

Background: Post-translational modifications of CRMP2 protein direct its regulation of effector proteins.
Results: Destruction of a CRMP2 SUMOylation site reduces surface expression and current density of sodium channel NaV1.7.
Conclusion: CRMP2 SUMOylation choreographs NaV1.7, but not NaV1.1 or NaV1.3, trafficking.
Significance: Learning how neuronal NaV1.7 trafficking is modulated by CRMP2 is important for understanding the mechanism of action of NaV-targeted anti-epileptic and anti-nociceptive drugs.
Keywords: Patch Clamp Electrophysiology, Signaling, Sodium Channels, Sumoylation, Trafficking, CRMP2, NaV1.1, NaV1.7, Post-translational Modification
Abstract
Voltage-gated sodium channel (NaV) trafficking is incompletely understood. Post-translational modifications of NaVs and/or auxiliary subunits and protein-protein interactions have been posited as NaV-trafficking mechanisms. Here, we tested if modification of the axonal collapsin response mediator protein 2 (CRMP2) by a small ubiquitin-like modifier (SUMO) could affect NaV trafficking; CRMP2 alters the extent of NaV slow inactivation conferred by the anti-epileptic (R)-lacosamide, implying NaV-CRMP2 functional coupling. Expression of a CRMP2 SUMOylation-incompetent mutant (CRMP2-K374A) in neuronal model catecholamine A differentiated (CAD) cells did not alter lacosamide-induced NaV slow inactivation compared with CAD cells expressing wild type CRMP2. Like wild type CRMP2, CRMP2-K374A expressed robustly in CAD cells. Neurite outgrowth, a canonical CRMP2 function, was moderately reduced by the mutation but was still significantly higher than enhanced GFP-transfected cortical neurons. Notably, huwentoxin-IV-sensitive NaV1.7 currents, which predominate in CAD cells, were significantly reduced in CAD cells expressing CRMP2-K374A. Increasing deSUMOylation with sentrin/SUMO-specific protease SENP1 or SENP2 in wild type CRMP2-expressing CAD cells decreased NaV1.7 currents. Consistent with a reduction in current density, biotinylation revealed a significant reduction in surface NaV1.7 levels in CAD cells expressing CRMP2-K374A; surface NaV1.7 expression was also decreased by SENP1 + SENP2 overexpression. Currents in HEK293 cells stably expressing NaV1.7 were reduced by CRMP2-K374A in a manner dependent on the E2-conjugating enzyme Ubc9. No decrement in current density was observed in HEK293 cells co-expressing CRMP2-K374A and NaV1.1 or NaV1.3. Diminution of sodium currents, largely NaV1.7, was recapitulated in sensory neurons expressing CRMP2-K374A. Our study elucidates a novel regulatory mechanism that utilizes CRMP2 SUMOylation to choreograph NaV1.7 trafficking.
Introduction
Collapsin response mediator protein 2 (CRMP2)3 specifies axon/dendrite fate and axonal outgrowth (1). Mapping the CRMP2 interactome has unraveled novel targets that enable it to subserve roles in regulation of microtubule dynamics, protein endocytosis, vesicle recycling, and synaptic assembly within neurons (2). Trafficking of ligand- and voltage-gated calcium channels has been recently demonstrated as an additional role for CRMP2 (3–6). In addition to protein targets, CRMP2 is the presumptive secondary target of the anti-epileptic drug (R)-lacosamide ((2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide; LCM), which has its primary action on voltage-gated sodium channels (VGSCs) (7, 8). By stabilizing sodium channels in the slow-inactivated state, LCM is believed to reduce the pathological activity of hyperexcitable neurons typified by prolonged depolarizations, without affecting normal physiological activity (7). We have shown that mutations within putative LCM-binding pockets in CRMP2 reduce LCM-induced shifts in slow inactivation of sodium channels compared with channels in the neuronal model CAD cells expressing wild type CRMP2 (9). Our data have also demonstrated CRMP2 labeling by fluorescent analogs of LCM that could be competitively displaced by excess LCM in rat brain lysates, implying binding of LCM to CRMP2 (9). In contrast to our findings, radiolabeled LCM was not found to bind to lysates from CRMP2-injected oocytes (10). Additionally, CRMP2 was shown to not interact with NaV (9). Thus, the exact mechanism by which CRMP2 modulates LCM-induced slow inactivation of sodium channels remains unclear. CRMP2 modulation of voltage-gated sodium channels may be predicated on involvement of a tertiary protein or modification of either protein.
Our laboratory is interested in delineating the mechanisms by which CRMP2 is directed to its many effectors and how it regulates their functions. Previous studies have implicated post-translational modifications, particularly phosphorylation, in directing CRMP2 interactions and regulating the function of the partner proteins. For instance, phosphorylation by cyclin-dependent kinase 5 (Cdk5) enhances interaction between CRMP2 and voltage-gated calcium channels to modulate calcium influx (11). Phosphorylation by glycogen synthase kinase 3β or Rho-associated protein kinase (ROCK) lowers the capability of CRMP2 to bind to tubulin heterodimers leading to microtubule destabilization, culminating in axon retraction/growth cone collapse (12–14). Oxidation of CRMP2 has been shown to link the redox protein thioredoxin to regulation of CRMP2 phosphorylation and semaphorin 3A-induced growth cone collapse (15). Proteolysis of CRMP2 by calpains has also been reported and is believed to contribute to neurodegeneration and cell death (5, 16). In addition to these modifications, we recently reported that CRMP2 is subject to a novel modification, the small ubiquitin-like modifier (SUMO) (17).
SUMOylation is a covalent, reversible modification that can add one of three ∼11-kDa SUMO proteins to lysines within target proteins. As with ubiquitination, a cascade of three enzymes, E1-activating, E2-conjugating, and E3-ligase, produce an isopeptide bond between the C-terminal glycine of SUMO1–3 and an ϵ-amino group of a target lysine within a SUMO motif on the acceptor protein (18). A typical SUMO motif is characterized by a large hydrophobic amino acid (ψ) preceding a target lysine followed by a negatively charged amino acid two sites downstream (ψKX(E/D)) (18–20). The E2 enzyme Ubc9 may conjugate one or more of the three vertebrate SUMO proteins to a SUMO motif (21), whereas the sentrin/SUMO-specific protease (SENP) 1 and SENP2 removes SUMOs, thus reversing the modification (22). SUMOylation has been implicated in modulation of several ion channels and receptors, including inactivation of voltage-gated potassium channels Kv1.5 (23), activation of Kv2.1 (24), activity of two-pore domain potassium leak channel K2P1 (25, 26), and endocytosis of glutamate receptor isoform 6 (GluR6) (27). Glutamate receptor isoforms GluR7 and metabotropic GluR (mGluRs)-2, -4, -6, -7a, and -7b have also been shown to be targets of SUMOylation (28).
Based on our findings that CRMP2 expression can alter the action of a modulator (i.e. LCM) of VGSCs (9, 29), we postulated that CRMP2 modification by SUMOylation might modulate the effect of CRMP2 on VGSCs. We tested this hypothesis by examining sodium currents in CAD cells expressing a mutant CRMP2, wherein the SUMO-targeted lysine 374 in CRMP2 was mutated to alanine (CRMP2-K374A) or all three residues of the SUMO consensus motif were mutated to alanines (CRMP2AAA). The CRMP2 SUMO-incompetent mutant expressed robustly and remained functional and able to promote neurite outgrowth. Remarkably, whereas LCM-induced enhancement in slow inactivation was unchanged in CAD cells expressing CRMP2-K374A, their current density, carried via huwentoxin-IV-sensitive NaV1.7 channels, was significantly decreased. Biotinylation experiments confirmed the loss of surface NaV1.7. The effects of CRMP2-K374A expression on current density were recapitulated in a heterologous cell line expressing NaV1.7. In contrast, the current densities of NaV1.1 or NaV1.3 were unaffected by CRMP2-K374A expression. Notably, CRMP2-K374A expression reduced sodium currents in nociceptive neurons that express high levels of NaV1.7 (30). Thus, our results identify SUMOylation of CRMP2 as a novel mechanism for the modulation of NaV1.7 trafficking.
EXPERIMENTAL PROCEDURES
Plasmids and Antibodies
The following plasmids were from Addgene (Cambridge, MA): HA-SUMO-1, HA-SUMO-2, HA-SUMO-3, HA-Ubc9, FLAG-SENP1, and FLAG-SENP2. Mutations in mouse CRMP2 cDNA (Mus musculus, GenBankTM accession number NM_009955.3) were introduced by QuikChange II XL (Agilent Technologies, Santa Clara, CA) (11) and cloned into FLAG epitope containing pCDNA3.1 plasmid. The introduced alanine mutations were verified by DNA sequencing. Although typically arginine mutations have been used to investigate putative SUMOylation status of proteins, this is not always the case as illustrated by a study wherein the Lys to Arg mutation in the potassium leak channel K2P1 failed to increase potassium currents (31). For this reason and additional ones described under “Results,” we chose to mutate the lysine residue to an alanine. A polyclonal FLAG epitope antibody and a monoclonal β-tubulin antibody were purchased from Sigma; the monoclonal NaV1.7 was from NeuroMab (Davis, CA), and the polyclonal pan-NaV antibody was from Alomone Laboratories (Jerusalem, Israel).
Primary Cortical Neuron Cultures, Transfection, and Neurite Outgrowth Analyses
Embryonic day 19 cortical neurons were prepared exactly as described (5). Briefly, cortices were dissected, and cells suspensions were plated onto poly-d-lysine-coated 96-well plates. Cells were grown in Neurobasal medium containing 2% NuSerum, 5% NS21, supplemented with penicillin/streptomycin (100 units/ml; 50 μg/ml), 0.1 mm l-glutamine, and 0.4 mm l-GlutaMAX (Invitrogen). Forty eight hours after plating, cells were fed with media containing 5-fluoro-2′-deoxyuridine (1.5 μg/ml) (Sigma) to reduce the number of non-neuronal cells. At DIV4, cells were transfected with either EGFP, wild type CRMP2, or CRMP2-K374A + 10% EGFP via Lipofectamine 2000 (Invitrogen). Transfections were allowed to proceed for ∼3 h. At DIV6, cells were fixed with 4% paraformaldehyde (Sigma) and imaged using the ImageXpress Micro Widefield High Content Screening System (Molecular Devices). Multiple parameters involved in neurite outgrowth were examined via the neurite outgrowth application module within the MetA Xpress software. This analysis combines the following measurements: number of primary neurites, number of branches, mean process length, and maximum process length to determine a summary of total outgrowth per cell.
Culturing CAD Cells and Transfection
The neuronally derived CAD cells were grown at 37 °C and in 5% CO2 as described previously (9, 32, 33). CAD cells were transfected with 1 μg/μl of polyethyleneimine (Sigma) (34) and 2 μg of CRMP2, CRMP2-K374A, SUMO1–3, Ubc9, or SENP1/2 cDNAs plus EGFP plasmid (0.2 μg). Under these conditions, transfection efficiencies of ∼85–90% were routinely observed along with ∼5% cell death. Twenty four hours after transfection, cells were plated on 12-mm glass coverslips (Electron Microscopy Sciences, Hatfield, PA) coated with laminin (VWR, Randor, PA). Experiments were performed 48–72 h after transfection. Efficiency of CAD cell transfection was >80% with this method. Huwentoxin-IV (Alomone Laboratories) was used to isolate NaV1.7 currents in CAD cells. The toxin was used at 125 nm, ∼5 times the IC50 for NaV1.7 (35); at this concentration it does not block NaV1.1 or NaV.1.3, which account for less than 8% of the NaV mRNA in CAD cells (32).
Culturing Human Embryonic Kidney 293 (HEK293) Cells Expressing NaV1.1, NaV1.3, and NaV1.7 and Transfection
These cell lines were obtained from Dr. Theodore R. Cummins (Indiana University School of Medicine). The cDNA gene encoding NaV1.1 was codon-optimized and synthesized using the open reading frame (GenBankTM accession number NC_000002.11) and subcloned into the vector pTarget. The cDNA genes encoding NaV1.3 from rat and NaV1.7 from human were subcloned into the vector pcDNA3.1-mod. The constructs were then transfected into HEK293 cells using the calcium phosphate precipitation technique. After 48 h, the cells were passaged into 100-mm dishes and treated with G418 (geneticin, Invitrogen) at 800 μg/ml to select for neomycin-resistant cells. After 2 weeks, colonies were picked and split. The colonies were then tested for channel expression with the whole cell patch clamp technique. The cell line was then maintained with 500 μg/ml G418. NaV1.1 (36), NaV1.3 (37), and NaV1.7 (38) stable cells were grown under standard tissue culture conditions (5% CO2 at 37 °C) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HEK293 cell transfections were preformed exactly as described above for CAD cells.
Culturing Primary Dorsal Root Ganglion (DRG) Neurons and Transfection
DRG neurons were isolated from 150 to 174 g female Sprague-Dawley rats using procedures developed previously (39–41). Isolated DRG neurons (∼1.5 × 106) were transfected with 0.5 μg of EGFP and 1.5 μg of wild type CRMP2 or CRMP2-374A cDNA using the rat neuron NucleofectorTM solution (program O-003; Amaxa Biosystems, Lonza Cologne, Germany) and plated on a 12-mm BD BioCoatTM poly-d-lysine/laminin-coated glass coverslips (BD Biosciences) and maintained at 37 °C and 3% CO2 in DMEM supplemented with nerve growth factor (30 ng/ml) (42, 43). Under these conditions, transfection efficiencies of ∼20–25% were routinely observed along with ∼10–15% cell death.
