

Background: Signaling mechanisms regulating rat embryonic stem cell (rESC) pluripotency are understood poorly.
Results: Inhibition of PKC signaling promotes rESC self-renewal without compromising developmental potency.
Conclusion: PKC signaling contributes to the balance of self-renewal versus differentiation of rESCs.
Significance: PKC signaling could be targeted to derive rat pluripotent stem cells to establish transgenic models and for regenerative studies.
Keywords: Chromatin Modification, Embryonic Stem Cell, NF-κB, PKC, Rat
Abstract
Embryonic stem cell (ESC) pluripotency is orchestrated by distinct signaling pathways that are often targeted to maintain ESC self-renewal or their differentiation to other lineages. We showed earlier that inhibition of PKC signaling maintains pluripotency in mouse ESCs. Therefore, in this study, we investigated the importance of protein kinase C signaling in the context of rat ESC (rESC) pluripotency. Here we show that inhibition of PKC signaling is an efficient strategy to establish and maintain pluripotent rESCs and to facilitate reprogramming of rat embryonic fibroblasts to rat induced pluripotent stem cells. The complete developmental potential of rESCs was confirmed with viable chimeras and germ line transmission. Our molecular analyses indicated that inhibition of a PKCζ-NF-κB-microRNA-21/microRNA-29 regulatory axis contributes to the maintenance of rESC self-renewal. In addition, PKC inhibition maintains ESC-specific epigenetic modifications at the chromatin domains of pluripotency genes and, thereby, maintains their expression. Our results indicate a conserved function of PKC signaling in balancing self-renewal versus differentiation of both mouse and rat ESCs and indicate that targeting PKC signaling might be an efficient strategy to establish ESCs from other mammalian species.
Introduction
Because of physiological and behavioral resemblances with humans, the rat is an excellent animal model to study human diseases (1), and recent success in establishing germ-line competent rESCs4 (2–4) has opened up new possibilities of using gene-targeted or transgenic rat models (5–7) to understand the genetic basis of human diseases. Germ line-competent rESCs can be established using the small molecule inhibitors CHIR99021 and PD032590 (henceforth mentioned as 2i), which inhibit glycogen synthase kinase 3 (GSK3) and MEK, respectively (2, 3). The addition of leukemia inhibitory factor (LIF) along with 2i further boosts self-renewal of rESCs (2, 3). Another study implemented inhibition of Rho-associated kinase and TGF-β signaling along with 2i to establish pluripotent rat stem cells (6). Both 2i and 2i/LIF culture conditions also efficiently maintain pluripotency in mESCs (8). However, unlike mESCs, rESCs that are established and maintained in 2i and 2i/LIF express relatively high levels of trophoblast stem cell (TSC)-specific factors, like CDX2 (4, 9), and have a propensity for genomic instability (2). Curiously, rESCs grown with Rho-associated kinase and TGF-β inhibitors also showed much higher levels of CDX2 expression (9). Also, the efficiency of germ line competence in 2i- or 2i/LIF-cultured rESCs is lower than that of mESC and random. Thus, to maximize the potential of rESCs as tools for genetic research, a better understanding of molecular mechanisms that modulate rESC pluripotency versus differentiation is necessary.
In an earlier study (10), we showed that inhibition of PKC signaling by a selective PKC inhibitor, 3-[1-[3-(dimethylamino)propyl]-5-methoxy-1H-indol-3-yl]-4-(1H-indol-3-yl)-1H-pyrrole-2,5-dione (Gö6983, henceforth mentioned as PKCi) is sufficient to maintain, derive, and propagate pluripotent mESCs. Our mechanistic analyses indicated that, among the PKC isoforms, the atypical PKC isoform PKCζ is crucial for inducing multilineage differentiation in mESCs. Furthermore, we also demonstrated that the PKCi culture condition facilitates reprogramming of differentiated mouse cells to iPSCs. Therefore, in this study, we tested whether inhibition of PKC signaling by PKCi maintains rESC self-renewal and pluripotency. We found that the PKCi culture condition maintains self-renewal of established rESCs without affecting their complete developmental potential. We also found that the PKCi culture condition enables the derivation of new ESC lines from rat blastocysts and reprogramming of differentiated rat cells to riPSCs. Also, our molecular analyses revealed that, unlike the 2i/LIF culture condition, PKCi-maintained rESCs do not express TSC-specific genes. Collectively, our results indicate that PKC signaling is an important regulatory pathway in balancing self-renewal versus lineage commitment in rESCs and could be exploited to establish and maintain germ line-competent pluripotent rat stem cells.
EXPERIMENTAL PROCEDURES
Inhibitors
PKCi (Gö6983) was purchased from Tocris Biosciences (catalog no. 2285, Ellisville, MO) and was used at a concentration of 5 μm unless stated otherwise. PD0325901 (1 μm, catalog no. 444966) and CHIR99021 (3 μm, catalog no. 04-0004) were purchased from Stemgent (Cambridge, MA). LIF (used at 100 IU/ml) was purchased from Millipore (ESGRO, Millipore, Temecula, CA).
ESC Culture
For regular maintenance, derived rESCs were cultured with either 2i/LIF or PKCi on irradiated feeder REF in N2B27 medium containing DMEM/F12 (Invitrogen, catalog no. 10565), neurobasal medium (catalog no. 21103, Invitrogen), 1% B27 supplement (catalog no. 17504-044, Invitrogen), 0.5% N2 supplement (catalog no. 17502-048, Invitrogen), 25 μg/ml BSA fraction V (catalog no. 15260, Invitrogen), and 100 μm 2-mercaptoethanol (catalog no. M7522-100ML, Sigma). For in vitro differentiation studies, rESCs were cultured without PKCi in monolayer culture for 5–6 days or allowed to form embryoid bodies (EBs).
Quantitative RT-PCR Analysis
RNA was extracted from different cell samples with TRIzol reagent (Invitrogen). cDNA was prepared by annealing RNA (1 μg) with 250 ng of a 5:1 mixture of random and oligo(dT) primers heated at 68 °C for 10 min. This was followed by incubation with Moloney murine leukemia virus reverse transcriptase (50 units) (Invitrogen) combined with 10 mm DTT, RNasin (Promega, Madison, WI), and 0.5 mm dNTPs at 42 °C for 1 h. Reactions were diluted to a final volume of 100 μl and heat-inactivated at 97 °C for 5 min. 20-μl PCR reactions contained 2 μl of cDNA, 10 μl of SYBR Green Master Mix (Applied Biosystems, Foster City, CA), and corresponding primer sets. Relative expression levels were determined from a standard curve generated from serial dilution of rat universal reference cDNA samples and were normalized to the expression of 18 S. At least three independent experiments were done for each set of data. Primers used in this study are mentioned in the supplemental information.
