCs. Antigen-pulsed NPC1−/− DCs are able to effectively activate antigen-specific CD4 T cells in vitro, and immunization of NPC1−/− mice reveals surprisingly normal CD4 T cell activation in vivo. Our data thus reveal that the localization of MHC-II on the intraluminal vesicles of multivesicular antigen processing compartments is not required for efficient antigen presentation by DCs.
Introduction
Antigen-presenting cells (APCs)2 initiate an immune response that recognizes and specifically eliminates foreign pathogens. Dendritic cells (DCs) are a specialized subset of APCs that are capable of stimulating immunologically naïve T cells. Stimulation is promoted by the capacity of DCs to endocytose, proteolyze, and display foreign protein antigens (Ags) on MHC class II (MHC-II) molecules on their cell surfaces. These MHC-II-Ag complexes bind to their cognate T cell receptor on the surface of CD4 T cells and elicit a cascade of activation events following recognition, including T cell proliferation and differentiation into effector cells, B cell-mediated antibody responses, and the generation of immunological memory against invading pathogens.
For MHC-II to be loaded with foreign antigenic peptides, MHC-II must traffic to compartments that contain these degraded protein fragments. MHC-II transit to these organelles occurs through interaction with the chaperone protein Invariant chain (Ii) in the endoplasmic reticulum where Ii serves to stabilize the MHC-II molecule and inhibit premature Ag binding to the MHC-II Ag binding groove (1). The MHC-II-Ii complex traffics through the trans-Golgi network and out to the plasma membrane where it is rapidly internalized into the endocytic pathway through the recognition of sorting signals in the cytoplasmic tail of Ii by components of the clathrin-coated vesicle machinery (2). Newly synthesized Ii-associated MHC-II moves along the endocytic pathway until it comes to reside in specialized late endosomal/prelysosomal compartments referred to as antigen processing compartments. These compartments are acidic and contain a wide variety of proteolytic enzymes that degrade MHC-II-associated Ii as well as internalized self- and foreign antigens. It is also in this compartment that the Ii-derived peptide, termed CLIP, is removed from the peptide binding groove of MHC-II by the enzymatic activity of the MHC-II peptide editor H2-DM (1). One particularly intriguing feature of the antigen processing compartment in APCs is that they have a multivesicular morphology, consisting of a large limiting membrane filled with small (50-nm) intraluminal vesicles (ILVs). Curiously, whereas H2-DM is localized primarily (but not exclusively) on the limiting membrane of the multivesicular body (MVB), most nascent (peptide-free) MHC-II is present on the ILVs themselves (3). Curiously, FRET studies have shown that MHC-II/H2-DM interactions occur only when each molecule is localized on the ILV and perturbing MVB morphology (by chronic treatment with chloroquine or infection with Salmonella) inhibits peptide binding to MHC-II (4).
MVB biogenesis and the formation of ILVs are regulated in large part by protein-mediated organization of the membrane lipids lysobisphosphatidic acid and cholesterol to obtain the membrane curvature required for the inward-budding of the MVB limiting membrane. In this study we have examined APC function in mice that possess a genetic defect in MVB lipid content. Niemann-Pick type C1 (NPC1) is a protein that regulates the egress of cholesterol from late endosomes/lysosomes (5, 6). We now report that the net effect of NPC1 deletion is that the formation of ILVs in DC MVBs is perturbed. In DCs isolated from NPC1 mutant mice MHC-II localized almost exclusively on the MVB limiting membrane. Surprisingly, the mislocalization of MHC-II to the limiting membrane of antigen processing compartments had little effect on the ability of MHC-II to bind antigenic peptides and function as stimulators of naïve CD4 T cells, suggesting that localization of MHC-II on ILVs of MVB is important for a function that is not directly related to efficient antigen processing and presentation.
EXPERIMENTAL PROCEDURES
Mice and Genotyping
C57BL/6 (H-2b) mice were obtained from Charles Rivers Laboratories (NCI-Frederick Animal Production Area, Frederick, MD). B10.BR (H-2k) and 3A9 TCR transgenic mice were from Jackson Laboratory. NPC1+/− mice on a BALB/c background were obtained from Dr. Robert Erickson (University of Arizona, Tucson). NPC1+/− mice were back-crossed onto an H-2b background through standard mating to C57BL/6 mice and onto an H-2k background through standard mating with B10.BR mice. NPC1−/− mice were obtained by mating heterozygous mice, and NPC1+/− littermates were used as controls in all experiments. Genotyping was performed as described (6). OTII TCR (CD45.2) transgenic mice were maintained by interbreeding homozygous mice in our colony, and OTII TCR transgenic mice on the CD45.1 background were generated by breeding OTII mice (CD45.2) with B6.SJL mice (CD45.1).