Patch Clamp Electrophysiology
Whole cell voltage clamp recordings were performed at room temperature on CAD, HEK293, or DRG cells using an EPC10 amplifier (HEKA Electronics, Germany). Electrodes were pulled from thin walled borosilicate glass capillaries (Warner Instruments, Hamden, CT) with a P-97 electrode puller (Sutter Instrument, Novato, CA) such that final electrode resistances were 1–2 megohms when filled with internal solutions. The internal solution for recording Na+ currents contained (in mm) the following: 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATP Na2-ATP, and 25 HEPES (pH 7.2) (290–310 mosm/liter). The external solution contained (in mm) the following: 100 NaCl, 10 tetraethylammonium chloride, 1 CaCl2, 1 CdCl2, 1 MgCl2, 10 d-glucose, 4 4-aminopyridine, 0.1 NiCl2, 10 HEPES (pH 7.3) (310–315 mosm/liter). Whole cell capacitance and series resistance were compensated with the amplifier. Series resistance error was always compensated to be less than ±3 mV. Linear leak currents were digitally subtracted by P/4. Signals were filtered at 10 kHz and digitized at 10–20 kHz. Analysis was performed using FitMaster (HEKA Electronics, Germany) and Origin8.6 (OriginLab Corp., Northampton, MA).
Cell Surface Biotinylation
Biotinylation was performed as described previously (5, 44, 45). CAD cells were transfected with CRMP2 or CRMP2-K374A alone or with SENP1 plus SENP2 and processed for biotinylation experiments 48 h later. Briefly, live cells were incubated with sulfosuccinimidyl 2-(biotinamido)ethyl-1,3′dithiopropionate (EZ-link Sulfo NHS-SS-biotin; 1 mg/mg protein, Pierce) for 30 min at 4 °C in cold PBS (pH 8.0). Excess biotin was quenched with PBS containing 100 mm glycine and washed three times with ice-cold PBS, and the pellet was resuspended in RIPA lysis buffer. The resuspended pellet was triturated 10 times (with a 25-gauge needle) and centrifuged at 100,000 × g for 20 min. The biotinylated proteins were separated from clear solubilizate by adsorption onto streptavidin-agarose beads (Novagen) for 2–4 h at 4 °C. Beads were washed 3–5 times with RIPA buffer, and bound biotinylated proteins were gently eluted off the beads with RIPA buffer containing 2% Triton X-100 and 650 mm NaCl by end-over-end incubation for 1 h at 30 °C. The biotinylated and total fractions were subjected to immunoblotting with β-tubulin and pan-NaV or NaV1.7 antibodies.
Immunocytochemistry, Confocal Microscopy
48–72 h after transfection, CAD cells were fixed with 4% paraformaldehyde (diluted in 0.1 mm PBS) for 10 min at room temperature, permeabilized with 0.2% Triton X-100 for 10 min, and then washed three times with 0.01 mm PBS. Cells were then preincubated with 10% bovine serum albumin (diluted in 0.1 mm PBS) for 1 h at RT to block nonspecific binding with the primary antibody. FLAG primary monoclonal antibody (Neuromab, Davis, CA) was diluted (in 0.1 mm PBS) to 1:500 and applied to the cells. After incubation for 2 h at room temperature, the CAD cells were washed again with PBS, and secondary antibodies (anti-mouse Alexa 488, 1:500; Molecular Probes, Eugene, OR) were incubated in blocking solution for 45 min at RT. Coverslips were mounted in Prolong Gold Antifade mounting media (Molecular Probes). CAD cells were imaged on a Nikon Ti swept-field confocal microscope using a ×60, 1.4 NA lens with a cooled Cascade 512B digital camera (Photometrics, Tucson, AZ). Z stack image pairs were captured through the sample. Images were deblurred off line by an iterative deconvolution protocol (Nikon Elements version 3.0) using a theoretical point-spread function.
Data Analysis
All data points are shown as mean ± S.E., and n is presented as the number of the separate experimental cells. Steady-state activation and inactivation curves were fitted using the Boltzmann equation as follows: y = 1/(1 + exp(V½ − V)/k), in which V½, V, and k represented midpoint voltage of kinetics, test potential, and slope factor, respectively. Statistical differences between control and experimental conditions were determined by using ANOVA with a Tukey's post hoc test or a Student's t test when comparing only two conditions. Values of p < 0.05 were judged to be statistically significant.
RESULTS
Identification of NDSM SUMOylation Motif in CRMP2
An analysis of the CRMP2 protein sequence revealed the presence of a single putative SUMOylation motif (GKMD; amino acids 373–376 of CRMP2) conforming to the canonical ψKX(E/D) SUMOylation motif (13). Further sequence alignments revealed that the motif also conformed to the more stringent negatively charged SUMOylation motif (NDSM) typified by the presence of a negatively charged acidic patch downstream of the canonical motif, which together promote E2-conjugating enzyme Ubc9-mediated addition of SUMO (Fig. 1A) (28). Sequence comparisons revealed full conservation across human, rodent, bovine, and fish species (data not shown) as well as across other CRMP family members (Fig. 1A). Lys-374, at the heart of the NDSM motif, lies on an accessible surface of CRMP2 monomers with likely unhindered access to Ubc9 and SUMO (Fig. 1B). To evaluate the effect of CRMP2 SUMOylation in vivo, we created a CRMP2 construct in which the SUMO target Lys-374 was substituted for alanine (CRMP2-K374A). It has been previously documented that mutation of SUMOylated lysine to either arginine or alanine blocks SUMO addition by Ubc9 (46–50). The CRMP2-K374A protein expressed robustly in heterologous CAD cells at levels similar to wild type CRMP2 (Fig. 2). A CRMP2 mutant harboring triple alanine mutations in CRMP2's putative SUMOylation consensus motif (KMD) was also created and expressed robustly (17).
FIGURE 1.
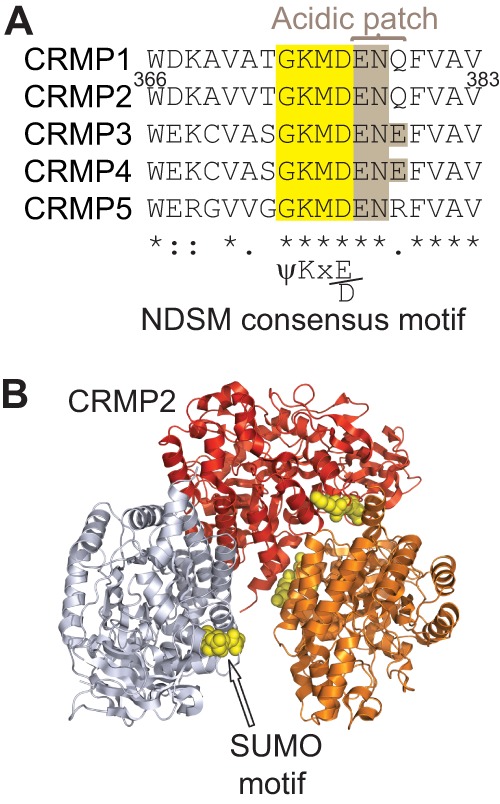
CRMP2 contains a conserved NDSM consensus motif. A, alignment of a short region of rodent CRMP1–5 sequences with the SUMOylation consensus motif of CRMPs highlighted in yellow. The presence of a negatively charged acidic patch (brown) flanking the canonical motif conforms to the more stringent negatively charged SUMOylation motif (NDSM). The numbers refer to the amino acid residues of rat CRMP2. B, structural representation of CRMP2 (Protein Data Bank code 2GSE (54)) with SUMOylation motif residues identified by yellow spheres (arrow). For clarity, only three monomers of the CRMP2 tetramer are shown.
FIGURE 2.
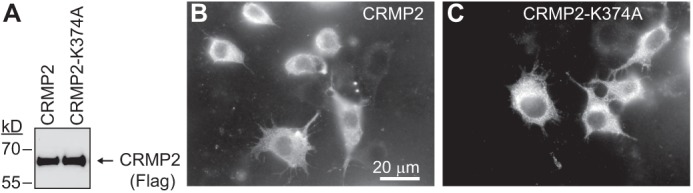
CRMP2-K374A mutant expresses robustly in CAD cells. A, immunoblot with anti-FLAG antibody from CAD lysates transfected with wild type (CRMP2) or the putative single site SUMOylation conjugation site mutant (CRMP2-K374A) showing comparable levels of expression of the proteins. Representative monochrome images of wild type CRMP2 (B) or mutant CRMP2–374A (C) expressed in CAD cells. Forty eight hours after transfection, the cells were permeabilized and then labeled with a mouse monoclonal antiserum directed against the FLAG epitope and visualized by incubation with fluorescent-conjugated Alexa 488 goat anti-mouse secondary antibody. Both proteins showed similar patterns of expression throughout the cytoplasm of the transfected cells. Scale bar, 20 μm.
CRMP2-K374A Mutant, Like Wild Type CRMP2, Augments Axonal Outgrowth
We and others have previously shown that CRMP2 expression specifies axonal ramification and dendritic complexity of neurons (1, 29). To test whether mutation of a putative SUMOylation motif affects this canonical function of CRMP2, we tested axonal outgrowth in DIV6 cortical neurons expressing EGFP, CRMP2-K374A, or wild type CRMP2. EGFP co-expression was used to identify transfected cells for analyses. Compared with EGFP-expressing neurons, both wild type CRMP2 and CRMP2-K374A increased the number of processes per neuron, the number of branches per neuron, the mean and maximum process lengths, and overall total outgrowth (Fig. 3). On average, the processes per neuron were increased by 21 and 29% for CRMP2 (n = 538) and CRMP2-K374A (n = 669), respectively, compared with EGFP-expressing neurons (4.6 ± 0.1, n = 682, p < 0.05). Branches per neuron were increased by 134 and 90% for CRMP2 and CRMP2-K374A, respectively, compared with EGFP-expressing neurons (25.7 ± 1.1, n = 682, p < 0.05). The mean and maximum process lengths per neuron were increased by 75 and 120% for CRMP2 and 18 and 76% for CRMP2-K374A, respectively, compared with EGFP-expressing neurons (191.6 ± 7.9, n = 682, p < 0.05 and 613.1 ± 28.8, n = 682, p < 0.05). Finally, overall total outgrowth, a sum of all the above parameters, was 113 and 74% greater in CRMP2 (n = 538)-expressing and CRMP2-K374A (n = 669)-expressing neurons, respectively, as compared with EGFP-expressing neurons (Fig. 3D). We have previously reported that elimination of the entire SUMO consensus motif, CRMP2AAA, also increases neurite outgrowth parameters compared with EGFP-expressing neurons (17). Collectively, these results suggest that replacement of CRMP2's lysine (residue 374) or all three residues within the putative SUMO motif maintains CRMP2-mediated axonal specification and dendritic complexity implying that the mutant protein is functional.
FIGURE 3.
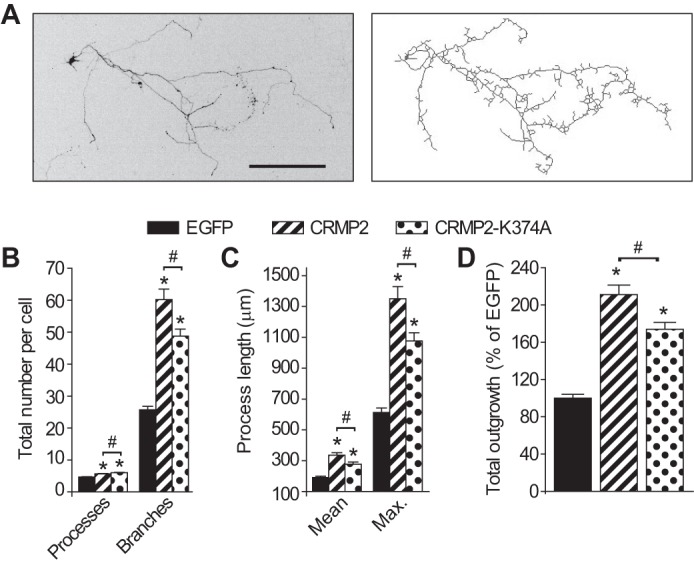
CRMP2 and CRMP2-K374A mutant increase neurite growth in neurons. A, representative inverted black and white image (left) and tracings (right) of a cortical neuron expressing CRMP2-K374A + 10% EGFP at 4× magnification. Scale bar, 200 μm. B, number of processes and branches; C, mean and maximum (max.) process length; and D, normalized total outgrowth for cells expressing EGFP alone (n = 538 from 8 wells), co-expressing EGFP and CRMP2 (n = 682 from 8 wells), or co-expressing EGFP and CRMP2-K374A (n = 669 from 8 wells). Asterisk indicates statistical significance compared with EGFP-transfected cells (p < 0.05, one-way ANOVA with Tukey's post hoc test). Hash mark represents statistical significance compared with wild type CRMP2-transfected cells (p < 0.05, one-way ANOVA with Tukey's post hoc test).