Western Blot Analysis
Whole cell lysates were prepared in SDS gel loading buffer, and Western blot analyses were performed following procedures described earlier (10). Antibodies used for this study are included in the supplemental information.
Immunostaining and Confocal Microscopy
Immunostaining to detect expression of OCT4, NANOG, and CDX2 in rESCs was performed using standard protocols (10). Briefly, rESCs were cultured on feeder layer-coated coverslips under different culture conditions. After 5 days, cells were fixed with 3.7% paraformaldehyde and permeabilized with 0.5% Triton X-100 in Dulbecco's phosphate-buffered saline. Nonspecific binding was blocked with 1% BSA, and cells were incubated overnight with primary antibody at a dilution of 1:200. Fluorescent conjugated secondary antibodies (Alexa Fluor 488 and Alexa Fluor 568, Molecular Probes and Invitrogen) were used at a 1:200 dilution. Coverslips were mounted on slides with DAPI mounting media (Invitrogen) and observed using a confocal microscope (Carl Zeiss).
EB Formation
To generate EBs, rESCs and riPSCs were grown in the absence of PKCi/LIF/2i in a differentiation medium containing 15% FBS (Stemcell Technologies, Vancouver, BC), 1% l-glutamine, 1% ascorbic acid (Stemcell Technologies), and 3 μl/ml methylthioglycolate (Sigma). Cells were washed with PBS, trypsinized (0.05% trypsin EDTA for 5 min), and made into single-cell suspensions. To generate day 5–6 EBs, 4000 cells/ml were added to differentiation medium in a low-adherent tissue culture plate (catalog no. 3471 Costar, Corning Inc., Corning, NY) with intermittent shaking.
Teratoma Formation Analyses
For teratoma formation, 1–2 million riPSCs were mixed with 30% Matrigel (BD Biosciences), injected into the kidneys of 6-week-old male SCID-beige mice (Charles River Laboratories), and tumors were collected 3–4 weeks post-injection. Tumor tissues were fixed with 10% Formalin overnight, embedded in paraffin, cut into 5-μm serial sections, and H&E-stained.
Chimera Generation and Germ Line Transmission
Six-week-old female Sprague-Dawley (SD) rats (Harlan Laboratories, Indianapolis, IN) were mated overnight to intact SD stud males. Uteri were collected 4.5 days post-coitum and flushed with M2 medium (Millipore) for the collection of blastocysts. Ten to 12 ESCs were injected into the blastocoel of each blastocyst. After injection, the blastocysts were transferred surgically into recipient SD females that were pseudopregnant by mating with vasectomized males. Chimeras with DA rESCs were determined from coat color. Chimeras were generated from F344 rESCs, and iPSCs were determined via microsatellite analyses. Male chimeras, generated from PKCi-maintained DA rESCs, were mated with SD female adult rats to test for germ line transmission.
De Novo Derivation of rESCs with PKCi
Rat ESCs were derived using our protocol described previously (5) with modifications. Briefly, blastocysts from F344 and DA rats were isolated and plated on mouse embryonic fibroblasts (Globalstem, Rockville, MD) or an REF feeder layer in medium containing either 2i/LIF, PKCi, or PKCi/PD0325901. As a control, medium without any inhibitors or with PD0325901 alone was also used. To obtain rESC colonies, blastocyst outgrowths were disaggregated either mechanically or using trypsin and replated on the feeder layer. rESC colonies were expanded by replating with PKCi at clonal or higher density. For in vitro analysis, cells were cultured following the same protocol mentioned earlier.
Isolation of REFs and Reprogramming to riPSCs
REFs were reprogrammed using a lentiviral vector expressing Oct4, Sox2, Klf4, or c-Myc (OSKM) in a single virus (polycistronic expression from a single promoter) (11) under the control of the doxycycline (Dox)-inducible tetO operator. REFs derived from embryos collected at days 14–16 of pregnancy were isolated from F344 and DA rats, expanded in DMEM containing 10% FBS (Invitrogen), and passaged at confluency using standard methods. At passage 3, REFs were infected overnight with lentiviral particles expressing OSKM and tetracycline-controllable transactivator, and then the culture medium was replaced with PKCi (5 μm) or PKCi + PD032801 and Dox (2 μg/ml). After 48 h, cells were cultured in medium without Dox. 12 days after Dox removal, several nascent iPSC-like colonies were observed. A few of those colonies were analyzed for endogenous OCT4 and NANOG expression. Other colonies were cultured up to 21 days. For further analysis, iPSC colonies were picked manually at different days, starting on day 18 after Dox removal. Cells were trypsinized and replated at clonal or higher density in medium with PKCi. Expression of the pluripotent markers in riPSCs, cultured for multiple passages with PKCi, was analyzed by qRT-PCR as well as immunofluorescence.
Analysis of NF-κB5X-Luc Reporter Activation
For NF-κB activity analysis, rESCs (5 × 104) were transfected with 1 μg of a plasmid expressing a luciferase reporter under the control of canonical NF-κB binding motifs (PathDetect NF-κB cis-Reporting System, Stratagene, catalog no. 219077). Transfection was performed using a nucleofector device (Lonza) following the protocol of the manufacturer and seeded on feeder REF on 24-well plates with rESC medium with or without PKCi. After 48 h, rESC colonies were picked up manually, whole cell lysates were prepared, and luciferase activity was measured in a Veritas microplate luminometer using the luciferase assay buffer (Promega). Luciferase activity was calibrated relative to total protein present in the whole cell lysate.
miRNA Expression Analysis
miRNA expression was determined using a miRCURY LNATM Universal RT microRNA PCR system (Exicon, Woburn, MA) using the protocol of the manufacturer. Total RNA was isolated from rESCs, riPSCs, and REFs using an RNA isolation kit (catalog no. 74104, Qiagen, Valencia, CA). cDNA was synthesized using a miRCURY LNATM Universal RT microRNA PCR, polyadenylation, and cDNA synthesis kit (catalog no. 203300, Exicon). Expression of a specific miRNA was measured using UniRT LNATM PCR primer sets. For miR-21, hsa-miR-21 primer sets (catalog no. 204230, Exicon, target sequence UAGCUUAUCAGACUGAUGUUGA) were used, and for miR-29a, hsa-miR-29a primer sets (catalog no. 204698, Exicon, target sequence UAGCACCAUCUGAAAUCGGUUA) were used. Endogenous 18 S rRNA was used as the control.