Cells and Reagents
Mouse DCs were generated by growing bone marrow cells in medium containing GM-CSF in 6-well dishes as described previously (7). When indicated, ovalbumin protein antigen (1 mg/ml) was added on day 5 of culture and removed by washing on day 6. Lipopolysaccharide (LPS), if provided, was added to cultures on day 6 at 1 μg/ml to activate cells.
The following antibodies were used in this study: rat anti-I-Ab (M5/114.15.2), rat anti-H2-DM, and rat anti-CD74 (In-1) were from BD Biosciences. Rabbit anti-I-A α-chain antiserum was a gift from R. Germain. Rabbit anti-H2 antiserum was a gift from J. Strominger, and mouse anti-I-Ab-CLIP mAb 15G4 was a gift from A. Rudensky. The rabbit anti-I-A β-chain antiserum (8) has been described previously. All chemicals were obtained from Sigma-Aldrich unless noted otherwise.
Microscopy
DCs were harvested, washed with cold Hanks' Balanced Salt Solution (Mediatech, Manassas, VA), and then placed on poly-l-lysine-coated glass coverslips for 20 min at 4 °C and processed for immunofluorescence microscopy essentially as described previously (9). If stained for cholesterol, cells were incubated for 90 min at room temperature with 50 μg/ml filipin complex diluted in PBS prior to mounting. All cells were visualized using a Zeiss Axiovert 200M confocal microscope coupled to a LSM510 laser module containing 405 nm/diode, argon ion, HeNe1, and HeNe2 lasers as described previously (9). For standard electron microscopy, DCs were fixed in paraformaldehyde/glutaraldehyde/polyvinylpyrrolidone, postfixed with osmium tetroxide/uranyl acetate, and embedded in EPON as described previously (10). Ultrathin sections were analyzed using a JEOL 1200EX transmission electron microscope. For immunogold labeling studies the cells were fixed in paraformaldehyde/polyvinylpyrrolidone in 0.2 m phosphate buffer (pH 7.4) and processed for ultrathin cryosectioning and staining as described (11).
CD4 T Cell Proliferation Assays
T cells were harvested from isolated lymph nodes and spleens using a MACS CD4 T Cell Isolation kit (Miltenyi Biotec, Auburn, CA) and labeled with CFSE as described previously (8). 4 × 105 CD4 T cells were mixed with 4 × 104 DCs in a final volume of 200 μl of medium in 96-well plates, and proliferation was assessed after 72 h. Flow cytometry was used to assess the percentage of CD4 cells undergoing division by examining the loss of CFSE signal.
For in vivo T cell proliferation studies, 4 × 105 CFSE-labeled OTII (CD45.1) cells were adoptively transferred into CD45.2 recipient mice. One day after transfer the mice were immunized intraperitoneally with 1 mg of ovalbumin in alum. After 72 h the mice were sacrificed, spleens were removed, and the dilution of CFSE dye on CD45.1 CD4 T cells was determined by FACS analysis.
Flow Cytometry and Cell Sorting
For surface staining, cells were washed with ice-cold FACS buffer (HBSS containing 2% FBS) and kept on ice. For intracellular staining, cells were fixed and permeabilized as described for indirect immunofluorescence microscopy before washing with FACS buffer. Cells were incubated with primary antibody (4 × 106 cells/ml) on ice for 40 min and then washed three times with FACS buffer. Alexa Fluor 488, 546, or 633-conjugated secondary antibodies were then incubated with the cells for 40 min on ice and washed three times with FACS buffer. Cells were either run immediately or fixed with 0.5% paraformaldehyde overnight at 4 °C and run the next day.
In some experiments, ovalbumin-pulsed and LPS-stimulated DCs were stained on ice with phycoerythrin-labeled anti-MHC-II mAb M5/114.15.2 and MHC-IIhigh cells were isolated by cell sorting. The DCs were then counted and mixed with CFSE-labeled naïve OTII CD4 T cells for in vitro T cell proliferation experiments. Control experiments revealed that anti-MHC-II labeling had little effect on the ability of the DCs to stimulate OTII T cells. Spleen DCs were isolated on ice using a MACS CD11c+ DC Isolation kit and processed for immunofluorescence microscopy as described above.
Immunoprecipitation and Immunoblot Analysis
MHC-II and Ii were immunoprecipitated from Triton X-100-solubilized DCs using mAb M5/114.15.2 and In-1, respectively, and analyzed by immunoblotting as described previously (12). Quantitation was performed using a Molecular Dynamics Densitometer and Total Lab software version 2003.03 (Nonlinear Dynamics, Durham, NC).