CRMP2AAA or CRMP2-K374A Mutants Do Not Alter LCM-induced Enhancement of Slow Inactivation
LCM stabilizes NaVs in a slow-inactivated state (7, 8, 51). We have previously shown that mutating an LCM-coordinating pocket within CRMP2 reduces LCM-dependent enhancement of slow inactivation (9). How this CRMP2 mutation changes LCM's efficacy toward the sodium channel is unknown. Because NaVs do not interact with CRMP2 directly (9), it is possible that a tertiary protein may be involved or modification of CRMP2 or the channel itself may facilitate the change. As we have recently identified SUMOylation of CRMP2 as a novel post-translational modification (17), here we tested whether SUMOylation of CRMP2 modifies LCM-induced enhancement of slow inactivation. Using the whole cell patch clamp configuration, we analyzed the effects of LCM on VGSCs in the presence of overexpressed wild type CRMP2, CRMP2AAA, and CRMP2-K374A. The E2-conjugating enzyme Ubc9, which adds SUMO1–3 (28), and SUMO2 were also expressed; SUMO2 was transfected as preliminary studies revealed no differences between any of the SUMOs on NaV current densities. Transfected CAD cells were conditioned to potentials ranging from −110 to +20 mV (in +10-mV increments) for 5 s, and then fast-inactivated channels were allowed to recover for 150 ms at a hyperpolarized pulse to −120 mV, and the fraction of channels available was tested by a single depolarizing pulse, to 0 mV, for 15 ms (Fig. 4A). Addition of 100 μm LCM to cells expressing wild type CRMP2, CRMP2AAA, and CRMP2-K374A significantly decreased the fraction of current available compared with those in the absence of LCM. For comparison, representative current traces at −50 mV are highlighted (Fig. 4B). At this potential, the channels are predominantly undergoing slow inactivation, as it is near the action potential threshold (9, 32). The slow inactivation versus voltage curves for the transfected cells in the absence and presence of 100 μm LCM are plotted in Fig. 4C and illustrate a marked reduction in current available (i.e. an enhancement in slow inactivation) in the presence of drug that was not different between the conditions tested (p > 0.05; ANOVA with Tukey's post hoc test). At −50 mV, ∼48% (n = 8) (calculated as 1 minus the normalized INa) of the Na+ current was available in LCM-treated CRMP2-expressing cells compared with 45% in CRMP2AAA and 43% in CRMP2-K374A-expressing cells (Fig. 4D; p > 0.05; ANOVA with Tukey's post hoc test). These results demonstrate that disruption of the SUMOylation site in CRMP2 does not affect LCM-induced enhancement of sodium channel slow inactivation.
FIGURE 4.
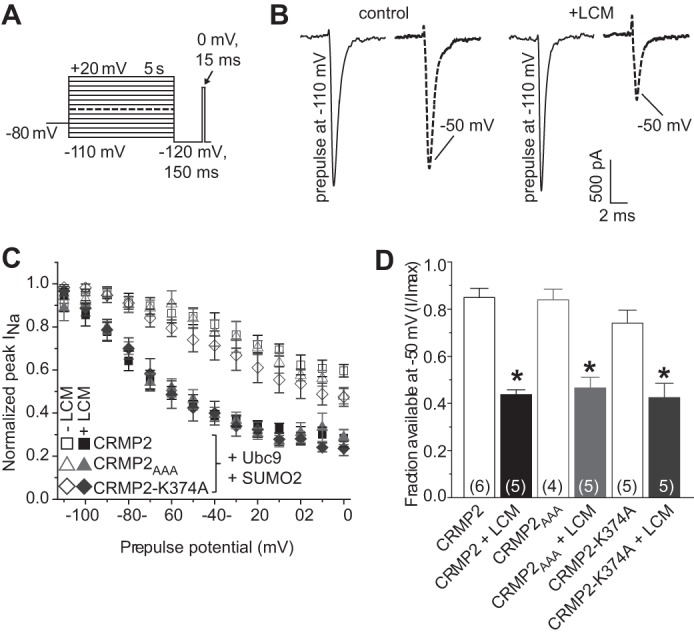
Disruption of CRMP2 SUMO motif does not affect LCM-induced enhancement of slow inactivation of sodium currents. A, voltage protocol for slow inactivation: currents were evoked by 5-s prepulses between −110 and +20 mV and then allowed to recover from fast inactivation for 150 ms at −110 mV. The fraction of available current was analyzed by a 15-ms test pulse to 0 mV. Cells were treated for 10 min with either 100 μm LCM or vehicle, 0.1% DMSO prior to whole cell patch clamp analysis. B, representative current traces from CAD cells expressing CRMP2 with Ubc9 and SUMO2 in the absence (control, 0.1% DMSO, left) or presence of 100 μm LCM (+LCM, right). To highlight the extent of slow inactivation, the maximum elicited current (prepulse to −110 mV) is compared with the trace elicited by a prepulse to −50 mV. The scale bar applies to all traces. C, summary of steady-state slow inactivation curves for CAD cells expressing both Ubc9 and SUMO2 and either CRMP2 (squares), CRMP2AAA (triangles), or CRMP2-K374A (diamonds) in the absence (open symbols) or presence (closed symbols) of 100 μm LCM. Some error bars are smaller than symbols. In this and subsequent figures, all data are represented as means ± S.E. D, summary of the fraction of available current at −50 mV for the same conditions presented in C. Asterisks indicate statistically significant differences in fraction between LCM-treated and untreated CAD cells expressing the indicated constructs (p < 0.05, one-way ANOVA with Tukey's post hoc test). Numbers in parentheses represent number of cells tested per condition.
CRMP2AAA and CRMP2-K374A Mutants Impair Macroscopic Sodium Current Density in CAD Cells
During the course of the slow inactivation experiments, we observed a diminution of macroscopic currents in CAD cells transfected with CRMP2 mutant constructs. Thus, we next tested if overexpression of wild type and CRMP2 mutant constructs could alter sodium current density. Current-voltage relationships in transfected CAD cells were examined by the application of 15-ms step depolarizations ranging from −70 to +80 mV (in +10-mV increments) from a holding potential of −80 mV (Fig. 5A). The transient inward current in CAD cells activated between −40 and −30 mV and reached its peak at 0 to +10 mV (Fig. 5, B and C). Peak inward Na+ currents were measured and expressed as peak current density (pA/pF) to account for variations in cell size. CAD cells expressing wild type CRMP2 exhibited a peak current density of −63.7 ± 9.8 pA/pF (n = 11), which was not significantly different from cells expressing EGFP alone (−60.1 ± 19.3 pA/pF (n = 9; p > 0.05, Student's t test)). In contrast, sodium currents were reduced to 28.0 ± 7.7% (n = 10) in CAD cells expressing CRMP2AAA and to 30.2 ± 8.7% (n = 10) in CAD cells expressing CRMP2-K374A (Fig. 5, B–D). The majority of these currents was via NaV1.7 as 81.3 ± 6.2% (n = 5) of the total current was blocked by the NaV1.7-selective agent huwentoxin-IV (125 nm) (35). Boltzmann parameters of voltage dependence of activation, half-activation voltage (V½), and slope factor (k) were not different across the various transfected conditions with the exception of V½ of activation being shifted by ∼5.7 mV in the hyperpolarizing direction in CAD cells expressing CRMP2-K374A when compared with those expressing EGFP (Table 1, p < 0.05, ANOVA with Tukey's post hoc test).
FIGURE 5.
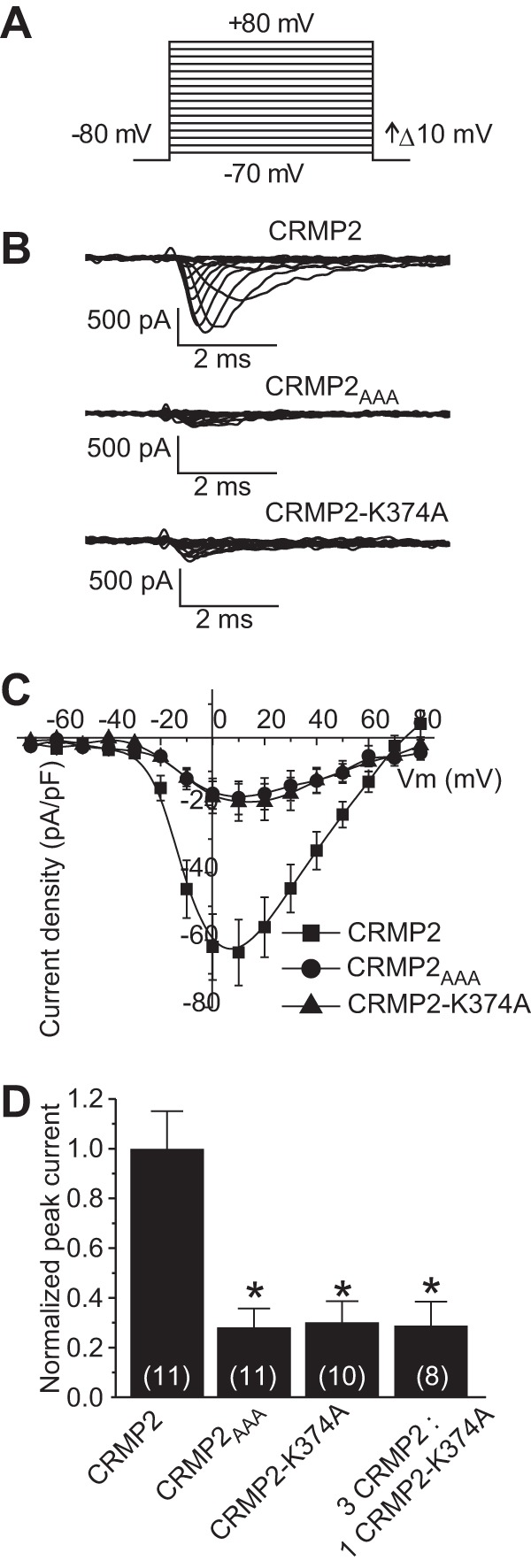
Disruption of CRMP2 SUMO motif reduces NaV1.7 current density. A, voltage protocol for current-voltage relationship: currents were evoked by 5-ms voltage steps from −70 to +80 mV in 10-mV increments. B, representative traces from CAD cells expressing CRMP2, CRMP2AAA, or CRMP2-K374A. Sodium currents were predominantly NaV1.7 as the NaV1.7-selective huwentoxin-IV blocked 81 ± 6.2% of total macroscopic sodium currents in these cells (n = 5, data not shown). C, current-voltage (I-V) relationships for the voltage step protocol shown in A. Peak currents were normalized to the cell capacitance. D, peak current density (pA/pF) measured at 0 to + 10 mV for wild type CRMP2-, CRMP2AAA-, CRMP2-K374A-, and CRMP2/CRMP2-K374A (3:1)-transfected CAD cells. Numbers in parentheses represent numbers of cells tested. Asterisks indicate statistically significant differences between CRMP2 and all other conditions tested (p < 0.05, one-way ANOVA with Tukey's post hoc test).
TABLE 1.
Comparative current densities, cell capacitances, and Boltzmann fits of voltage dependence of channel activation for the respective transfection conditions in CAD cells
Values for V½, the voltage of half-maximal activation, and slope were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean ± S.E. As two cells were tested for the voltage dependence of activation for the condition with 3 CRMP2/1 CRMP2-K374A, only the average data are presented. N values are indicated in parentheses. All transfection conditions contained EGFP for identification of construct expression.
| Transfection condition | Peak current density | Cell capacitance | Voltage dependence of activation |
|
|---|---|---|---|---|
| V1/2 | Slope | |||
| pA/pF | pF | mV | mV/e-fold | |
| EGFP | 60.1 ± 19.3 (9) | 16.0 ± 2.5 (9) | −12.0 ± 1.0 (8) | 7.0 ± 0.7 (8) |
| CRMP2 | 63.7 ± 9.8 (11) | 21.9 ± 1.8 (11) | −9.9 ± 0.9 (11) | 5.7 ± 0.5 (11) |
| CRMP2-K374A | 19.1 ± 5.5 (10)a | 19.5 ± 2.0 (10) | −6.3 ± 1.6 (7)b | 7.6 ± 1.3 (7) |
| 3 CRMP2/1 CRMP2-K374A | 9.1 ± 3.1 (8)a,b | 15.7 ± 1.2 (8) | −13.7 (2) | 6.0 (2) |
| CRMP2AAA | 17.8 ± 4.9 (11)a,b | 17.9 ± 1.4 (11) | −10.6 ± 2.6 (4) | 7.2 ± 1.4 (4) |
| CRMP2 | 63.7 ± 9.8 (11) | 21.9 ± 1.8 (11) | −9.9 ± 0.9 (11) | 5.7 ± 0.5 (11) |
| CRMP2 + SENP1 | 16.0 ± 3.7 (10)a | 16.1 ± 1.8 (10) | −8.4 ± 1.1 (3) | 7.1 ± 0.9 (4) |
| CRMP2 + SENP2 | 14.6 ± 2.8 (11)a | 20.4 ± 1.4 (11) | −13.3 ± 1.6 (4) | 6.0 ± 1.2 (4) |
| CRMP2 + SENP1 + SENP2 | 20.0 ± 7.9 (12)a | 17.3 ± 1.6 (12) | −9.9 ± 1.7 (4) | 8.4 ± 1.0 (4) |
| CRMP2-K374A | 19.1 ± 5.5 (10) | 19.5 ± 2.0 (10) | −6.3 ± 1.6 (7) | 7.6 ± 1.3 (7) |
| CRMP2-K374A + SENP1 | 29.0 ± 12.2 (8) | 18.8 ± 3.2 (8) | −4.2 ± 1.9 (5) | 13.2 ± 1.2 (5)a |
| CRMP2-K374A + SENP2 | 14.7 ± 4.3 (8) | 16.2 ± 1.3 (8) | −3.5 ± 1.5 (3) | 9.2 ± 0.6 (3) |
| CRMP2-K374A + SENP1 + SENP2 | 26.9 ± 9.5 (9) | 21.4 ± 1.5 (9) | −3.4 ± 1.9 (3) | 11.0 ± 1.0 (3) |
| CRMP2AAA | 17.8 ± 4.9 (11) | 17.9 ± 1.4 (11) | −10.6 ± 2.6 (4) | 7.2 ± 1.4 (4) |
| CRMP2AAA + SENP1 + SENP2 | 16.0 ± 3.7 (11) | 15.4 ± 1.0 (11) | 1.2 ± 2.9 (4)* | 8.6 ± 1.6 (4) |
| EGFP | 60.1 ± 19.3 (9) | 16.0 ± 2.5 (9) | −12.0 ± 1.0 (8) | 7.0 ± 0.7 (8) |
| Ubc9 + SUMO1 | 49.8 ± 8.7 (10) | 14.4 ± 0.6 (10) | −9.8 ± 1.6 (9) | 8.3 ± 0.9 (9) |
a Data represent statistically significant differences as compared with control within the tested groups (p < 0.05, ANOVA with Tukey's post hoc test or Student's t test).
b Data denote statistical difference from EGFP control (p < 0.05, ANOVA with Tukey's post hoc test).