ChIP
Real time PCR-based quantitative ChIP analysis was performed using our protocol described previously (12, 13). Briefly, cells were trypsinized, cross-linked with formaldehyde (1%), and sonicated to generate chromatin fragments. Antibodies were used to immunoprecipitate protein-DNA cross-linked fragments. Precipitated complexes were eluted and reverse-cross-linked. Enrichment of chromatin fragments was measured by quantitative PCR using SYBR Green (Applied Biosystems) fluorescence relative to a standard curve of input chromatin. The primer sequences and antibody information are included in the supplemental information.
Transient Transfection Assay
The promoter regions of Rno-miR-21 (-242 to +56 bp) and Rno-miR-29a (-413 to +15 bp) were cloned with or without deletion of conserved putative NF-κB motifs in the pGL3 basic vector (Promega) containing a luciferase reporter gene. For transient transfection analysis, F344 rESCs were transfected with an equal amount of each plasmid (3 μg) using a nucleofector (Lonza). Cells were transferred on REF feeders and cultured without PKCi for 72–96 h. Cell lysates were harvested, and luciferase activity was measured in a Veritas microplate luminometer using the luciferase assay buffer (Promega). The luciferase activity for each sample was normalized to the protein concentration of the lysate. At least three independent preparations of each plasmid were analyzed, and the results were averaged.
RNA Interference and Expression of Pre-miRNAs
shRNAs targeting mouse PKCζ mRNA were cloned in pLKO1 (Addgene, Cambridge, MA). Lentiviral supernatants were produced in HEK293T cells as described previously (14, 15). F344 rESCs were infected with lentiviral supernatants and selected by the addition of 1 μg/ml puromycin (Sigma). A construct with the target sequence ATCCCGGTAAGTTCTGTTG, corresponding to the 3′ UTR region of PKCζ mRNA, specifically knocked down PKCζ expression. For pre-miRNA expression, rat pre-miR-21 (Rno-miR-21, ENSEMBL gene ENSRNOG00000035480) and pre-miR-29a (Rno-miR-29a, ENSEMBL gene ENSRNOG00000035458) were cloned in pLKO1. For transduction, lentiviral particles were produced in HEK293T cells, and rESCs were infected with viral particles. The transduced cells were cultured further for 5 days without PKCi. Cells from colonies were picked up manually, and RNA was isolated for gene expression analyses.
Bisulfite Sequencing
Genomic DNA was isolated from cells cultured in PKCi or 2i/LIF. Isolated DNA was modified with a CpGenome Fast DNA modification kit (catalog no. S7824, Millipore) to convert the unmethylated cytosine to uracil according to the instructions of the manufacturer. The promoter region of Oct4 and Nanog was amplified by PCR using Taq polymerase and primers described earlier (16) (also mentioned in the supplemental information). PCR products were cloned into the pGMT vector (Promega) and sequenced with M13 reverse primer.
RESULTS
Inhibition of PKC Signaling Maintains rESCs in an Undifferentiated State without Affecting Their Complete Developmental Potential
To determine whether PKCi could maintain previously established rESC lines in an undifferentiated state, we cultured an F344 rESCs line that expresses EGFP under the control of a 3.1-kb portion of the rat Oct4 promoter/enhancer region (4, 17). We found that PKCi culture efficiently maintains undifferentiated colony morphology and OCT4-EGFP reporter expression of established rESC lines (Fig. 1A) when they are cultured with PKCi in the absence of 2i or 2i/LIF. Furthermore, immunofluorescence analyses in another F344 rESC line (without the OCT4-EGFP reporter) showed strong expression of endogenous OCT4 and NANOG proteins when cultured for five consecutive passages with PKCi (Fig. 1B). Thus, the morphology, the expression of OCT4-EGFP reporter, and the expression of endogenous pluripotency factors confirmed that culturing F344 rESC lines with PKCi could maintain their undifferentiated state.
FIGURE 1.

Inhibition of PKC signaling supports rESC self-renewal. A, micrographs of an OCT4-EGFP reporter expressing F344 rESCs showing undifferentiated colony morphology and reporter GFP expression after they were cultured for five consecutive passages with PKCi. Scale bars = 250 μm. B, immunofluorescence images show OCT4 and NANOG expression in F344 rESCs that were maintained with PKCi for seven consecutive passages. C, micrographs showing undifferentiated colony morphology (a), presence of alkaline phosphatase (b), and OCT4 expression in the nuclei (c and d) in DA rESCs after culturing for seven consecutive passages with PKCi. Scale bars = 100 μm. D, plot showing relative mRNA expression of the pluripotency genes Oct4, Nanog, and Sox2 in DA rESCs after they were maintained for seven passages in PKCi and 2i/LIF culture conditions. DA REFs were used as negative controls. Gene expressions were measured by qRT-PCR analyses (mean ± S.E., three independent experiments). *, p ≤ 0.05. E, micrograph showing day 5 EBs that were formed from DA rESCs after PKCi withdrawal. Scale bar = 250 μm. F, differentiation potency in PKCi-maintained DA rESCs was determined by measuring mRNA expression (mean ± S.E., three independent experiments) in day 5 EBs. Undifferentiated DA rESCs were used as a control. The plot shows significant (p ≤ 0.01) induction in lineage-specific markers (Gata4 and Gata6 for endoderm, Bmp4 and T for mesoderm, and Nestin and Otx2 for ectoderm) after a 5-day absence of PKCi and EB formation. G, chimeric rats generated with DA rESCs that were cultured with PKCi. H, germ line offspring (white arrow) from the DA chimeras shown in G.
To test whether the PKCi culture condition inhibits differentiation of rESCs irrespective of rat strains, we cultured rESCs that were derived from DA rats. We found that, similar to F344 rESCs, the PKCi culture condition maintained an undifferentiated state and expression of pluripotency markers in DA rESCs (Fig. 1, C and D) for seven consecutive passages, indicating that PKCi-mediated maintenance of rESC self-renewal is not dependent upon rat strains.