RESULTS AND DISCUSSION
Cholesterol Accumulation Alters the Structure of Antigen Processing Compartments and Causes Mislocalization of MHC-II to the Limiting Membrane
To examine whether DC MVB morphology is affected by altered lysosomal lipid content, we made use of DCs derived from NPC1−/− mice. NPC1−/− DCs were harvested, fixed, and stained with filipin, a fluorescent compound that specifically binds to unesterified cholesterol (13). Confocal microscopy revealed the dramatic accumulation of cholesterol in defined structures in NPC1−/− DCs compared with control (NPC1+/−) DCs (Fig. 1A). This phenotype was observed in all NPC1−/− DCs examined. These data agree with previous studies showing that fibroblasts isolated from NPC1-deficient mice accumulate aberrant levels of lipids, such as cholesterol and sphingolipids, in late endosomal/lysosomal organelles (14, 15). Colocalization studies revealed that these compartments were late endosomal/lysosomal in nature, containing the peptide editor H2-DM (Fig. 1A) and were significantly enlarged compared with control DC MVBs (Fig. 1, A and B). Electron microscopy analysis of endosomal compartments in NPC1−/− DCs indicate that cholesterol accumulation generates a phenotype very similar to that observed in other cell types (16–18) including (i) the appearance of grossly enlarged late endosomes (above 2 μm) mostly void of ultrastructurally normal inner vesicles, (ii) enlargement of late endosomal MVBs associated with disorganized inner vesicles (if present), and (iii) increased number of autophago-lysosomes and lysosomes with a multilamellar morphology which contain an increased number of compact lamellae embedded in electron-dense accumulated cholesterol (Fig. 1C and supplemental Fig. 1).
FIGURE 1.
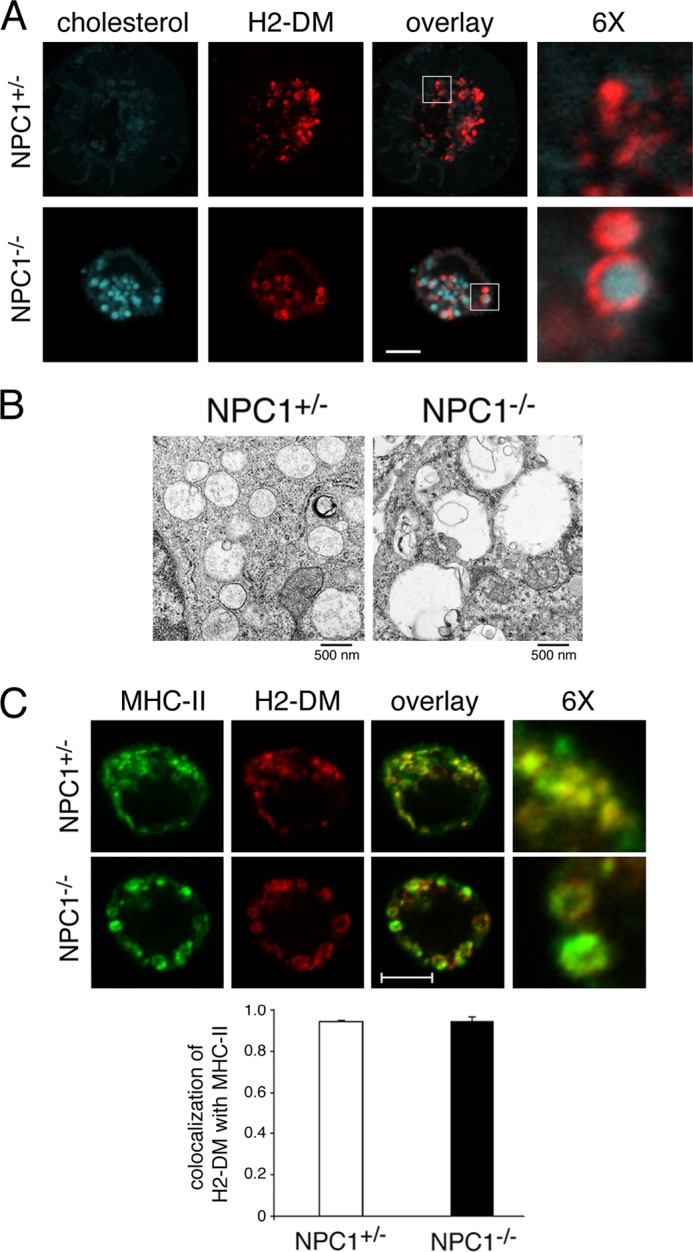
MVB ultrastructure and MHC-II localization are disturbed in lipid-overloaded MVBs. A, control or NPC1−/− DCs were harvested after 7 days of growth in medium and attached to polylysine-coated coverslips prior to fixation, permeabilization, and staining with filipin to detect unesterified cholesterol (blue) and a mAb recognizing H2-DM (red). 6× magnified images of the indicated regions of the merged image are shown. Scale bar represents 5 μm. B, DCs were fixed and sectioned for electron microscopy. Scale bar represents 500 nm. C, immature DCs were fixed, permeabilized, and stained with mAbs recognizing MHC-II (M5/114, green) or H2-DM (red). 6× magnified images of the indicated regions of the merged image are shown. Scale bar represents 5 μm. The extent of colocalization of H2-DM+ red pixels with MHC-II+ green pixels was determined using software provided with the LSM510 confocal microscope.