CRMP2-K374A Mutant Impairs NaV1.7 Surface Trafficking
Changes in current density can be attributed to either a change in the number of channels within the cell membrane or a change in channel gating (i.e. open probability). Cell surface biotinylation was used to explore the first possibility, as described previously (5, 44, 45, 52). Immunoblotting with NaV1.7 for streptavidin-enriched complexes from biotinylated CAD cells showed decreased NaV1.7 surface expression in cells expressing CRMP2-K374A versus wild type CRMP2 (Fig. 6A, top blot, compare 1st with 3rd lanes). Surface NaV1.7 levels, normalized to total tubulin levels, in CRMP2-K374A-expressing CAD cells were 15.1 ± 2.5% of those in cells expressing wild type CRMP2 (Fig. 6B, n = 4, p < 0.05, ANOVA with Tukey's post hoc test). Increasing deSUMOylation with the sentrin/SUMO-specific proteases SENP1 and SENP2 (22) decreased levels of surface NaV1.7 to 14.2 ± 3.9% of those in cells expressing wild type CRMP2 (Fig. 6B, n = 4, p < 0.05, ANOVA with Tukey's post hoc test). Under these conditions, surface NaV1.7 levels in CRMP2-K374A-expressing CAD cells were reduced to 7.9 ± 2.1% of those in cells expressing wild type CRMP2 (Fig. 6B, n = 4, p < 0.05, ANOVA with Tukey's post hoc test). Additional experiments with a pan-NaV antibody also demonstrated a substantial reduction in surface-expressed NaV channels in CAD cells expressing CRMP2-K374A (Fig. 6, C and D; n = 4, p < 0.05, Student's t test). These results are consistent with the reduction in NaV1.7 current density in CRMP2-K374A-expressing CAD cells observed earlier. Thus, reduced CRMP2 SUMOylation likely accounts for NaV1.7 surface trafficking impairment.
FIGURE 6.
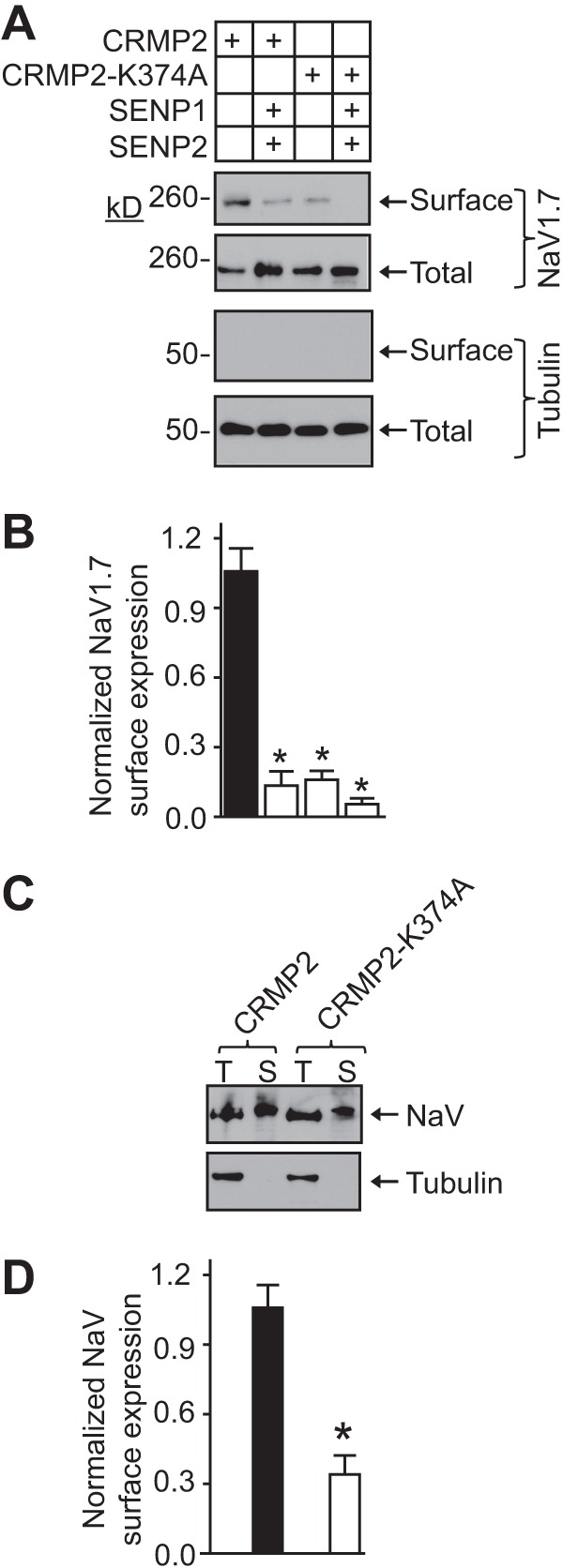
CRMP2-K374A co-expression and increasing deSUMOylation reduces NaV1.7 surface expression. A, cell surface expression of NaV1.7 was monitored using a biotinylation assay. CAD cells expressing wild type CRMP2 or CRMP2-K374A alone or in the presence of deSUMOylating proteases SENP1 and SENP2 were biotinylated; the cell surface proteins were harvested from the cell lysates, and the precipitates were analyzed by immunoblotting with NaV1.7 (top 2 blots) and βIII-tubulin (bottom 2 blots) antibodies. Equal amounts of samples were used for the precipitation of biotinylated proteins. Of these samples, the entire biotinylated fraction (surface) and 10% of the nonbiotinylated (total) fraction were loaded. CRMP2-K374A overexpression decreased the amount of NaV1.7 at the cell surface compared with CRMP2-transfected cells. No change was observed in the total levels of NaV1.7 or tubulin. B, averaged NaV1.7 surface expression, normalized to total NaV1.7 and then to total tubulin. Asterisks indicate statistically significant differences between CRMP2 and all other conditions tested (p < 0.05, one-way ANOVA with Tukey's post hoc test). C, representative blots of total (T) and surface (S) biotinylated fractions from CAD cells expressing wild type CRMP2 or CRMP2-K374A immunoblotted with a pan-NaV antibody (top) or tubulin (bottom). D, averaged NaV surface expression, normalized to total NaV and then to total tubulin. Asterisks indicate statistically significant differences between CRMP2 and CRMP2-K374A (n = 4; p < 0.05, Student's t test).
Increasing deSUMOylation Reduces NaV1.7 Current Density
Our earlier results showed the following: (i) CRMP2 mutants with disrupted SUMO motifs reduced NaV1.7 currents and (ii) promoting deSUMOylation with the sentrin/SUMO-specific proteases SENP1 and SENP2 reduced trafficking of NaV1.7 in cells expressing both wild type and CRMP2 mutants. Here, we tested how boosting the deSUMOylation machinery would affect NaV1.7 current density. NaV1.7 currents in CAD cells expressing wild type CRMP2 were reduced to 25.2 ± 5.8% (n = 10) in cells co-expressing SENP1, 23.0 ± 4.4% (n = 11) in cells co-expressing SENP2, and 31.5 ± 12.3% (n = 12) in cells co-expressing SENP1 and SENP2 compared with cells expressing only wild type CRMP2 (Fig. 7A, p < 0.05 versus CRMP2 alone, ANOVA with Tukey's post hoc test). NaV1.7 current densities were reduced by ∼72 and ∼70% in CAD cells expressing CRMP2AAA or CRMP2-K374A, respectively (Fig. 7B). Expression of either protease alone or in combination did not result in a further reduction in NaV1.7 current density (Fig. 7B, p > 0.05, ANOVA with Tukey's post hoc test). Boltzmann parameters of voltage dependence of activation were not different across the transfected conditions with the exception of k in CAD cells co-expressing CRMP2-K374A and SENP1 compared with CAD cells expressing CRMP2-K374A alone (Table 1, p < 0.05, ANOVA with Tukey's post hoc test). These results suggest that the SUMOylation state of CRMP2 can affect NaV1.7 current density.
FIGURE 7.
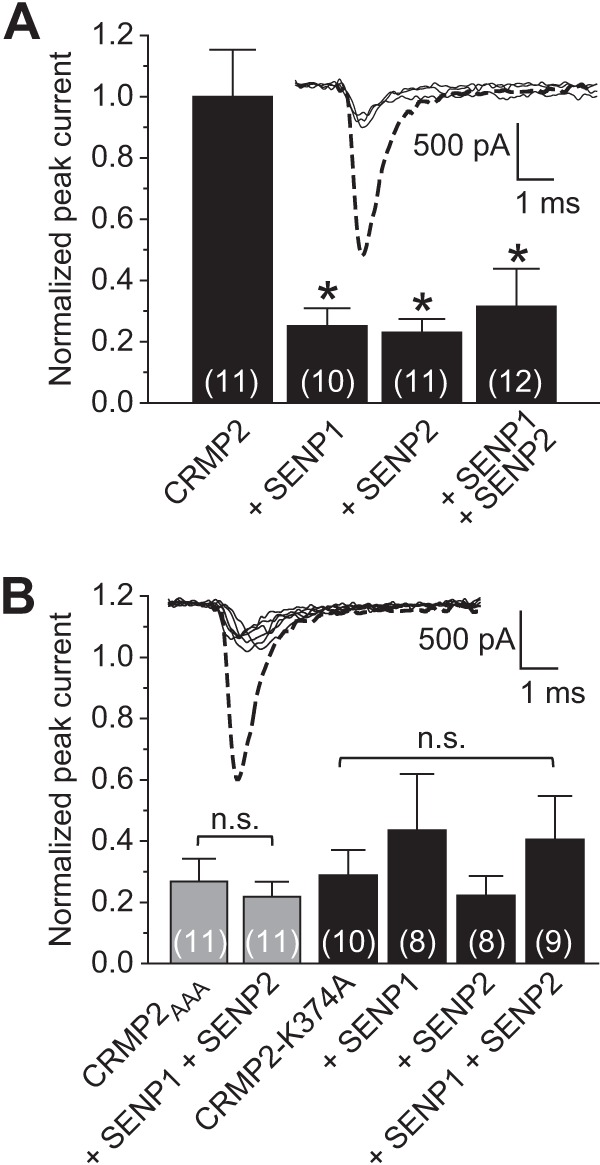
Removal of SUMO by SENP1 and SENP2 reduces NaV1.7 current density. Summary of normalized peak currents in the presence or absence of SUMO-specific proteases SENP1 and/or SENP2 in CAD cells expressing CRMP2 (A) or CRMP2AAA/CRMP2-K374A (B). Peak currents were normalized to the cell capacitance. Exemplar peak current traces (at 0 mV) are shown in each panel with the thicker dashed trace representing the CRMP2-transfected condition. Because there were no differences between the amplitudes, the other traces shown are not labeled. Numbers in parentheses represent numbers of cells tested. Asterisks indicate statistically significant differences between CRMP2 and all other conditions tested (p < 0.05, one-way ANOVA with Tukey's post hoc test). n.s., not significant.
That increasing deSUMOylation did not result in a further decrement in NaV1.7 current density beyond that observed with the genetic loss of CRMP2 SUMOylation (i.e. CRMP2 SUMO-incompetent mutants) implies that the CRMP2 SUMO mutants may be acting in a dominant negative fashion. Because CRMPs are tetrameric proteins (53), we hypothesized that introduction of a single non-SUMOylatable mutant monomer within the tetramer may be sufficient to confer loss of current density. We tested this hypothesis by transfecting CAD cells with a molar 3:1 ratio of wild type CRMP2/CRMP2-K374A cDNAs and assessing NaV1.7 currents. Peak NaV1.7 currents in these cells were reduced to 28.5 ± 9.6% (n = 8) of wild type CRMP2-expressing CAD cells (see Fig. 5D, p < 0.05 compared with wild type CRMP2, ANOVA with Tukey's post hoc test) and were no different from CRMP2-K374A-expressing cells (see Fig. 5D, p > 0.05 compared with wild type CRMP2, ANOVA with Tukey's post hoc test). Overexpression of molar 3:1 ratio of wild type CRMP2/CRMP2-K374A cDNAs enhanced cortical neuron neurite outgrowth to near identical levels as wild type CRMP2 alone (99.5% ± 5.1 (n = 269–297 cells from 8 wells), p > 0.05, Student's t test). Overexpression of a molar 3:1 ratio of wild type CRMP2/CRMP2-K374A cDNAs in CAD cells did not affect Boltzmann parameters of voltage dependence of activation (Table 1, p < 0.05, ANOVA with Tukey's post hoc test). These results suggest that the presence of a single non-SUMOylatable CRMP2 monomer is sufficient to bring about a reduction in current density without affecting the axonal growth promoting capabilities of CRMP2.
Increasing SUMOylation Does Not Enhance NaV1.7 Current Density in CAD Cells Expressing Endogenous CRMP2
That destruction of the SUMO site in CRMP2 resulted in a decrease in NaV1.7 currents suggests that wild type SUMO-competent CRMP2 may be important for constitutive trafficking of NaV1.7. If so, then boosting the SUMOylation machinery with overexpression of Ubc9 and SUMO1 would be predicted to increase endogenous sodium currents. However, we found that this condition yielded sodium currents that were 78.2 ± 13.7% (n = 10) of CAD cells expressing wild type CRMP2 (Table 1, p > 0.05, Student's t test). This is perhaps not surprising as CAD cells express endogenous levels of the SUMOylation machinery as detected by quantitative RT-PCR experiments (data not shown). As expected, increasing SUMOylation machinery with overexpression of Ubc9 plus SUMO1–3 was unable to recover the loss of sodium current densities in CAD cells expressing CRMP2 mutants (data not shown). This facile finding confirms that a single SUMOylation site in CRMP2 is responsible for CRMP2 SUMOylation-dependent modification of NaV1.7 trafficking.