Next, we tested whether rESCs, cultured for multiple passages with PKCi, have the multilineage differentiation potential in vitro and complete developmental potential in vivo. Upon withdrawal of PKCi, rESCs readily formed EBs (Fig. 1E) with induction of lineage-specific genes (F). Furthermore, DA rESCs that were maintained for five to eight passages with PKCi readily yielded chimeric rats when injected into SD blastocysts (Fig. 1G and supplemental Table 1). Upon further crossing, those chimeras produced germ line offspring (Fig. 1H). These results confirmed that rESCs maintained in PKCi for multiple passages were pluripotent stem cells.
Inhibition of PKC Signaling Facilitates de Novo Derivation of rESCs
To test whether inhibition of PKC signaling is sufficient for de novo derivation of rESCs, we isolated blastocysts at embryonic day 4.5 from F344 and DA females and cultured them on an REF feeder layer in the PKCi culture condition. We readily established several F344 rESC lines (Fig. 2A) and successfully propagated them for multiple passages. However, with the PKCi culture condition, the rate of proliferation of ESC-like colonies from DA blastocysts was poor and reduced further upon passaging (data not shown). Intriguingly, when we added the MEK inhibitor PD0325901 along with PKCi, we readily derived rESC colonies from DA blastocysts (Fig. 2B). On the contrary, with PD0325901 alone, we were unable to establish new rESC lines from either F344 or DA blastocysts (supplemental Table 1). Furthermore, when propagated with PKCi alone for multiple passages, the newly derived rESC lines successfully maintained undifferentiated colony morphology (Fig. 2C), expressed pluripotency markers (D), and maintained chromosomal stability (E). Our results indicate that PKC inhibition alone is sufficient to derive new rESCs lines from certain rat strains (such as F344). However, a combined inhibition of PKC and MEK signaling might be more efficient to derive new rESC lines from other rat strains.
FIGURE 2.

PKC inhibition facilitates de novo derivation of rESCs. A, passage 1 ESC colony (red border) derived from F344 rat blastocyst with PKCi. Scale bar = 250 μm. B, passage 1 ESC colony derived from DA rat blastocyst with PKCi and the MEK inhibitor PD0325901. Scale bar = 250 μm. C, newly derived DA rESCs of B were cultured for five consecutive passages with PKCi alone. The image shows that newly derived DA rESCs maintain undifferentiated ESC colony morphology in the PKCi-alone culture condition. Scale bar = 250 μm. D, confocal images showing expression of OCT4 and NANOG in passage 6, PKCi-maintained, newly derived DA rESCs. E, karyotype analysis in F344 rESCs that were derived and maintained for five passages with PKCi. F, micrograph showing EBs (day 5) developed from newly derived, PKCi-maintained (passage 6) DA rESCs after PKCi withdrawal. Scale bar = 250 μm. G, analyses of chimera generation with PKCi-derived and maintained (passage five) F344 rESCs. The F344 ESCs were injected into SD blastocysts, genomic DNA was isolated from pups that were born from ESC-injected blastocysts (lanes 1–7), and chimera generation was confirmed (pups. 1, 3, and 4, blue stars) by analyzing rat microsatellite D17rat165 via PCR and gel electrophoresis. Lanes c1, c2, and c3 show microsatellite analyses with DNAs isolated from known control rat strains (F344 and SD cross, SD, and F344 strains, respectively). Lane M indicates the DNA ladder. H, chimeric rats generated with PKCi and MEK inhibitor-derived DA ESCs after maintaining in PKCi for six passages.
We also tested the differentiation potential of PKCi culture-derived F344 and DA rESC lines. We found that, in the absence of PKCi, the newly derived rESCs readily formed EBs (Fig. 2F) with induction of lineage-specific genes (data not shown). Furthermore we found that both F344 and DA rESCs, which were maintained for five and six consecutive passages, respectively, with PKCi alone, could generate chimeras upon blastocyst injection (Fig. 2, G and H). These results confirmed the multilineage developmental potency of PKCi-derived and -maintained rESCs, suggesting maintenance of their pluripotent state.
PKC Inhibition Facilitates Reprogramming of Differentiated Rat Cells to riPSCs
We tested the efficacy of the PKCi culture condition in the derivation of iPSCs from REFs. For this analysis, we isolated REFs from the F344 and DA strains. We transduced REFs with lentiviral particles containing a tetracycline-inducible OKSM construct (Fig. 3A). Transduced REFs were transiently treated with Dox to induce the expression of pluripotency factors from the lentiviral construct. Later, cells were maintained without Dox in the PKCi culture condition. We also used the 2i/LIF culture condition as a positive control because earlier studies reported the successful establishment of riPSC cells with the 2i/LIF culture condition from differentiated rat cells (16, 18). We found that the efficiency to derive rat iPSCs in the 2i/LIF condition is very low (two of five attempts were successful in the Weiss laboratory and zero of four attempts in the Paul laboratory, data not shown). However, in the PKCi culture condition, we readily derived iPSCs from both F344 and DA REFs. We found that in the PKCi culture condition, nascent iPSC colonies with expression of endogenous pluripotency factors appeared from F344 REFs ∼12–14 days after Dox removal (Fig. 3, B and C), and matured iPSC colonies with ESC-like morphology were observed after 18–21 days (B). We have also been able to efficiently establish iPSC colonies from DA REFs with PKCi (Fig. 3D). Established DA iPSC lines were maintained successfully with PKCi for multiple passages with sustained expression of pluripotent markers (Fig. 3, E and F).
FIGURE 3.