Ultrastructural analyses have shown that most MHC-II is present on the ILVs of MVBs whereas the peptide editor H2-DM is localized primarily on the MVB-limiting membrane (3). In wild-type DCs, confocal microscopy revealed that MHC-II and H2-DM colocalized in the small punctate Ag processing compartments present throughout the cytosol of immature cells (Fig. 1C). Whereas MHC-II did indeed colocalize with H2-DM in NPC1−/− DCs, both H2-DM and MHC-II were found to be restricted to the limiting membrane of these swollen organelles as evidenced by the readily discernible ring structures (Fig. 1C). Analysis by immunoelectron microscopy confirmed this finding and revealed an almost complete lack of intraluminal vesicles of MVB in NPC1−/− DCs and localization of MHC-II and H2-DM to the limiting membranes of these structures (supplemental Fig. 2). These results reveal that failure to maintain the proper lipid balance in MVBs leads to a change in the ultrastructure of the compartment and an alteration in the localization of MHC-II to the limiting membrane of these disorganized MVBs.
MVB Integrity Is Not Essential for MHC-II-Peptide Complex Formation
The characteristic multivesicular morphology of the MHC-II antigen processing and peptide loading compartments has been proposed to be essential for efficient MHC-II/H2-DM interactions and high affinity peptide binding (4). Because NPC1−/− DCs possess severely disorganized MVBs, we began to ask whether cells with perturbed MVB structures were capable of functioning as APCs in vitro and in vivo. Bone marrow-derived DCs obtained from NPC1−/− mice expressed surface MHC-II when the cells were in the resting (immature) state, and addition of LPS to these cells resulted in an increase in MHC-II surface expression like that observed in control DCs (Fig. 2A and supplemental Fig. 3, A and B). Curiously, the DCs cultures from NPC1−/− mice routinely contained more “MHC-II low” cells both before and after maturation with LPS (supplemental Fig. 3C). As a consequence of this, the total amount of MHC-II associated with DCs isolated from 10-week-old NPC1−/− mice was approximately half that of DCs isolated from wild-type mice (Fig. 2B). Analysis of these MHC-II molecules revealed that they had bound high affinity peptides, as evidenced by their SDS stability in SDS-polyacrylamide gels (19). Thus, whereas NPC1−/− DCs possess less total MHC-II than their wild-type controls, the proportion of MHC-II in these DCs that have bound antigenic peptides is similar to that in the control DCs, demonstrating that there is no selective defect in peptide binding to MHC-II in NPC1−/− DCs whose MVB are devoid of normal ILV.
FIGURE 2.