CRMP2-K374A Mutant Reduces NaV1.7 Currents in a HEK293 NaV1.7 Cell Line in a Ubc9-dependent Manner
Because we could not pharmacologically or genetically isolate NaV1.1 and NaV1.3 currents, which account for <15% of the CAD cell sodium current, we chose to test the importance of CRMP2-SUMOylation in NaV1.7 trafficking in a HEK293 cell line expressing only NaV1.7 channels (38). Representative traces of NaV1.7 currents from these cells are illustrated in Fig. 8, A and C. Cells expressing SUMO1 plus wild type or mutant CRMP2 were not different from cells expressing EGFP: 104.0 ± 26.9% for SUMO1 + CRMP2 (n = 8) and 91.0 ± 17.1% (n = 10) for SUMO1 + CRMP2-K374A (Table 2 and Fig. 8B; p > 0.05, ANOVA with Tukey's post hoc test versus EGFP). Because these results seemed at odds with our earlier findings in CAD cells, we asked if the endogenous SUMOylation machinery could possibly be different between the two cell types. quantitative RT-PCR data revealed an almost 10-fold higher level of Ubc9 mRNA in CAD cells compared with HEK293 cells (data not shown). Therefore, we repeated the above experiments in the additional presence of the E2-conjugating enzyme Ubc9 to determine whether it was needed to recapitulate the loss of sodium currents observed in our earlier findings. Compared with HEK293 NaV1.7 cells expressing wild type CRMP2, Ubc9, and SUMO1, substitution with mutant CRMP2-K374A reduced NaV1.7 current density to 44.9 ± 10.6% (Fig. 8D, n = 8, p < 0.05, ANOVA with Tukey's post hoc test). A similar reduction was observed with a substitution with CRMP2AAA (Table 2, 56.7 ± 12.1%, n = 11, p < 0.05, ANOVA with Tukey's post hoc test). Biophysical properties of fast inactivation (Fig. 8, E and F, circles) or steady-state activation (see Fig. 8, E and F, squares) of NaV1.7 currents were not different between HEK293 NaV1.7 cells expressing SUMO1 + CRMP2 or SUMO1 + CRMP2-K374A in the absence (Fig. 8E) or presence of Ubc9 (Fig. 8F; values listed in Table 2). Thus, our results in HEK293 NaV1.7 cells phenocopy our earlier finding of diminution of sodium currents in CAD cells expressing CRMP2 mutants.
FIGURE 8.
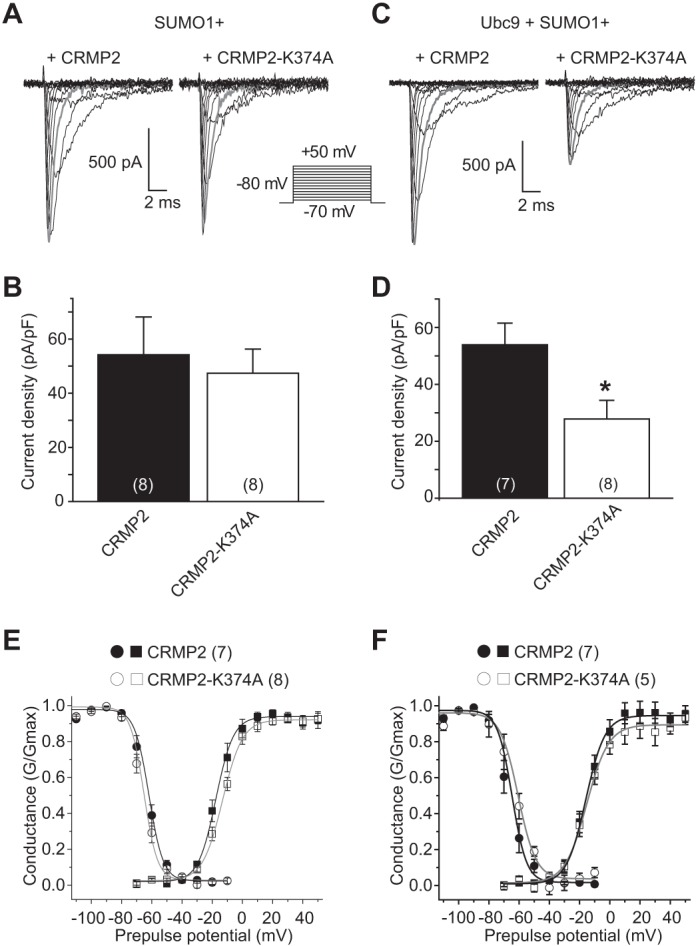
CRMP-K374A mutant reduces NaV1.7 current density in a Ubc9-dependent fashion. Exemplar family of current traces, in response to the voltage protocol shown, from HEK293-expressing NaV1.7 cells also expressing SUMO1 and CRMP2 (A, left) or SUMO1 and CRMP2-K374A (A, right) in the additional presence of Ubc9 (C). B and D, summary of normalized peak currents recorded from NaV1.7-HEK293 cells transfected with the indicated constructs. Numbers in parentheses represent numbers of cells tested. There was no difference between current densities in cells when CRMP2 constructs were co-expressed with SUMO1 (B) (p > 0.05, Student's t test). Addition of the E2-conjugating enzyme Ubc9 produced a marked decrease in current density of cells expressing CRMP2-K374A and SUMO1 (p < 0.05, Student's t test) (D). E and F, representative Boltzmann fits for activation and steady-state inactivation for NaV1.7-HEK293 cells transfected with the indicated constructs are shown. The calculated values for V½ and k of activation and steady-state inactivation for all conditions tested and the associated statistics are presented in Table 2.
TABLE 2.
Comparative current density, cell capacitance, and Boltzmann parameters of voltage dependence of channel activation and steady-state fast inactivation curves for the respective transfection conditions in HEK293 cells expressing NaV1.7, NaV1.1, or NaV1.3 channels
Values for V½, the voltage of half-maximal activation, and slope were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean ± S.E. N values are indicated in parentheses. All transfection conditions contained EGFP for identification of construct expression.
| Transfection condition | Peak current density | Cell capacitance | Voltage dependence of activation |
Voltage dependence of fast inactivation |
||
|---|---|---|---|---|---|---|
| V1/2 | Slope | V1/2 | Slope | |||
| pA/pF | pF | mV | mV/e-fold | mV | mV/e-fold | |
| NaV1.7 | ||||||
| EGFP | 52.1 ± 8.2 (5) | 19.8 ± 2.0 (5) | −14.0 ± 1.0 (5) | 6.6 ± 0.5 (5) | −64.0 ± 1.1 (4) | 6.0 ± 0.4 (4) |
| SUMO1 + CRMP2 | 54.2 ± 14.0 (8) | 18.1 ± 1.9 (8) | −17.6 ± 1.0 (7)a | 5.8 ± 0.5 (7) | −61.9 ± 2.6 (7) | 4.7 ± 1.6 (7) |
| SUMO1 + CRMP2-K374A | 47.4 ± 8.9 (8) | 18.5 ± 1.8 (8) | −13.1 ± 0.6 (7) | 6.6 ± 0.4 (7) | −65.2 ± 3.2 (8) | 4.5 ± 1.9 (8) |
| Ubc9 + SUMO1 + CRMP2 | 53.9 ± 7.6 (7) | 20.0 ± 1.8 (7) | −15.9 ± 0.7 (7) | 7.7 ± 0.9 (7) | −64.6 ± 3.5 (7) | 5.9 ± 1.8 (7) |
| Ubc9 + SUMO1 + CRMP2-K374A | 24.2 ± 5.7 (8)a | 18.9 ± 2.3 (8) | −15.2 ± 1.1 (5) | 6.4 ± 0.3 (5) | −60.6 ± 2.7 (5) | 4.8 ± 2.0 (5) |
| Ubc9 + SUMO1 + CRMP2AAA | 30.6 ± 6.5 (11)a | 19.3 ± 2.1 (11) | −17.7 ± 0.7 (10) | 7.9 ± 0.4 (10) | −67.6 ± 2.7 (8) | 5.7 ± 1.3 (8) |
| NaV1.1 | ||||||
| EGFP | 78.6 ± 12.7 (5) | 18.1 ± 2.7 (5) | −16.7 ± 1.9 (5) | 6.6 ± 0.9 (5) | −57.9 ± 2.2 (4) | 6.0 ± 1.1 (4) |
| Ubc9 + SUMO1 + CRMP2 | 80.8 ± 28.4 (7) | 19.5 ± 2.6 (7) | −15.5 ± 0.9 (7) | 6.8 ± 0.4 (7) | −60.1 ± 1.9 (7) | 6.2 ± 1.1 (7) |
| Ubc9 + SUMO1 + CRMP2-K374A | 75.3 ± 19.2 (7) | 18.4 ± 1.3 (7) | −15.4 ± 1.8 (7) | 6.2 ± 1.0 (7) | −63.7 ± 3.5 (7) | 5.0 ± 2.1 (7) |
| NaV1.3 | ||||||
| EGFP | 377.9 ± 59.0 (5) | 14.6 ± 2.6 (5) | −21.2 ± 1.4 (3) | 5.3 ± 0.5 (3) | −59.0 ± 1.2 (3) | 6.6 ± 0.6 (3) |
| Ubc9 + SUMO1 + CRMP2 | 382.7 ± 54.3 (6) | 11.2 ± 2.3 (6) | −22.6 ± 3.2 (6) | 4.1 ± 0.8 (6) | −54.1 ± 1.8 (6) | 5.0 ± 0.9 (6) |
| Ubc9 + SUMO1 + CRMP2-K374A | 347.2 ± 80.8 (6) | 11.1 ± 1.5 (6) | −25.0 ± 0.6 (5) | 3.7 ± 0.2 (5) | −56.9 ± 1.0 (5) | 6.0 ± 0.6 (5) |
a Data represent statistically significant differences as compared with control (i.e. EGFP condition) within each tested group (p < 0.05, ANOVA with Tukey's post hoc test).
CRMP2-K374A Mutant Does Not Impair NaV1.1 or NaV1.3 Current Density
The NaV1.1 and NaV1.3 isoforms account for most of the remaining NaV mRNA in CAD cells (32). Thus, we next asked whether the CRMP2-K374A mutant could affect NaV1.1 or NaV1.3 current densities in HEK293 cell lines expressing NaV1.1 or NaV1.3. Representative peak traces for NaV1.1 currents in response to depolarizing steps from −70 mV to +50 mV are illustrated in Fig. 9A. Compared with EGFP-transfected HEK293 NaV1.1 cells, expression of wild type CRMP2 or CRMP2-K374 did not affect current density as follows: 102.3 ± 36.1% (n = 7) and 95.8 ± 24.4% (n = 7), respectively, compared with EGFP (Fig. 9B and Table 2; p > 0.05, ANOVA with Tukey's post hoc test). There were no differences in the properties of fast inactivation (Fig. 9C, circles) or steady-state activation (see Fig. 9C, squares) of NaV1.1 currents across any of the conditions tested (Table 2).
FIGURE 9.
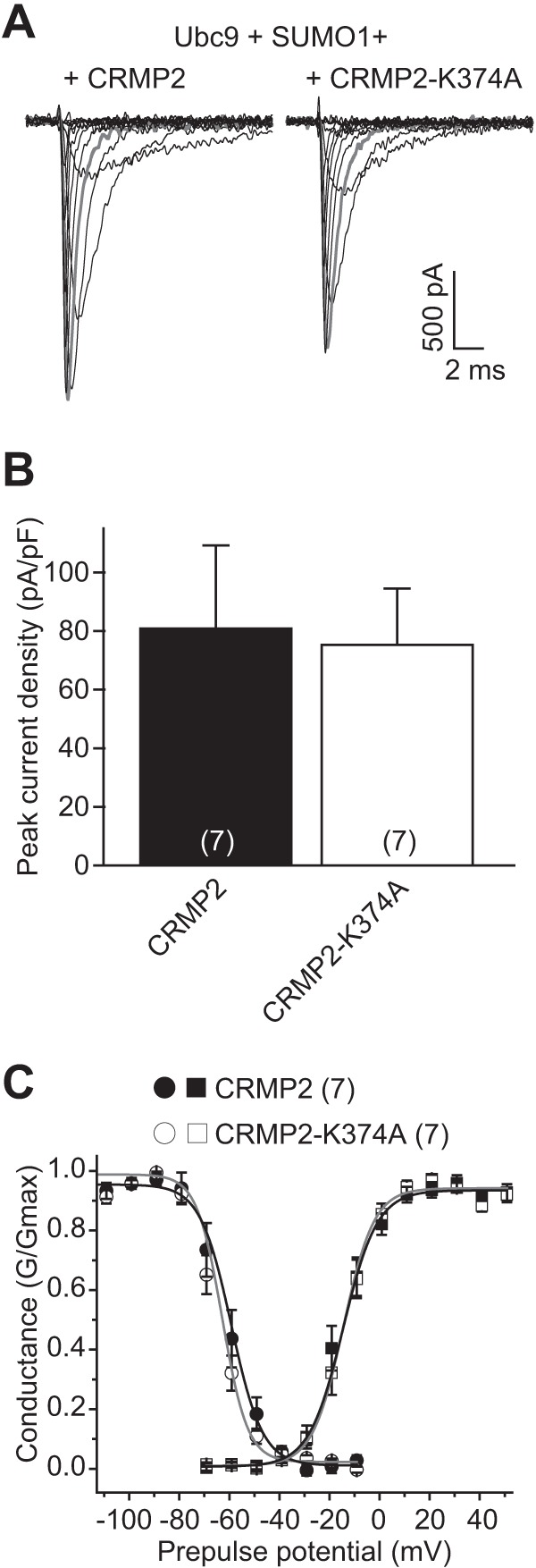
NaV1.1 current density is unaffected by CRMP2-K374A expression. A, exemplar family of current traces, in response to the voltage protocol shown in Fig. 8, from HEK293 cells expressing NaV1.1 and Ubc9, SUMO1 and CRMP2 (left), or CRMP2-K374A (right). B, summary of normalized peak currents recorded from NaV1.1-HEK293 cells expressing Ubc9, SUMO1, and CRMP2 (black bar) or CRMP2-K374A (white bar). Numbers in parentheses represent numbers of cells tested. There was no difference between the two conditions tested (p > 0.05, Student's t test). C, representative Boltzmann fits for activation and steady-state inactivation for NaV1.1-HEK293 cells transfected with the indicated constructs are shown. The calculated values for V½ and k of activation and steady-state inactivation for all conditions tested and the associated statistics are presented in Table 2.