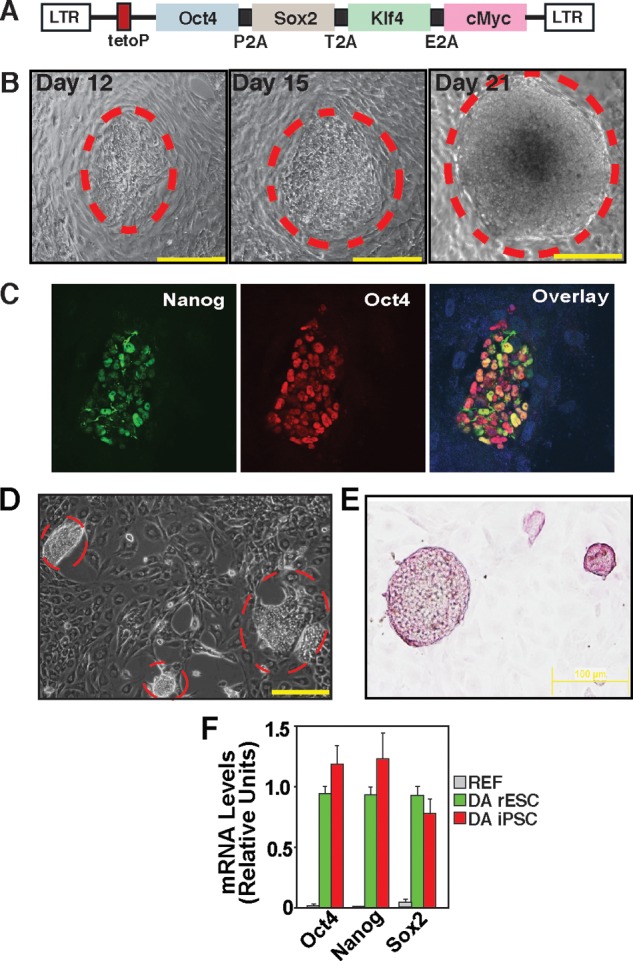
Reprogramming of differentiated rat cells to riPSCs in the PKCi culture condition. A, schematic of the Dox-inducible, OSKM-expressing, lentiviral vector (11). LTR, long terminal repeat. B, micrographs showing an F344 riPSC colony in the PKCi culture condition at days 12, 15, and 21. Scale bars = 250 μm. C, confocal images showing expression of endogenous OCT4 and NANOG in a F344 REF-derived nascent (day 13) iPSC colony in the PKCi culture condition. D, micrograph showing riPSC colonies derived from DA REFs in the PKCi culture condition (day 21). Scale bar = 250 μm. E, micrograph showing alkaline phosphatase activity in PKCi-derived and maintained (passage 5) DA iPSC colonies. F, plot showing relative mRNA expression of pluripotency genes in newly derived DA rESCs and DA riPSCs after they were cultured for six passages with PKCi. DA REFs were used as negative control cells.
Next, we tested the differentiation and developmental potency of PKCi-derived F344 and DA riPSC lines. We propagated two F344 and two DA iPSC lines for seven consecutive passages in the PKCi culture condition and on the basis of a stable euploid karyotype (data not shown) and selected one iPSC line for each of the strains for further analyses of multilineage differentiation and developmental potency in vivo. We found that PKCi-derived riPSCs readily formed EBs (Fig. 4A) in the absence of PKCi with the induction of lineage-specific genes (B) at comparable levels with PKCi-derived rESCs. Furthermore, the PKCi-derived riPSCs readily developed teratomas when injected into the immunocompromised mice (Fig. 4C) and also generated chimeras upon blastocyst injection (D). Collectively, these results suggest that PKCi-derived riPSCs maintain their multilineage developmental potency.
FIGURE 4.
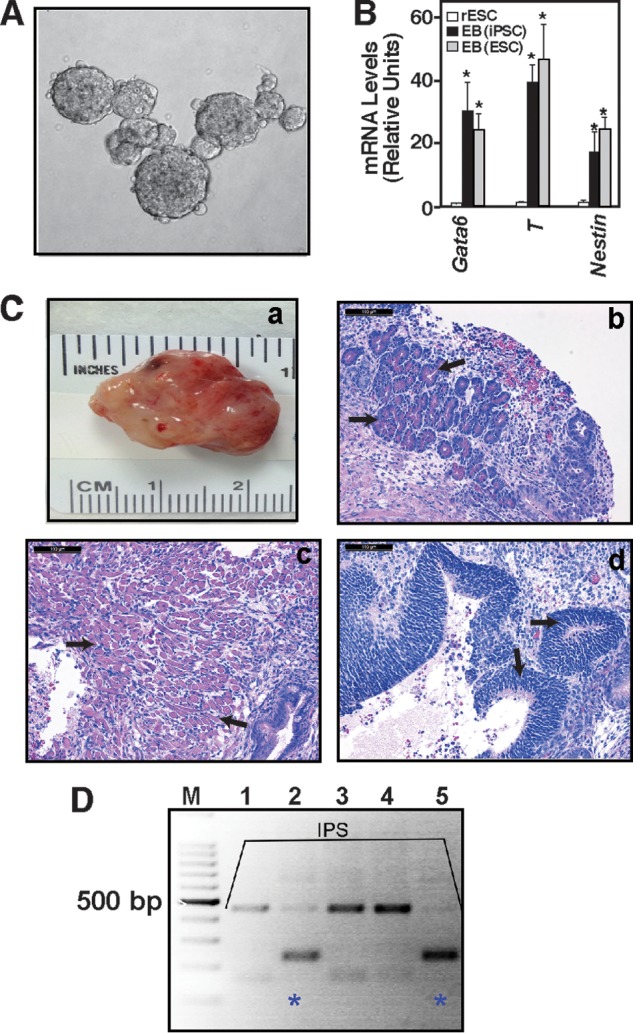
PKCi culture-derived riPSCs maintain in vivo developmental potency. A, micrograph showing EBs (day 5) developed from newly derived, PKCi-maintained (passage 6) DA riPSCs after PKCi withdrawal. B, expression of differentiation markers was analyzed in day 5 EBs generated from newly derived DA rESCs and DA riPSCs (qRT-PCR analyses, mean ± S.E., three independent experiments). The plot shows significant (*, p ≤ 0.01) induction of lineage-specific markers in EBs from both rESCs and riPSCs compared with undifferentiated DA rESCs. C, the in vivo developmental potency of DA riPSCs that were maintained for six passages in PKCi was assessed via teratoma formation analyses. The micrographs show isolated teratomas (a) with pancreatic tissue (endoderm, black arrows, b), muscle (mesoderm, black arrows, c), and neuronal rosette (ectoderm, black arrows, d) characterized by H&E staining. D, the in vivo developmental potency of F344 rIPSCs was assessed via chimera generation. F344 rIPSCs were injected into SD blastocysts, genomic DNA was isolated from pups that were born from transferred blastocysts (lanes 1–5), and chimera generation was confirmed (pups 2 and 5, blue stars) by analyzing rat microsatellite D17rat165 via PCR.
Successful derivation of new rESC and iPSC lines from both the F344 and DA rat strains strongly indicate that PKC signaling is an important pathway to balance self-renewal versus differentiation in pluripotent rat cells. However, to further characterize the PKCi culture condition, we performed two different experiments. We asked whether proper epigenetic signatures at pluripotency and developmental genes are maintained and whether expressions of TSC-specific factors are reduced in rESCs/riPSCs upon culturing in the PKCi condition.