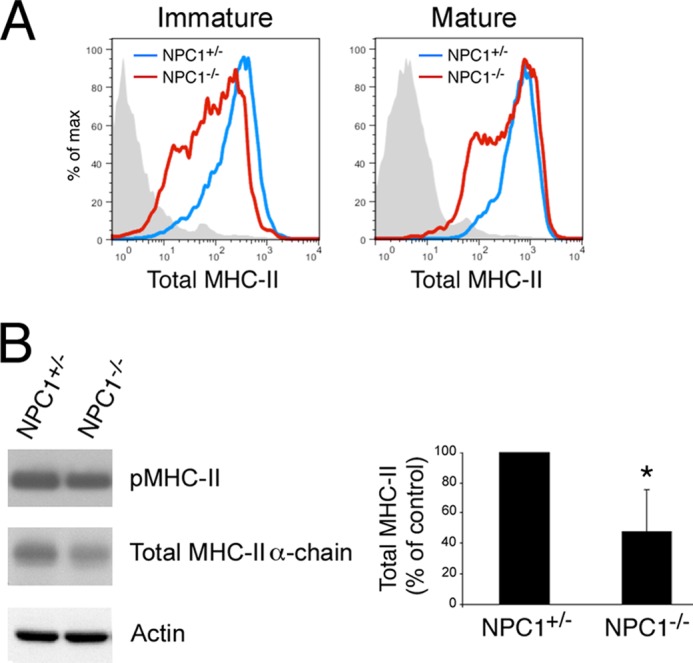
NPC1−/− DCs are activated normally by LPS and generate MHC-II-peptide complexes. A, immature or LPS-matured control (blue lines) or NPC1−/− (red lines) DCs were monitored by flow cytometry for surface expression of total surface MHC-II using mAb M5/114. B, LPS-matured control or NPC1−/− DCs were harvested and lysed in Triton X-100. Aliquots of the lysates were incubated with SDS-PAGE sample buffer and were either left at room temperature for 30 min or boiled for 3 min prior to SDS-PAGE. SDS stable pMHC-II complexes (53 kDa), total MHC-II α-chain (35 kDa), and actin (41 kDa) were detected by immunoblotting. The total amount of MHC-II present in each sample was quantitated by densitometry, normalized to the amount of actin present in the sample, and expressed as a percentage of total MHC-II present in control DCs.
Proteolysis Is Unaffected in NPC1−/− DCs
The ability of NPC1−/− DCs to generate SDS stable MHC-II-peptide complexes suggests that MVB integrity is not essential for antigen proteolysis. To examine this directly, control DCs and NPC1−/− DCs were incubated with different amounts of a dye-quenched form of the model protein antigen OVA, DQ-OVA. This form of OVA is fluorescent only after proteolytic cleavage. Quantitative analysis of DQ-OVA fluorescence revealed that OVA proteolysis was essentially identical in control and NPC1−/− DCs (Fig. 3A), showing that proteolytic cleavage of protein antigens is equally efficient in control and NPC1−/− DCs.
FIGURE 3.

Antigen uptake and proteolysis as well as Ii association and CLIP removal from MHC-II are normal in NPC1−/− DCs. A, immature NPC1+/− (control, blue) and NPC1−/− (red) DCs were incubated with the indicated amount of DQ-OVA for 1.5 h at 37 °C. Uptake and degradation were monitored by flow cytometry. The mean fluorescence intensity of DQ-OVA taken up and processed in NPC1−/− DCs was expressed relative to control DCs. B, mature NPC1+/− (control) and NPC1−/− DCs were lysed in Triton X-100, and immunoprecipitations were performed using isotype control (IgG2b) and anti-MHC-II mAb. Portions of each immunoprecipitate were analyzed by immunoblotting for the presence of intact Ii or MHC-II β-chain. The amount of Ii present in the MHC-II immunoprecipitate from NPC1−/− DCs was expressed as a percentage of Ii present in control DCs. Error bars, S.D. C, mature NPC1+/− (solid lines) and NPC1−/− DCs (dashed lines) were incubated with the MHC-II-CLIP mAb 15G4, washed, and counterstained with the pan-MHC-II mAb M5/114. The stained cells were analyzed by FACS, and cells in the MHC-II low and MHC-II high gates were analyzed separately for expression of total MHC-II and MHC-II-CLIP complexes.
Antigenic peptides are unable to bind to nascent MHC-II molecules until the MHC-II-associated Ii is proteolytically degraded and the remaining CLIP-fragment of Ii is removed from the MHC-II peptide binding groove by H2-DM (20). Analysis of anti-MHC-II immunoprecipitates revealed that association of intact Ii with MHC-II is normal in NPC1−/− DCs (Fig. 3B). We also analyzed the MHC-II low and MHC-II high populations in both control and NPC1−/− DCs to examine the presence of MHC-II-CLIP complexes in these DCs. Total MHC-II and MHC-II-CLIP expression on the surface of control and NPC1−/− DCs were essentially identical when the MHC-II low and MHC-II high population were analyzed separately (Fig. 3C). Taken together, these data reveal that whereas genetic deletion of NPC1 profoundly alters MVB morphology and almost completely eliminates the ILV from the MVB in DCs, this mutation has no significant effect on antigen proteolysis, Ii binding to MHC-II, the generation of MHC-II CLIP complexes, and high affinity peptide binding to MHC-II.