Similarly, compared with EGFP-transfected HEK293 NaV1.3 cells, expression of wild type CRMP2 or CRMP2-K374 did not affect current density as follows: 101.3 ± 14.5% (n = 6) and 91.9 ± 21.4% (n = 6), respectively, compared with EGFP (Fig. 10, A and B and Table 2; p > 0.05, ANOVA with Tukey's post hoc test). NaV1.3 currents in HEK293 cells typically display a slowly inactivating, persistent current (37). This property was evident in our recordings (see representative traces in Fig. 10A), where we observed an incomplete inactivation over the course of the depolarizing voltage steps. To assess if wild type or mutant CRMP2 expression can affect this component of NaV1.3, we assessed the amount of current that persisted near the end of the depolarizing voltage step to 0 mV (Fig. 10A, arrow at ∼15 ms) and expressed it as a percent of peak current at this voltage. There was no difference between wild type of the CRMP2 and CRMP2-K374A conditions, 6.6 ± 0.9% (n = 6) and 7.1 ± 1.2% (n = 6) current remaining, respectively (Fig. 10B, p > 0.05, Student's t test). Finally, there were also no differences in the properties of fast inactivation (Fig. 10C, circles) or steady-state activation (see Fig. 10C, squares) of NaV1.3 currents across any of the conditions tested (Table 2). Thus, these results show that impairment in CRMP2 SUMOylation selectively affects the trafficking of NaV1.7, but not NaV1.1 or NaV1.3.
FIGURE 10.
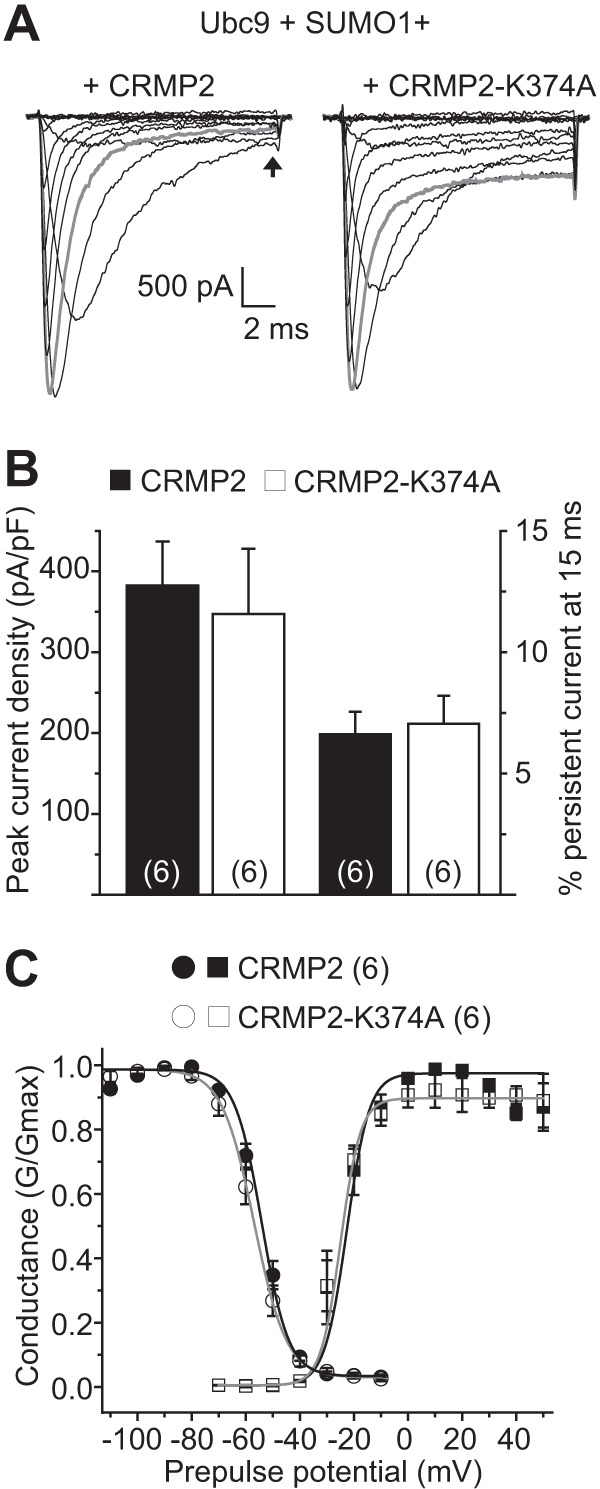
NaV1.3 current density is unaffected by CRMP2-K374A expression. A, exemplar family of current traces, in response to the voltage protocol shown in Fig. 8, from HEK293 cells expressing NaV1.1 and Ubc9, SUMO1 and CRMP2 (left), or CRMP2-K374A (right). B, summary of normalized peak currents (left axis) or persistent current remaining at 15 ms (see arrow in A) as a percent of peak current (right axis) recorded from NaV1.3-HEK293 cells expressing Ubc9, SUMO1, and CRMP2 (black bars) or CRMP2-K374A (white bars). Numbers in parentheses represent numbers of cells tested. There was no difference between the two conditions tested for either peak current of % persistent current (p > 0.05, Student's t test). C, representative Boltzmann fits for activation and steady-state inactivation for NaV1.3-HEK293 cells transfected with the indicated constructs are shown. The calculated values for V½ and k of activation and steady-state inactivation for all conditions tested and the associated statistics are presented in Table 2.
CRMP2-K374A Mutant Reduces Current Density in DRG Neurons
Our previous results demonstrated that an impairment in CRMP2 SUMOylation blunts trafficking of NaV1.7 in two cell lines. Here, we tested the effects of overexpression of the SUMO-incompetent CRMP2 on sodium currents in native neurons isolated from DRG as these cells express abundant NaV1.7 (55). Sodium currents were examined in DRGs expressing either wild type CRMP2 or CRMP2-K374A; Ubc9 was not transfected as DRGs express endogenous Ubc9 mRNA at levels even higher than CAD cells (data not shown). A representative family of current traces from DRGs transfected with wild type CRMP2 or CRMP2-K374A are illustrated in Fig. 11A. The recorded peak current density in DRGs expressing CRMP2-K374A was reduced to 54.4 ± 9.7% that of wild type CRMP2-expressing controls (Fig. 11B, n = 9, p < 0.05, Student's t test). Importantly, the peak current density of DRGs was not augmented by CRMP2 alone compared with EGFP as follows: 390.9 ± 70.6 (n = 8) versus 378.3 ± 57.2 (n = 8), respectively (Fig. 11B, p > 0.05, Student's t test). Biophysical properties of steady-state activation of sodium currents were not different between the conditions (see Fig. 11C, squares); however, cells expressing CRMP2-K374A exhibited a depolarizing shift of ∼9 mV in V½ of fast inactivation (Fig. 11C, circles; p < 0.05, Student's t test) without a commensurate change in k of fast inactivation (p > 0.05, Student's t test).
FIGURE 11.

CRMP2-K374A mutant reduces sodium current density in DRG neurons but does not impair CRMP2-mediated neurite outgrowth. A, representative sodium currents recorded in dissociated rat DRG neurons 48 h after transfection with EGFP and CRMP2 (left) or EGFP and CRMP2-K374A (right). B, summary bar graph showing peak sodium current in each condition. Numbers in parentheses represent numbers of cells tested. There was a significant reduction in peak current density in DRG neurons expressing CRMP2-K374A compared with those expressing wild type CRMP2 (p < 0.05, Student's t test). C, representative Boltzmann fits for activation and steady-state inactivation for DRG neurons transfected with the indicated constructs are shown. The calculated values for V½ and k of activation were not significantly different between the conditions as follows: −19.2 ± 0.8 mV and 3.9 ± 0.4 mV/e-fold (n = 6) for EGFP plus CRMP2-transfected cells versus −18.5 ± 1.3 mV and 5.5 ± 0.7 mV/e-slope (n = 4) for EGFP plus CRMP2-K374A-transfected cells (p > 0.05 for both parameters, Student's t test). CRMP2-K374A produced a significant ∼9-mV depolarizing shift in V½ of steady-state inactivation: −59.9 ± 2.2 mV for EGFP plus CRMP2-transfected cells versus −50.7 ± 1.8 mV for EGFP and CRMP2-K374A expressing cells (n = 4 for both conditions, p < 0.05, Student's t test). Values for k of steady-state inactivation were not different between conditions: 13.2 ± 1.3 mV/e-fold for EGFP plus CRMP2-transfected cells versus 10.6 ± 2.0 for EGFP plus CRMP2-K374A-transfected cells (n = 4 for both conditions, p > 0.05, Student's t test). D, example image showing the overlay of EGFP fluorescence (green) and nuclear staining with Hoechst (blue) together with the brightfield Hoffman image of transfected DRG neurons. Scale bar equals 50 μm and applies to both panels. Note the similar degree of neurite complexity between the wild type and mutant CRMP2 conditions. E, number of processes and branches; F, mean and maximum (max.) process length, and G, normalized total outgrowth for cells co-expressing EGFP and CRMP2 (n = 54 from four wells) or co-expressing EGFP and CRMP2-K374A (n = 72 from four wells). There were no statistical differences for any of the parameters between the two conditions (p > 0.05, Student's t test).
We also examined if DRG neurite growth promoting activity of CRMP2 was affected by destruction of the CRMP2 SUMOylation site using neurite outgrowth analysis as described earlier. Compared with wild type CRMP2, expression of CRMP2-K374A did not alter the number of processes per neuron, the number of branches per neuron, the mean and maximum process lengths, and overall total outgrowth (Fig. 11, D–G). On average, the number of processes per neuron was 10.8 ± 0.5 and 10.1 ± 0.5 for cells expressing CRMP2 (n = 54) and CRMP2-K374A (n = 72) (p > 0.05), respectively. The number of branches per neuron was 88.9 ± 7.9 and 86.2 ± 5.8 for cells expressing CRMP2 and CRMP2-K374A (p > 0.05), respectively. The mean and maximum process lengths per neuron were 251.8 ± 17.9 and 1416.7 ± 126.6 μm for CRMP2 and 273.3 ± 14.4 and 1316.6 ± 82.0 μm for CRMP2-K374A (p > 0.05). Finally, overall total outgrowth, a sum of all the above parameters, in DRG neurons expressing CRMP2-K374A was 97.3 ± 5.6% of those expressing wild type CRMP2 (100.0 ± 7.8%, p > 0.05) (Fig. 11F).
Collectively, our findings in DRGs recapitulate the main finding of reduced current density observed in CAD cells and HEK293 cells expressing NaV1.7 and identify SUMOylation of CRMP2 as a modulatory mechanism of NaV1.7 trafficking.
DISCUSSION
Accumulating evidence indicates that the CRMP2 proteome is far greater than previously appreciated (2). It is also becoming evident that post-translational modifications, in particular phosphorylation, can influence CRMP2's interactions and in doing so bestow upon CRMP2 the ability to engage different signaling pathways (17). Here, we describe a previously undescribed modification for CRMP2, SUMOylation, which can alter the trafficking of a voltage-gated sodium channel. The trafficking of NaVs is an understudied but emerging field (56). Examples of proteins involved in trafficking of NaV1s include the Alzheimer disease-related secretases β-site amyloid precursor protein-cleaving enzyme 1 and presenilin/γ-secretase, which regulate NaV1 function by cleaving auxiliary subunits of the channel complex (57, 58); mutants of NaV1.1 linked to familial epilepsy are trafficking-defective, and their function can be restored by incubation at temperature <30 °C, as well as through protein-protein interactions with modulatory proteins or drugs (59). There is at least one report linking microtubule dynamics in the trafficking of cardiac NaV1.5 (60). Trafficking of the tetrodotoxin-resistant NaV1.8 isoform is believed to occur via masking of an endoplasmic retention motif in NaV1.8 by the auxiliary β3 (61) with help from annexin II light chain (62). Post-translational regulation and direct, as well as indirect, protein-protein interactions have been advanced as possible regulatory mechanisms for NaV trafficking (56, 63). However, very little is currently known about the mechanisms regulating NaV1.7 cell surface expression.
In studying CRMP2-mediated regulation of the enhancement of the slow-inactivated state of sodium channels by the anti-epileptic drug LCM, we discovered that CRMP2 did not elicit this effect via direct interaction with the sodium channel (9). Despite an initial report of LCM binding to CRMP2 expressed in oocyte membranes (64) and two other reports demonstrating binding of a chemically modified fluorescent version of LCM to CRMP2 (65), a recent study using radioligand binding and biophysical and biochemical approaches reported that LCM does not bind specifically to CRMP2 (10). If correct, these findings would argue for the existence of regulatory pathway(s) that provide a nexus between CRMP2, LCM, and sodium channels. Further support of a CRMP2-LCM nexus comes from the demonstration that LCM, perhaps independent of its action on VGSCs, can inhibit CRMP2-mediated post-traumatic axon sprouting (29). Therefore, we tested if SUMOylation of CRMP2 could affect the manner by which LCM affects sodium channels. The functional regulation of CRMP2 by SUMOylation was studied by the following: (i) creating constructs in which the target lysine in the SUMOylation motif as well as adjoining residues were mutated to alanine, and (ii) by manipulating expression of the SUMOylation machinery with overexpression of Ubc9 and SUMOs to boost SUMOylation and overexpression of SENP1 and SENP2 to decrease SUMOylation. Although putative SUMO motifs are typically investigated by mutating the target lysine to arginine (31, 46–50), this is not always the case. For instance, mutation of the SUMO target lysine in the potassium leak channel K2P1 to an arginine had no effect on channel activity, although an alanine mutation increased activity consistent with an increase observed in the presence of SENP1 (26). For this reason as well as the fact that alanine residues have been the preferred choice in demonstrating SUMOylation of several proteins, including Ran GTPase-activating protein 1 (50), acute promyelocytic leukemia protein (47), the polycomb protein Pc2/Cbx4 (46), and basic Kruppel-like factor/Kruppel-like factor 3 (BKLF), a zinc finger transcription factor (49), we chose to mutate the CRMP2 SUMO target lysine to alanine.