PKC Inhibition Maintains ESC-specific Chromatin Marks at the Pluripotency Genes
Because epigenetic mechanisms modulate ESC self-renewal versus differentiation (19), we tested whether inhibition of PKC function contributes to the maintenance of the ESC-specific chromatin structure in rESCs/rIPSCs, thereby preventing the induction of developmental regulator genes and maintaining the expression of pluripotency genes. One of the epigenetic components, repressive trimethylation on lysine 27 of histone H3 (H3K27Me3) by the polycomb repressor 2 (PRC2) complex at the developmental regulator genes, has been implicated in stem cell pluripotency (20, 21). We tested whether PKCi treatment maintains PRC2-mediated H3K27 methylation at developmental regulator genes. Our quantitative ChIP analyses revealed that PKCi treatment maintained high levels of H3K27Me3 and chromatin occupancy of enhancer of zeste 2 (EZH2), the catalytic histone methyl transferase subunit (22) of the PRC2 complex at the Gata6, T, and Nestin genes (markers for endoderm, mesoderm, and ectoderm differentiation, respectively) (Fig. 5A). In contrast, an H3K27Me3 mark and EZH2 recruitment was not detected at the chromatin domain of the pluripotency genes Oct4 and Nanog (Fig. 5A).
FIGURE 5.
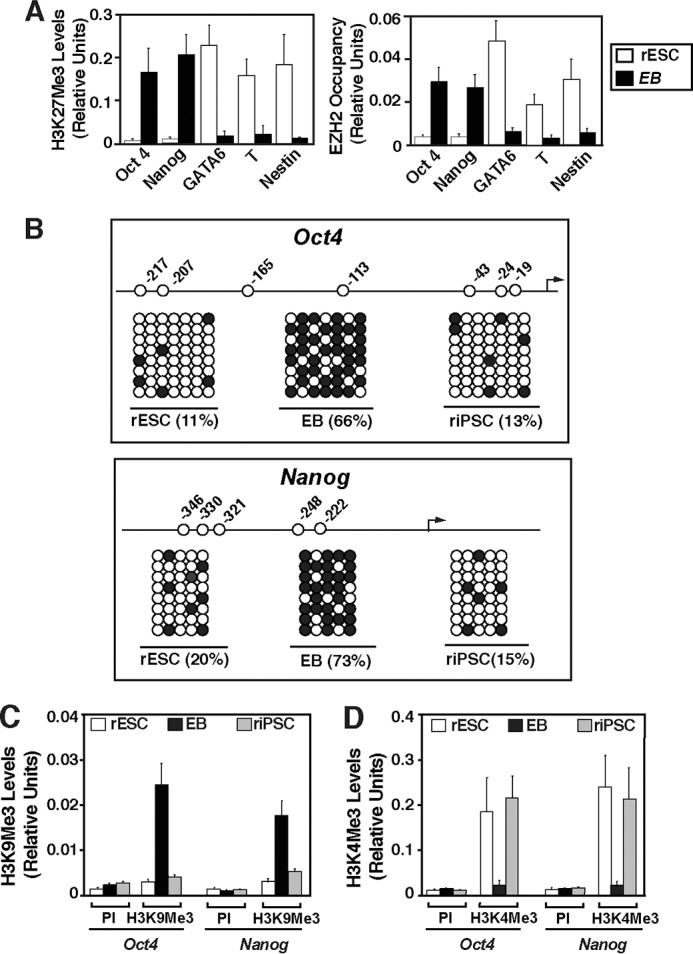
PKC inhibition maintains ES cell-specific epigenetic modifications at the pluripotency genes. A, quantitative ChIP analysis (mean ± S. E., three independent experiments) showing low levels of H3K27Me3 (left panel) and EZH2 (right panel) at the promoter regions of the Oct4 and Nanog genes in F344 rESCs derived and cultured with PKCi. The plots also show that the promoter regions of lineage-specific genes have high levels of H3K27Me3 and EZH2 in undifferentiated rESCs. These patterns reversed in EBs after 6 days of PKCi removal. B, plots showing the position of CpG sites in the rat Oct4 (top panel) and Nanog (bottom panel) promoter and the extent of DNA methylation (●) at the promoter regions of the Oct4 and Nanog loci. F344 rESCs and riPSCs derived and cultured with PKCi and day-6 EBs derived from F344 rESCs were analyzed. Black arrows, transcription start sites. C, and D, quantitative ChIP analyses (mean ± S.E., three independent experiments) showing lack of H3K9Me3 but enrichment of H3K4Me3, respectively, at the Oct4 and Nanog promoters in PKCi-treated F344 rESCs and riPSCs analyzed in B. Preimmune serum (PI) was used as a negative control for ChIP analyses.
To further validate the ESC-specific chromatin signature at the pluripotency genes in PKCi-treated rESCs, we analyzed epigenetic mechanisms that are implicated in the regulation of Oct4 and Nanog expression. During ESC differentiation, silencing of the Oct4 and Nanog genes is associated with de novo DNA methylation at their regulatory regions (23, 24). We tested whether PKCi treatment prevents DNA methylation at the regulatory regions of these genes by bisulfite sequencing. For our analyses, we cultured F344 rESCs with PKCi or generated EBs after PKCi removal. We found that in the presence of PKCi, the promoter regions of Oct4 and Nanog remain largely unmethylated (Fig. 5B). Earlier experiments indicated that, during ESC differentiation, trimethylation at the lysine 9 residue of histone H3 (H3K9Me3) by the histone methyltransferase G9A precedes the DNA methylation at the Oct4 locus (23). H3K9 methylation is also implicated in silencing Nanog expression during ESC differentiation (25). Therefore, we tested whether PKCi treatment prevents H3K9 trimethylation (H3K9Me3) at the Oct4 and Nanog loci in rESCs. PKCi treatment inhibits H3K9Me3 deposition at those loci (Fig. 5C). In contrast, in PKCi-treated rESCs, the Oct4 and Nanog loci contain high levels of H3K4 trimethylation (H3K4Me3) (Fig. 5D), a mark for transcriptionally active genes. Collectively, these results indicate that the PKCi culture condition maintains an ESC-specific epigenetic signature at the pluripotency genes, thereby maintaining their transcription in rESCs.