NPC1−/− DCs Function as APCs in Vitro
To determine whether the profound block in MVB formation and the aberrant localization of MHC-II to the MVB limiting membrane affect APC function, we assessed the ability of NPC1−/− DCs to stimulate Ag-specific T cells. Unsorted bone marrow-derived DCs from control and NPC1−/− mice were pulsed with intact OVA protein, and the ability of the cells to stimulate the proliferation of OVA-specific OTII CD4 T cells was determined. Antigen presentation of OVA peptide requires the activity of the late endosomal/lysosomal proteinases cathepsin D (21) and the expression of newly synthesized (and not recycling) MHC-II (22) and is dependent on the presence of Ii and the activity of H2-DM in these late endocytic compartments (23). Unsorted NPC1−/− DCs were poor stimulators of OTII T cells compared with control DCs (Fig. 4A). However, when MHC-II high cells isolated by FACS were used as APCs we found no significant alteration in APC function in NPC1−/− DCs compared with control DCs (Fig. 4, B and C). A dose-response titration revealed that NPC1−/− DCs were as efficient as wild-type DCs in activating OTII T cells when pulsed with 0.02, 0.10, 0.5, or 1.0 mg/ml OVA protein antigen (data not shown). Curiously, the MHC-II low cells, isolated either from control DCs or NPC1−/− DCs, were unable to activate OTII T cells under the conditions of this assay, revealing that the difference in T cell proliferation between unsorted control DCs and NPC1−/− DCs is due to the difference in the proportion of MHC-II high cells in the population. The sorted MHC-II high cells obtained from NPC1−/− mice were also specifically analyzed by immunofluorescence microscopy which confirmed that these cells possessed enlarged cholesterol-laden MHC-II compartments like that shown in Fig. 1 (data not shown). NPC1−/− mice were also back-crossed onto the H-2k background, and NPC1−/− DCs obtained from these mice were just as effective as control DCs in activating HEL-specific 3A9 CD4 T cells when pulsed with either high or low dose HEL protein antigen (supplemental Fig. 4). These data show that NPC1−/− DCs are effective APCs when one controls for the total amount of MHC-II on their surface.
FIGURE 4.

NPC1−/− DCs effectively activate antigen-specific naïve CD4 T cells in vitro. A and B, immature NPC1+/− (control) and NPC1−/− DCs were incubated with ovalbumin protein, washed, and activated with LPS overnight. The cells were then stained with pan-MHC-II mAb, and MHC-II high cells were sorted by FACS. Aliquots of the unsorted cells (A) or isolated MHC-II high cells (B) were analyzed for total MHC-II expression by FACS and were incubated with CFSE-labeled naïve OTII CD4 T cells at a 1:10 ratio. CFSE dilution was examined 72 h later by FACS analysis. C, the percentage of CFSE-labeled OTII T cells proliferating more than once when incubated with either unsorted DCs or the isolated MHC-II high DCs was calculated. The data shown are the mean ± S.D. (error bars) from three independent experiments.
APCs from NPC1−/− Mice Function in Vivo
Because NPC1−/− DCs were able to function in vitro as effective APCs, we asked whether APC function in living NPC1−/− mice was normal. DCs are the primary stimulators of naïve CD4 T cells in vivo, and therefore antigen-specific CD4 T cell proliferation in immunized NPC1−/− mice was used as a measure of in vivo APC activity. To control for the fact that NPC1-deficient CD4 T cells themselves could have altered function, we transferred purified OVA-specific naïve CD4 T cells into either control or NPC1−/− mice 1 day prior to immunization with OVA protein. OVA-specific T cells proliferated in NPC1−/− mice to nearly the same extent as in control mice (Fig. 5A). There was routinely a small (but statistically insignificant) reduction in T cell proliferation in immunized NPC1−/− mice; however, this could be readily explained by the slight reduction in total CD11c+ DCs present in the spleens of NPC1−/− mice compared with control mice (Fig. 5B). Importantly, when CD11c+ DCs were isolated from NPC1−/− mice we found that these DCs also possessed cholesterol-laden intracellular compartments, and some of these compartments still possessed accumulated MHC-II (Fig. 5C). These data thus demonstrate that the significant alterations in MVB morphology present in DCs obtained from NPC1−/− mice do not impair antigenic peptide loading and argue against an important role for multivesicular morphology in APC function.
FIGURE 5.

NPC1−/− DCs effectively activate antigen-specific naïve CD4 T cells in vivo. A, CFSE-labeled naïve OTII CD4 T cells (CD45.1) were transferred into NPC1+/− (control) and NPC1−/− mice (CD45.2) and followed by immunization 24 h later with ovalbumin protein. After 3 days CD45.1+ CD4 T cells from the spleen were isolated, and CFSE dilution was examined by FACS analysis. B, the absolute number of CD11chigh MHC-II+ DCs present in the spleens of NPC1+/− (control) and NPC1−/− mice was determined by FACS analysis. C, CD11c+ DCs were isolated from the spleens of NPC1+/− (control) and NPC1−/− mice by immunomagnetic purification. Live cells were attached to polylysine-coated coverslips prior to fixation, permeabilization, and staining with MHC-II mAb (M5/114, green) and counterstaining with filipin to detect unesterified cholesterol (red).