Both CRMP2 SUMO mutants, CRMP2AAA and CRMP2-K374A, produced functional proteins as assessed by their propensities to increase neurite outgrowth relative to EGFP-transfected neurons. That augmented neurite outgrowth was recapitulated to near wild type CRMP2 levels suggests that the mutant CRMP2 proteins are neither misfolded nor unable to oligomerize despite the placement of the SUMO target lysine 374 within the oligomerization domain (amino acids 8–134 and 281–435) of CRMPs (53). Neither of the CRMP2 mutants affected LCM-induced shifts in slow inactivation of sodium channels in CAD cells, suggesting that removal of SUMOylation does not affect the biophysical property of slow inactivation. For proteins that are directly SUMOylated, such as ion channels, fine-tuning of channel function has been reported for potassium channels Kv1.5 (23) and Kv2.1 (24), and loss of Kv1.5 SUMOylation, by either disruption of the conjugation sites or expression of the SUMO protease SENP2, leads to a selective ∼15-mV hyperpolarizing shift in the voltage dependence of steady-state inactivation (23), whereas SUMOylation of Kv2.1 in hippocampal neurons was shown to regulate firing by shifting the half-maximal activation voltage of channels up to 35 mV (24).
While interrogating the potential effects of CRMP2 SUMO mutants on LCM-induced enhancement of slow inactivation, we noticed a clear reduction in macroscopic sodium currents in CAD cells. As these cells express mRNAs for NaV1.7 (>90% of total NaV mRNA), NaV1.1, and NaV1.3 (32), using huwentoxin-IV, we confirmed that ∼85% of the CAD cell sodium current was carried via NaV1.7. This current was dramatically reduced by expression of either CRMP2 SUMO mutant in CAD cells, suggesting a contribution for SUMOylation of CRMP2 in trafficking of NaV1.7. Expression of deSUMOylating enzymes, SENP1 or SENP2, forced a decrement in NaV1.7 current in cells expressing wild type CRMP2. Cell surface biotinylation under the same conditions revealed reduced NaV1.7 surface expression, corroborating the reduction in current density and suggesting that CRMP2 SUMOylation reduces current density likely via reduced trafficking similar to what has been reported for the kainate receptor subunit GluR6 wherein SUMOylation facilitates endocytosis of this receptor (66). Therefore, by genetic or enzymatic manipulation, the removal of CRMP2 SUMOylation leads to reduced NaV1.7 current. Co-expression of deSUMOylating enzymes SENP1 or SENP2 did not further reduce NaV1.7 current densities in CAD cells expressing CRMP2 SUMO mutants, suggesting that the mutants may serve in a dominant negative manner for NaV1.7 trafficking. To further examine this hypothesis, we co-expressed both wild type CRMP2 and CRMP2-K374A at a 3:1 stoichiometry and determined that the presence of a single mutant was sufficient to force a reduction in NaV1.7 currents. Furthermore, this stoichiometric ratio of CRMP2 constructs did not impair CRMP2-dependent enhancement of neurite growth in cortical neurons.
The NaV1.7 isoform is preferentially expressed in the peripheral nervous system within ganglia related to nociceptive pain, including dorsal root ganglia, trigeminal ganglia, and sympathetic ganglia (30). In nociceptive neurons responsible for the transduction of pain signals, the channel modulates current threshold required to fire action potentials in response to stimuli (67, 68). Gain-of-function mutations, i.e. those that lower NaV1.7 current threshold for initiation of action potentials, produce allodynia, a lowered stimulus threshold for pain. Such mutations are the cause of pain syndromes, including erythromelalgia, paroxysmal extreme pain disorder, and small fiber neuropathy (67). A loss-of-function mutation of NaV1.7 can cause equally detrimental modifications in pain sensation, where stimuli never reach threshold to propagate pain. Patients with such mutations display a complete loss of pain sensation (69). Increased surface expression of NaV1.7 has been associated with pain resulting from diabetic neuropathy (70) and inflammation (71). Although NaV1.7 is the predominant voltage-gated sodium channel isoform found in CAD cells, mRNA expression data suggest a secondary contribution by the NaV1.1 and NaV1.3 isoforms (32). Remarkably, in stark contrast to NaV1.7, NaV1.1 and NaV1.3 current densities were not affected by expression of CRMP2-K374A, suggesting that CRMP2 SUMOylation-dependent modulation of sodium current density may be specific to particular sodium channel isoforms. Of the tetrodotoxin-sensitive voltage-gated sodium channels present in the nervous system, only NaV1.7 lacks a putative SUMOylation motif (72). Thus, any modification of the channel by SUMOylation is likely due to indirect modification of accessory proteins, such as CRMP2 demonstrated in this study. Mechanisms that specifically or preferentially regulate NaV1.7 are of particular relevance due to the role of NaV1.7 in transduction of peripheral pain.
The NaV1.1 isoform is the most commonly mutated gene in epilepsy, with over 600 known mutations (73, 74). The channel is targeted to the axon initial segment within CNS inhibitory neurons where, much like NaV1.7 in the periphery, NaV1.1 channels underlie the generation of action potentials (75). Loss-of-function mutations lead to a loss of inhibition within excitatory circuits, an underlying mechanism of genetic epilepsy syndromes (73, 75). Although sequence analysis of NaV1.1 and NaV1.3 reveals a putative SUMOylation site (72), it is unlikely to be SUMOylated given placement of the target lysines within the S5-S6 linker of domain IV. Importantly, NaV1.1 and NaV1.3 are present in small diameter dorsal root ganglion neurons responsible for nociception, but to a much lesser extent than NaV1.7 (55). The NaV1.3 isoform is primarily expressed during early development thereafter decreasing to almost undetectable levels in the normal adult nervous system (76). DRGs from mice harboring a heterozygous mutation in the neurofibromatosis type 1 gene exhibit increased mRNA expression of several NaV1 channels, including NaV1.1, NaV1.3, NaV1.7, and NaV1.8 (77). Spinal nerve ligation and axotomy both increase NaV1.3 mRNA levels in DRG neurons (78, 79). In DRGs, NaV1.1 and NaV1.7 isoforms share trafficking guidance by sodium channel β2 (β2) subunits, which promote surface expression of NaV1.1 and NaV1.7 via interactions with cell adhesion molecules and the cytoskeletal protein ankyrin (55, 80). β2 subunit expression is up-regulated in sensory neurons in models of neuropathic pain (81). Consequently, targeting voltage-gated sodium channel trafficking mediated by the β2 subunit will alter surface expression of both NaV1.1 and NaV1.7 channel isoforms and may exert control in both the peripheral and central nervous systems. However, such a strategy does not allow for selective control over individual NaV isoforms, and it is presently unclear as to how NaV1.3 trafficking may be regulated by the β2 subunit or other subunits for that matter. In this regard, post-translational regulation of an auxiliary subunit, such as SUMOylation of CRMP2 shown here, may provide a mechanism for selectively targeting individual isoforms of NaVs.
One potential mechanism underlying the different effects of CRMP2 SUMOylation on NaV1.7, but not NaV1.1 or NaV1.3, trafficking could be that CRMP2 SUMOylation may promote assembly with binding partners that are not shared between the NaV isoforms. That increasing the SUMOylation machinery did not increase NaV1.7 current density suggests that the SUMOylated CRMP2 may direct protein(s) that associate with the channel early in its biogenesis. Another possibility may be that other CRMP isoforms or heterotetramers of CRMP1–5 may be differentially SUMOylated and modulate different target NaVs. Studies aimed at uncovering the mechanistic underpinnings of these differences are presently underway in our laboratory.
The modification of CRMP2 SUMOylation represents a novel mechanism, whereby surface expression of expressed NaV1.7 could possibly be targeted exclusively. Given the unequivocal link between chronic pain and NaV1.7 and demonstrated increases in NaV1.7 following injury, these channels have emerged as an attractive target for drug discovery for the treatment of pain. Our findings identify manipulation of NaV1.7 trafficking mechanisms within primary DRG neurons as a novel strategy for design of antinociceptive therapeutics against these channels.
Acknowledgments
We thank our colleagues at the Stark Neurosciences Research Institute, Dr. Theodore R. Cummins (Pharmacology and Toxicology, Indiana University School of Medicine) for providing HEK293 cells expressing NaV isoforms, and Reesha Patel for comments on the manuscript. We also thank Dr. Joel M. Brittain for generation of the CRMP2-K374A construct; Dr. May Khanna (Biochemistry and Molecular Biology, Indiana University School of Medicine) for assistance with PyMOL and comments on the manuscript; and Stephanie Martinez for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Clinical and Translational Sciences Grant RR025761 from the Indiana Clinical and Translational Sciences Institute funded by NCRR Project Development Team. This work was also supported by the Indiana State Department of Health-Spinal Cord and Brain Injury Fund A70-9-079138 (to R. K.), National Scientist Development Grant SDG5280023 from the American Heart Association (to R. K.), and Neurofibromatosis New Investigator Award NF1000099 from the Department of Defense/Congressionally Directed Medical Research Program (to R. K.).
- CRMP2
- collapsin response mediator protein 2
- β2
- β2 subunit of voltage-gated sodium channels
- CAD
- catecholamine A differentiated
- CRMP2AAA
- CRMP2 mutant with lysine 374, methionine 375, and aspartate 376 mutated to alanines
- CRMP2-K374A
- CRMP2 mutant with lysine 374 mutated to alanine
- DIV
- days in vitro
- DRG
- dorsal root ganglion
- EGFP
- enhanced green fluorescent protein
- LCM
- lacosamide
- NaV1.x
- voltage-gated sodium channel isoform 1.x
- NDSM
- negatively charged SUMOylation motif
- SENP
- sentrin/SUMO-specific protease
- SUMO
- small ubiquitin-like modifier
- ANOVA
- analysis of variance
- VGSC
- voltage-gated sodium channel
- pF
- picofarad.