PKCi-maintained rESCs Express a Reduced Amount of Trophoblast Stem Cell-specific Factors
To date, the 2i/LIF culture condition is the most common strategy to maintain or derive rESCs. However, in rESCs, the 2i culture condition induces a surprisingly high amount of Cdx2, a trophectoderm/TSC-specific marker (4, 9). Because TSCs contribute to the placental development rather than the embryo proper, a strong expression of Cdx2 could further inhibit the developmental potency of rESCs. Thus, we tested whether PKCi-maintained rESCs show a reduced expression of TSC-specific genes compared with the 2i/LIF-cultured rESCs.
We compared the expression of trophoblast-specific markers in PKCi- and 2i/LIF-cultured rESCs. We maintained both F344 and DA rESCs in PKCi or 2i/LIF for five passages and determined the gene expression pattern in these cells. We found that expression of endoderm-, mesoderm-, and ectoderm-specific genes like Gata6, Brachyury (T), and Nestin were down-regulated in rESCs in both culture conditions (Fig. 6A). However, expression of the TSC-specific genes Cdx2 and Eomesodermin (Eomes) were highly up-regulated in the 2i/LIF culture condition (Fig. 6A) and strongly suppressed in PKCi-cultured rESCs (A). Immunofluorescence analyses of CDX2 expression (Fig. 6B) also confirmed the mRNA analysis. Suppression of Cdx2 and Eomes expression was also confirmed in PKCi culture condition-derived and -maintained rESCs and iPSCs (data not shown). Thus, although the 2i/LIF and PKCi culture conditions show a similar repression of endoderm-, mesoderm-, and ectoderm-specific genes, they differ significantly with respect to some key trophectoderm/TSC-specific genes, with the PKCi culture condition being a better suppressor for TSC-specific genes in rESCs.
FIGURE 6.
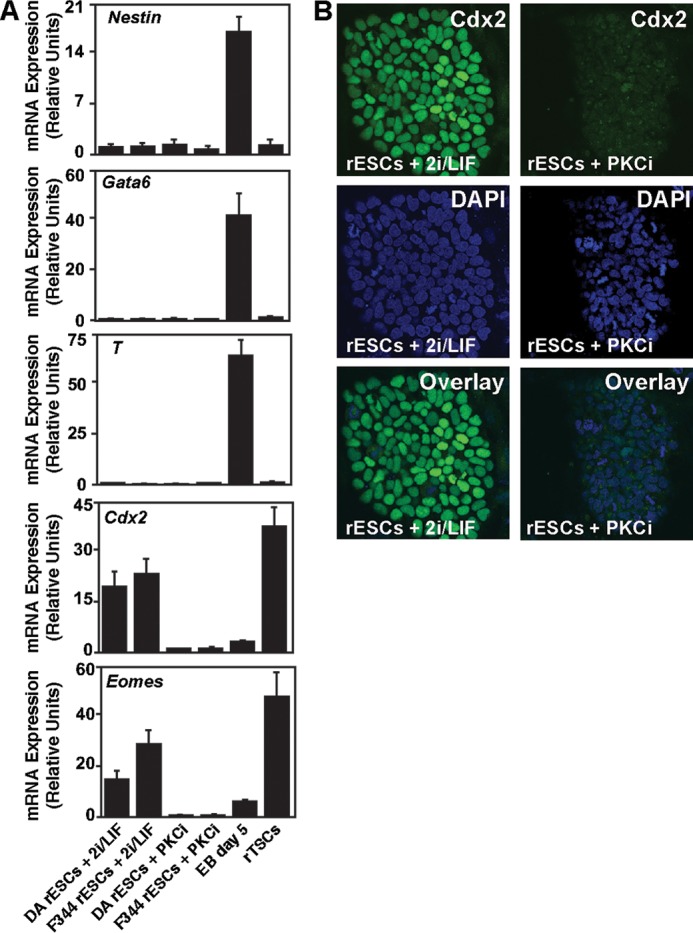
The PKCi culture condition represses TSC-specific gene expression in rESCs. A, qRT-PCR analysis (means ± S. E., three independent experiments) showing high-level expressions of the TSC-specific genes Cdx2 and Eomes in 2i/LIF-cultured rESCs. However, the PKCi culture condition strongly represses their expression. B, confocal images show high levels of CDX2 protein expression in 2i/LIF-cultured F344 rESCs but not in PKCi-cultured F344 rESCs.
PKCi Inhibits a PKCζ-NF-κB Axis in rESCs
Our earlier analyses in mESCs indicated that the atypical PKC isoform PKCζ is crucial for inducing multilineage differentiation in mESCs. Also, downstream of PKCζ, we implicated NF-κBp65 (RelA) function in inducing mESC lineage commitment (10). Thus, we tested whether a PKCζ -NF-κBp65 regulatory axis is also associated with the rESC lineage commitment and whether PKCi maintains rESC self-renewal by inhibiting that axis. We analyzed PKCζ phosphorylation and confirmed that PKCi inhibits PKCζ phosphorylation in rESCs (Fig. 7A). Furthermore, inhibition of RelA phosphorylation was also observed in PKCi-treated rESCs (Fig. 3A). To further understand the contribution of PKCζ in rESC differentiation, we knocked down PKCζ in F344 rESCs (Fig. 7B). Interestingly, when cultured without PKCi or 2i/LIF, the PKCζ knocked down (PKCζkd) rESC colonies maintained an undifferentiated rESC morphology for three to five passages (Fig. 7C), although a few PKCζkd rESC colonies could not maintain robust undifferentiated ESC colony morphology (C). Furthermore, analyses of mRNA expression showed that, compared with wild-type F344 rESCs, PKCζkd F344 rESCs maintained a high level of expression of pluripotency factors in the absence of PKCi (Fig. 7D). Collectively, the maintenance of undifferentiated colony morphology and high-level expressions of pluripotency factors in PKCζkd rESC strongly indicate that a PKCζ function contributes to the differentiation of rESCs. Therefore, we next asked whether a PKCζ-NF-κB signaling axis is associated with rESC differentiation.
FIGURE 7.