MHC-II molecules are primarily localized on the ILV of MVBs in APCs (3). Whereas the peptide editor H2-DM is localized primarily on the limiting membrane of MVBs, small amounts of H2-DM are routinely observed on the ILV (3, 24). It has been proposed that productive interactions of MHC-II and H2-DM, which are necessary for CLIP removal from MHC-II and high affinity peptide binding to MHC-II, only occur when MHC-II and H2-DM interact when each are present on the ILV of MVB (4). The cholesterol accumulation defect in NPC1−/− DCs results in severe perturbations of MVB morphology, and there are very few intact MVBs in these cells. Despite this fact, however, we find that MHC-II/H2-DM interactions are essentially normal when both MHC-II and H2-DM are restricted to the limiting (outer) membrane of MVBs. These data argue strongly that the localization of MHC-II to ILV is not essential for APC function.
If localization of MHC-II to ILV is not important for APC function, what then is the reason for this unique distribution observed in APCs? The ILVs of MVB give rise to the exosomes that are secreted from cells after fusion of the MVB with the plasma membrane (25), and antigen-specific T cell engagement can stimulate release of exosomes from APCs (8). In addition, the multivesicular nature of the MVB serves to increase the membrane surface area, allowing for an increased concentration of membrane proteins in this tightly packed organelle. Because exosome secretion is not likely to play an important role in the stimulation of naïve CD4 T cells observed here (8), we propose that the localization of MHC-II to the ILV serves primarily to package large amounts of this membrane protein in a restricted space, thereby allowing access of the “functional” region of MHC-II (the peptide binding groove) to the lumen of the organelle where antigenic peptides are located.
Acknowledgments
We thank Steve Shaw for critical reading of the manuscript; R. Erickson for providing NPC1−/− mice; and R. Germain, J. Strominger, and A. Rudensky for the antibodies.
This work was supported, in whole or in part, by the Intramural Research Program of the National Institutes of Health (to P. A. R.).

This article contains supplemental Figs. 1–4.
- APC
- antigen-presenting cell
- Ag
- antigen
- CFSE
- carboxyfluorescein succinimidyl ester
- DC
- dendritic cell
- Ii
- Invariant chain
- ILV
- intraluminal vesicle
- MVB
- multivesicular body
- NPC1
- Niemann-Pick type C1
- OVA
- ovalbumin.
REFERENCES
- 1. Berger A. C., Roche P. A. (2009) MHC class II transport at a glance. J. Cell Sci. 122, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCormick P. J., Martina J. A., Bonifacino J. S. (2005) Involvement of clathrin and AP-2 in the trafficking of MHC class II molecules to antigen-processing compartments. Proc. Natl. Acad. Sci. U.S.A. 102, 7910–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kleijmeer M., Ramm G., Schuurhuis D., Griffith J., Rescigno M., Ricciardi-Castagnoli P., Rudensky A. Y., Ossendorp F., Melief C. J., Stoorvogel W., Geuze H. J. (2001) Reorganization of multivesicular bodies regulates MHC class II antigen presentation by dendritic cells. J. Cell Biol. 155, 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zwart W., Griekspoor A., Kuijl C., Marsman M., van Rheenen J., Janssen H., Calafat J., van Ham M., Janssen L., van Lith M., Jalink K., Neefjes J. (2005) Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity 22, 221–233 [DOI] [PubMed] [Google Scholar]
- 5. Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Gu J., Rosenfeld M. A., Pavan W. J., Krizman D. B., Nagle J., Polymeropoulos M. H., Sturley S. L., Ioannou Y. A., Higgins M. E., Comly M., Cooney A., Brown A., Kaneski C. R., Blanchette-Mackie E. J., Dwyer N. K., Neufeld E. B., Chang T. Y., Liscum L., Strauss J. F., 3rd, Ohno K., Zeigler M., Carmi R., Sokol J., Markie D., O'Neill R. R., van Diggelen O. P., Elleder M., Patterson M. C., Brady R. O., Vanier M. T., Pentchev P. G., Tagle D. A. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277, 228–231 [DOI] [PubMed] [Google Scholar]
- 6. Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., Pentchev P. G., Pavan W. J. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277, 232–235 [DOI] [PubMed] [Google Scholar]