REFERENCES
- 1. Inagaki N., Chihara K., Arimura N., Ménager C., Kawano Y., Matsuo N., Nishimura T., Amano M., Kaibuchi K. (2001) CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4, 781–782 [DOI] [PubMed] [Google Scholar]
- 2. Khanna R., Wilson S. M., Brittain J. M., Weimer J. M., Sultana R., Butterfield A. D., Hensley K. (2012) Opening Pandoras' jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory, and motor neurons, and central disorders. Future Neurol. 7, 749–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chi X. X., Schmutzler B. S., Brittain J. M., Wang Y., Hingtgen C. M., Nicol G. D., Khanna R. (2009) Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 122, 4351–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brittain J. M., Piekarz A. D., Wang Y., Kondo T., Cummins T. R., Khanna R. (2009) An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brittain J. M., Chen L., Wilson S. M., Brustovetsky T., Gao X., Ashpole N. M., Molosh A. I., You H., Hudmon A., Shekhar A., White F. A., Zamponi G. W., Brustovetsky N., Chen J., Khanna R. (2011) Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 286, 37778–37792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bretin S., Rogemond V., Marin P., Maus M., Torrens Y., Honnorat J., Glowinski J., Prémont J., Gauchy C. (2006) Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J. Neurochem. 98, 1252–1265 [DOI] [PubMed] [Google Scholar]
- 7. Errington A. C., Stöhr T., Heers C., Lees G. (2008) The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169 [DOI] [PubMed] [Google Scholar]
- 8. Errington A. C., Coyne L., Stöhr T., Selve N., Lees G. (2006) Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology 50, 1016–1029 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y., Brittain J. M., Jarecki B. W., Park K. D., Wilson S. M., Wang B., Hale R., Meroueh S. O., Cummins T. R., Khanna R. (2010) In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein-2 identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 285, 25296–25307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolff C., Carrington B., Varrin-Doyer M., Vandendriessche A., Van der Perren C., Famelart M., Gillard M., Foerch P., Rogemond V., Honnorat J., Lawson A., Miller K. (2012) Drug binding assays do not reveal specific binding of lacosamide to collapsin response mediator protein 2 (CRMP-2). CNS Neurosci. Ther. 18, 493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brittain J. M., Wang Y., Eruvwetere O., Khanna R. (2012) Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 586, 3813–3818 [DOI] [PubMed] [Google Scholar]
- 12. Arimura N., Ménager C., Kawano Y., Yoshimura T., Kawabata S., Hattori A., Fukata Y., Amano M., Goshima Y., Inagaki M., Morone N., Usukura J., Kaibuchi K. (2005) Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell. Biol. 25, 9973–9984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149 [DOI] [PubMed] [Google Scholar]
- 14. Arimura N., Inagaki N., Chihara K., Ménager C., Nakamura N., Amano M., Iwamatsu A., Goshima Y., Kaibuchi K. (2000) Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 275, 23973–23980 [DOI] [PubMed] [Google Scholar]
- 15. Morinaka A., Yamada M., Itofusa R., Funato Y., Yoshimura Y., Nakamura F., Yoshimura T., Kaibuchi K., Goshima Y., Hoshino M., Kamiguchi H., Miki H. (2011) Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 4, ra26. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Z., Ottens A. K., Sadasivan S., Kobeissy F. H., Fang T., Hayes R. L., Wang K. K. (2007) Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J. Neurotrauma 24, 460–472 [DOI] [PubMed] [Google Scholar]
- 17. Ju W., Li Q., Wilson S. M., Brittain J. M., Meroueh L., Khanna R. (2013) SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels 7, 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 19. Wilkinson K. A., Nakamura Y., Henley J. M. (2010) Targets and consequences of protein SUMOylation in neurons. Brain Res. Rev. 64, 195–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anckar J., Sistonen L. (2007) SUMO: getting it on. Biochem. Soc. Trans. 35, 1409–1413 [DOI] [PubMed] [Google Scholar]
- 21. Wilkinson K. A., Henley J. M. (2010) Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukhopadhyay D., Dasso M. (2007) Modification in reverse: the SUMO proteases. Trends Biochem. Sci. 32, 286–295 [DOI] [PubMed] [Google Scholar]
- 23. Benson M. D., Li Q. J., Kieckhafer K., Dudek D., Whorton M. R., Sunahara R. K., Iñiguez-Lluhí J. A., Martens J. R. (2007) SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc. Natl. Acad. Sci. U.S.A. 104, 1805–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plant L. D., Dowdell E. J., Dementieva I. S., Marks J. D., Goldstein S. A. (2011) SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol. 137, 441–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Plant L. D., Dementieva I. S., Kollewe A., Olikara S., Marks J. D., Goldstein S. A. (2010) One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc. Natl. Acad. Sci. U.S.A. 107, 10743–10748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajan S., Plant L. D., Rabin M. L., Butler M. H., Goldstein S. A. (2005) Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121, 37–47 [DOI] [PubMed] [Google Scholar]
- 27. Konopacki F. A., Jaafari N., Rocca D. L., Wilkinson K. A., Chamberlain S., Rubin P., Kantamneni S., Mellor J. R., Henley J. M. (2011) Agonist-induced PKC phosphorylation regulates GluK2 SUMOylation and kainate receptor endocytosis. Proc. Natl. Acad. Sci. U.S.A. 108, 19772–19777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilkinson K. A., Nishimune A., Henley J. M. (2008) Analysis of SUMO-1 modification of neuronal proteins containing consensus SUMOylation motifs. Neurosci. Lett. 436, 239–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson S. M., Xiong W., Wang Y., Ping X., Head J. D., Brittain J. M., Gagare P. D., Ramachandran P. V., Jin X., Khanna R. (2012) Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein 2-mediated neurite outgrowth and tubulin polymerization. Neuroscience 210, 451–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dib-Hajj S. D., Yang Y., Black J. A., Waxman S. G. (2013) The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 [DOI] [PubMed] [Google Scholar]
- 31. Feliciangeli S., Bendahhou S., Sandoz G., Gounon P., Reichold M., Warth R., Lazdunski M., Barhanin J., Lesage F. (2007) Does sumoylation control K2P1/TWIK1 background K+ channels? Cell 130, 563–569 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Park K. D., Salome C., Wilson S. M., Stables J. P., Liu R., Khanna R., Kohn H. (2011) Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem. Neurosci. 2, 90–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y., Wilson S. M., Brittain J. M., Ripsch M. S., Salomé C., Park K. D., White F. A., Khanna R., Kohn H. (2011) Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem. Neurosci. 2, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson S. M., Schmutzler B. S., Brittain J. M., Dustrude E. T., Ripsch M. S., Pellman J. J., Yeum T. S., Hurley J. H., Hingtgen C. M., White F. A., Khanna R. (2012) Inhibition of transmitter release and attenuation of AIDS therapy-induced and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J. Biol. Chem. 287, 35065–35077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao Y., Bingham J. P., Zhu W., Moczydlowski E., Liang S., Cummins T. R. (2008) Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 283, 27300–27313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. King A. M., Yang X. F., Wang Y., Dustrude E. T., Barbosa C., Due M. R., Piekarz A. D., Wilson S. M., White F. A., Salomé C., Cummins T. R., Khanna R., Kohn H. (2012) Identification of the benzyloxyphenyl pharmacophore: a structural unit that promotes sodium channel slow inactivation. ACS Chem. Neurosci. 3, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cummins T. R., Aglieco F., Renganathan M., Herzog R. I., Dib-Hajj S. D., Waxman S. G. (2001) Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Theile J. W., Cummins T. R. (2011) Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol. Pharmacol. 80, 724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindsay R. M. (1988) Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J. Neurosci. 8, 2394–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winter J., Forbes C. A., Sternberg J., Lindsay R. M. (1988) Nerve growth factor (NGF) regulates adult rat cultured dorsal root ganglion neuron responses to the excitotoxin capsaicin. Neuron 1, 973–981 [DOI] [PubMed] [Google Scholar]
- 41. Chi X. X., Nicol G. D. (2007) Manipulation of the potassium channel Kv1.1 and its effect on neuronal excitability in rat sensory neurons. J. Neurophysiol. 98, 2683–2692 [DOI] [PubMed] [Google Scholar]
- 42. Leclere P. G., Panjwani A., Docherty R., Berry M., Pizzey J., Tonge D. A. (2005) Effective gene delivery to adult neurons by a modified form of electroporation. J. Neurosci. Methods 142, 137–143 [DOI] [PubMed] [Google Scholar]
- 43. Chi X. X., Schmutzler B. S., Brittain J. M., Hingtgen C. M., Nicol G. D., Khanna R. (2009) Regulation of N-type voltage-gated calcium (CaV2.2) channels and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 23, 4351–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joiner W. J., Khanna R., Schlichter L. C., Kaczmarek L. K. (2001) Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 276, 37980–37985 [DOI] [PubMed] [Google Scholar]
- 45. Brittain J. M., Duarte D. B., Wilson S. M., Zhu W., Ballard C., Johnson P. L., Liu N., Xiong W., Ripsch M. S., Wang Y., Fehrenbacher J. C., Fitz S. D., Khanna M., Park C. K., Schmutzler B. S., Cheon B. M., Due M. R., Brustovetsky T., Ashpole N. M., Hudmon A., Meroueh S. O., Hingtgen C. M., Brustovetsky N., Ji R. R., Hurley J. H., Jin X., Shekhar A., Xu X. M., Oxford G. S., Vasko M. R., White F. A., Khanna R. (2011) Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Merrill J. C., Melhuish T. A., Kagey M. H., Yang S. H., Sharrocks A. D., Wotton D. (2010) A role for non-covalent SUMO interaction motifs in Pc2/CBX4 E3 activity. PLoS One 5, e8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duprez E., Saurin A. J., Desterro J. M., Lallemand-Breitenbach V., Howe K., Boddy M. N., Solomon E., de Thé H., Hay R. T., Freemont P. S. (1999) SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J. Cell Sci. 112, 381–393 [DOI] [PubMed] [Google Scholar]
- 48. Zhu J., Zhu S., Guzzo C. M., Ellis N. A., Sung K. S., Choi C. Y., Matunis M. J. (2008) Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J. Biol. Chem. 283, 29405–29415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Perdomo J., Verger A., Turner J., Crossley M. (2005) Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol. Cell. Biol. 25, 1549–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sampson D. A., Wang M., Matunis M. J. (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 276, 21664–21669 [DOI] [PubMed] [Google Scholar]
- 51. Sheets P. L., Heers C., Stoehr T., Cummins T. R. (2008) Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 326, 89–99 [DOI] [PubMed] [Google Scholar]
- 52. Chan A. W., Khanna R., Li Q., Stanley E. F. (2007) Munc18: a presynaptic transmitter release site N type (CaV2.2) calcium channel interacting protein. Channels 1, 11–20 [PubMed] [Google Scholar]
- 53. Wang L. H., Strittmatter S. M. (1997) Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J. Neurochem. 69, 2261–2269 [DOI] [PubMed] [Google Scholar]
- 54. Stenmark P., Ogg D., Flodin S., Flores A., Kotenyova T., Nyman T., Nordlund P., Kursula P. (2007) The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J. Neurochem. 101, 906–917 [DOI] [PubMed] [Google Scholar]
- 55. Lopez-Santiago L. F., Pertin M., Morisod X., Chen C., Hong S., Wiley J., Decosterd I., Isom L. L. (2006) Sodium channel β2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J. Neurosci. 26, 7984–7994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shao D., Okuse K., Djamgoz M. B. (2009) Protein-protein interactions involving voltage-gated sodium channels: Post-translational regulation, intracellular trafficking and functional expression. Int. J. Biochem. Cell Biol. 41, 1471–1481 [DOI] [PubMed] [Google Scholar]
- 57. Kim D. Y., Ingano L. A., Carey B. W., Pettingell W. H., Kovacs D. M. (2005) Presenilin/γ-secretase-mediated cleavage of the voltage-gated sodium channel β2-subunit regulates cell adhesion and migration. J. Biol. Chem. 280, 23251–23261 [DOI] [PubMed] [Google Scholar]
- 58. Kim D. Y., Carey B. W., Wang H., Ingano L. A., Binshtok A. M., Wertz M. H., Pettingell W. H., He P., Lee V. M., Woolf C. J., Kovacs D. M. (2007) BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 9, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rusconi R., Scalmani P., Cassulini R. R., Giunti G., Gambardella A., Franceschetti S., Annesi G., Wanke E., Mantegazza M. (2007) Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J. Neurosci. 27, 11037–11046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Casini S., Tan H. L., Demirayak I., Remme C. A., Amin A. S., Scicluna B. P., Chatyan H., Ruijter J. M., Bezzina C. R., van Ginneken A. C., Veldkamp M. W. (2010) Tubulin polymerization modifies cardiac sodium channel expression and gating. Cardiovasc. Res. 85, 691–700 [DOI] [PubMed] [Google Scholar]
- 61. Takahashi N., Kikuchi S., Dai Y., Kobayashi K., Fukuoka T., Noguchi K. (2003) Expression of auxiliary β subunits of sodium channels in primary afferent neurons and the effect of nerve injury. Neuroscience 121, 441–450 [DOI] [PubMed] [Google Scholar]
- 62. Okuse K., Malik-Hall M., Baker M. D., Poon W. Y., Kong H., Chao M. V., Wood J. N. (2002) Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature 417, 653–656 [DOI] [PubMed] [Google Scholar]
- 63. Leterrier C., Brachet A., Fache M. P., Dargent B. (2010) Voltage-gated sodium channel organization in neurons: protein interactions and trafficking pathways. Neurosci. Lett. 486, 92–100 [DOI] [PubMed] [Google Scholar]
- 64. Beyreuther B. K., Freitag J., Heers C., Krebsfänger N., Scharfenecker U., Stöhr T. (2007) Lacosamide: a review of preclinical properties. CNS Drug Rev. 13, 21–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Park K. D., Stables J. P., Liu R., Kohn H. (2010) Proteomic searches comparing two (R)-lacosamide affinity baits: An electrophilic arylisothiocyanate and a photoactivated arylazide group. Org. Biomol. Chem. 8, 2803–2813 [DOI] [PubMed] [Google Scholar]
- 66. Martin S., Wilkinson K. A., Nishimune A., Henley J. M. (2007) Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat. Rev. Neurosci. 8, 948–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Estacion M., Han C., Choi J. S., Hoeijmakers J. G., Lauria G., Drenth J. P., Gerrits M. M., Dib-Hajj S. D., Faber C. G., Merkies I. S., Waxman S. G. (2011) Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol. Pain 7, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Momin A., Wood J. N. (2008) Sensory neuron voltage-gated sodium channels as analgesic drug targets. Curr. Opin. Neurobiol. 18, 383–388 [DOI] [PubMed] [Google Scholar]
- 69. Cox J. J., Reimann F., Nicholas A. K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y., Al-Gazali L., Hamamy H., Valente E. M., Gorman S., Williams R., McHale D. P., Wood J. N., Gribble F. M., Woods C. G. (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hong S., Morrow T. J., Paulson P. E., Isom L. L., Wiley J. W. (2004) Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 279, 29341–29350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Black J. A., Liu S., Tanaka M., Cummins T. R., Waxman S. G. (2004) Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247 [DOI] [PubMed] [Google Scholar]
- 72. Benson M., Iniguez-Lluhi J. A., Martens J. (2009) in SUMO Regulation of Cellular Processes (Wilson V. G., ed) pp. 117–136, Springer Science+Business Media [Google Scholar]
- 73. Catterall W. A., Kalume F., Oakley J. C. (2010) NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Escayg A., Goldin A. L. (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51, 1650–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vacher H., Trimmer J. S. (2012) Trafficking mechanisms underlying neuronal voltage-gated ion channel localization at the axon initial segment. Epilepsia 53, 21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Felts P. A., Yokoyama S., Dib-Hajj S., Black J. A., Waxman S. G. (1997) Sodium channel α-subunit mRNAs I, II, III, NaG, Na6, and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res. Mol. Brain Res. 45, 71–82 [DOI] [PubMed] [Google Scholar]
- 77. Hodgdon K. E., Hingtgen C. M., Nicol G. D. (2012) Dorsal root ganglia isolated from Nf1+/− mice exhibit increased levels of mRNA expression of voltage-dependent sodium channels. Neuroscience 206, 237–244 [DOI] [PubMed] [Google Scholar]
- 78. Kim C. H., Oh Y., Chung J. M., Chung K. (2001) The changes in expression of three subtypes of TTX-sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res. 95, 153–161 [DOI] [PubMed] [Google Scholar]
- 79. Waxman S. G., Kocsis J. D., Black J. A. (1994) Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 72, 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Malhotra J. D., Kazen-Gillespie K., Hortsch M., Isom L. L. (2000) Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J. Biol. Chem. 275, 11383–11388 [DOI] [PubMed] [Google Scholar]
- 81. Pertin M., Ji R. R., Berta T., Powell A. J., Karchewski L., Tate S. N., Isom L. L., Woolf C. J., Gilliard N., Spahn D. R., Decosterd I. (2005) Up-regulation of the voltage-gated sodium channel β2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980 [DOI] [PMC free article] [PubMed] [Google Scholar]