- 7. Inaba K., Turley S., Iyoda T., Yamaide F., Shimoyama S., Reis e Sousa C., Germain R. N., Mellman I., Steinman R. M. (2000) The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J. Exp. Med. 191, 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muntasell A., Berger A. C., Roche P. A. (2007) T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. 26, 4263–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walseng E., Bakke O., Roche P. A. (2008) Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J. Biol. Chem. 283, 14717–14727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tallóczy Z., Martinez J., Joset D., Ray Y., Gácser A., Toussi S., Mizushima N., Nosanchuk J., Goldstein H., Loike J., Sulzer D., Santambrogio L. (2008) Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PLoS Pathog. 4, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Potolicchio I., Chitta S., Xu X., Fonseca D., Crisi G., Horejsi V., Strominger J. L., Stern L. J., Raposo G., Santambrogio L. (2005) Conformational variation of surface class II MHC proteins during myeloid dendritic cell differentiation accompanies structural changes in lysosomal MIIC. J. Immunol. 175, 4935–4947 [DOI] [PubMed] [Google Scholar]
- 12. Poloso N. J., Muntasell A., Roche P. A. (2004) MHC class II molecules traffic into lipid rafts during intracellular transport. J. Immunol. 173, 4539–4546 [DOI] [PubMed] [Google Scholar]
- 13. Demel R. A., Van Deenen L. (1965) Penetration of lipid monolayers by polyene antibiotics: correlation with selective toxicity and mode of action. J. Biol. Chem. 240, 2749–2753 [PubMed] [Google Scholar]
- 14. Liscum L., Faust J. R. (1987) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 262, 17002–17008 [PubMed] [Google Scholar]
- 15. Zhang J. R., Coleman T., Langmade S. J., Scherrer D. E., Lane L., Lanier M. H., Feng C., Sands M. S., Schaffer J. E., Semenkovich C. F., Ory D. S. (2008) Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J. Clin. Invest. 118, 2281–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vanier M. T., Rodriguez-Lafrasse C., Rousson R., Duthel S., Harzer K., Pentchev P. G., Revol A., Louisot P. (1991) Type C Niemann-Pick disease: biochemical aspects and phenotypic heterogeneity. Dev. Neurosci. 13, 307–314 [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi T., Beuchat M. H., Lindsay M., Frias S., Palmiter R. D., Sakuraba H., Parton R. G., Gruenberg J. (1999) Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1, 113–118 [DOI] [PubMed] [Google Scholar]
- 18. Tang Y., Leao I. C., Coleman E. M., Broughton R. S., Hildreth J. E. (2009) Deficiency of Niemann-Pick type C1 protein impairs release of human immunodeficiency virus type 1 and results in GAG accumulation in late endosomal/lysosomal compartments. J. Virol. 83, 7982–7995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tulp A., Verwoerd D., Dobberstein B., Ploegh H. L., Pieters J. (1994) Isolation and characterization of the intracellular MHC class II compartment. Nature 369, 120–126 [DOI] [PubMed] [Google Scholar]
- 20. Busch R., Rinderknecht C. H., Roh S., Lee A. W., Harding J. J., Burster T., Hornell T. M., Mellins E. D. (2005) Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol. Rev. 207, 242–260 [DOI] [PubMed] [Google Scholar]
- 21. Rodriguez G. M., Diment S. (1992) Role of cathepsin D in antigen presentation of ovalbumin. J. Immunol. 149, 2894–2898 [PubMed] [Google Scholar]
- 22. St-Pierre Y., Watts T. H. (1990) MHC class II-restricted presentation of native protein antigen by B cells is inhibitable by cycloheximide and brefeldin A. J. Immunol. 145, 812–818 [PubMed] [Google Scholar]
- 23. Miyazaki T., Wolf P., Tourne S., Waltzinger C., Dierich A., Barois N., Ploegh H., Benoist C., Mathis D. (1996) Mice lacking H2-M complexes, enigmatic elements of the MHC class II peptide-loading pathway. Cell 84, 531–541 [DOI] [PubMed] [Google Scholar]
- 24. Stang E., Guerra C. B., Amaya M., Paterson Y., Bakke O., Mellins E. D. (1998) DR/CLIP (class II-associated invariant chain peptides) and DR/peptide complexes colocalize in prelysosomes in human B lymphoblastoid cells. J. Immunol. 160, 4696–4707 [PubMed] [Google Scholar]
- 25. Murk J. L., Stoorvogel W., Kleijmeer M. J., Geuze H. J. (2002) The plasticity of multivesicular bodies and the regulation of antigen presentation. Semin. Cell Dev. Biol. 13, 303–311 [DOI] [PubMed] [Google Scholar]