Maintenance of self-renewal in PKCζ-depleted rESCs. A, Western blot analyses showing the inhibition of phosphorylation of PKCζ at threonine (T) 410 and RelA at serine (S) 311) in PKCi-cultured F344 rESCs. B, Western blot analyses showing knockdown of PKCζ in F344 rESC with shRNA molecules targeted against the 3′ UTR region of PKCζ mRNA. shRNA2 but not shRNA1 showed strong RNAi. C, micrograph showing PKCζkd F344 rESC cells that were cultured in the absence of PKCi for four consecutive passages (20 days). The red borders indicate undifferentiated rESC colonies. The yellow border indicates a colony in which individual cells are visible, an indication of differentiation. Scale bar = 250 μm. D, relative mRNA expression of pluripotency genes was analyzed (mean ± S.E., three independent experiments). The plot shows significantly high (p ≤ 0.01) expression of pluripotency genes in PKCζkd F344 rESCs (passage 4) compared with expression levels in control F344 rESCs (passage 1) when cultured without PKCi. F344 rESCs, maintained in PKCi for four passages, were used as control cells. E, qRT-PCR analysis (mean ± S.E., three independent experiments) of NF-κB target gene (Plaur and Igfpb2) expression in F344 rESCs. F344 rESCs were cultured with PKCi for four passages and without PKCi for 5 days, and gene expression was compared with PKCζkd F344 rESCs cultured without PKCi for three passages. F, analysis (mean ± S.E., three independent experiments) of NFk5x-Luc reporter activation in cells analyzed in E. *, p ≤ 0.05.
To determine NF-κB activity in differentiating rESCs, we cultured F344 rESCs in the absence of PKCi and tested mRNA expression of NF-κB target genes, plasminogen activator (Plaur), and insulin-like growth factor binding protein 2 (Igfbp2) (10). We found that transcription of both Plaur and Igfbp2 is activated in F344 rESCs in the absence of PKCi (Fig. 7E). However, PKCζ depletion inhibited their activation (Fig. 3E). The impairment of NF-κB transcriptional activity was further assessed with a reporter plasmid in which Luciferase reporter gene expression is regulated by five NF-κB binding motifs (NFκ5x-Luc reporter). We found that reporter gene activation was strongly inhibited in PKCi-treated as well as PKCζkd rESCs (Fig. 7F). Collectively, our results indicated that differentiation of rESCs is associated with induction of a PKCζ-NF-kB signaling axis.
The PKCζ-NF-κB-miR-21/miR-29a Axis Contributes to rESC Differentiation
We wanted to test how the PKCζ-NF-κB signaling axis contributes to rESC differentiation. To that end, we asked whether the PKCζ-NF-κB axis regulates expression of miR-21, miR-29a, and miR-34a in rESCs/riPSCs. We focused on the regulation of certain miRNAs for two different reasons. First, suppression of miR-21, miR-29a, and miR-34a is implicated in iPSC derivation and maintaining an ESC-like phenotype (26–28), and second, studies in other cell types indicated that miR-21, miR-29a, and miR-34a transcription could be directly regulated by NF-κB (29–31).
We tested whether expression of miR-21, miR-29a, and miR-34a in PKCi-treated and PKCζkd F344 rESCs are altered compared with the expression levels in the F344 rESC lines that were differentiated after removal of PKCi. We found that expression of miR-21 and miR-29a were significantly down-regulated in both PKCi-cultured and PKCζkd rESCs (Fig. 8A) as well as in PKCi culture-derived riPSCs (B), whereas miR-34a expression in rESCs was not altered significantly upon PKCζ knockdown (A).
FIGURE 8.

Downstream to PKCζ, a NF-κB-miR-21/miR-29a regulatory axis contributes to rESC differentiation. A, plot showing qRT-PCR analyses of miRNA expression in F344 rESCs with or without PKCi treatment and PKCζ depletion (mean ± S.E., three independent experiments). Cells were cultured for 5 days before expression analysis. *, p ≤ 0.05. B, plot showing miR-21 and miR-29a expressions in F344 REFs and iPSCs (derived and cultured with PKCi). C, nucleotide sequences of rat miR-21 (Rno-miR-21) and miR-29a (Rno-miR-29a) genes showing conserved putative NF-κB binding motifs (red letters). Green letters indicate the pre-miR sequences. D, quantitative ChIP analyses showing RelA occupancy at the rat miR-21 and miR-29a loci in control F344 rESCs when cultured without PKCi. However, the RelA occupancy is lost in the PKCi culture condition or in PKCζ-depleted rESCs. E, F344 rESCs were transiently transfected with plasmids in which the miR-21 and miR-29a promoter regions (miR-21p and miR-29ap) were fused in front of a luciferase (Luc) reporter gene. In mutated (mt_miR-21p and mt_miR-29ap) constructs, conserved NF-κB motifs (mentioned in C) were deleted. All cells were cultured without PKCi. The plot depicts relative luciferase activity in the cell lysates normalized by the protein concentration of the lysates (mean ± S.E., three independent experiments). F, micrograph showing passage 2 PKCζkd F344 rESC colonies (red border) after 4 days of transduction with pre-miR-21- and pre-miR-29a-expressing lentiviral vectors. The image shows that the rESC colonies lost the undifferentiated ESC-like morphology. G, PKCζkd F344 rESCs were transduced either with pre-miR-21- or pre-miR-29a-expressing lentiviral vectors or empty vectors, total RNAs were isolated after 5 days, and qRT-PCR analyses were performed to compare expressions of miRNAs, pluripotency genes (Oct4, Nanog, Sox2), and lineage-specific genes (T, Gata6).
Next we tested whether transcriptional repression of miR-21 and miR-29a expression is due to an impairment of NF-κB-activity at the chromatin domains of the rat miR-21 (Rno-miR-21) and miR-29a (Rno-miR-29a) genes. Therefore, we tested RelA recruitment at those miRNA loci in PKCi culture and PKCζkd F344 rESCs. The promoter region of human miR-21 is located ∼(-)3.5 kb upstream of the pre-miR-21 sequence (32). We found that the corresponding region in the rat miR-21 locus is located at the ∼(-)2.6 kb region and contains a conserved putative NF-κB binding motif (GGR(A/G)R(A/G)NNY(C/T)Y(C/T)C) (33) (Fig. 4C, upper panel). Sequence analyses revealed the presence of a conserved NF-κB motif at ∼(-)30 bp to the transcriptional start sites to pre-miR-29a (Fig. 8C, lower panel). ChIP analyses revealed high levels of RelA recruitment at those conserved NF-κB sites in F344 rESCs upon PKCi withdrawal. However, RelA recruitment was impaired at those NF-κB sites in PKCζkd F344 rESCs (Fig. 8D). The functional importance of these conserved NF-κB motifs was validated by analyzing luciferase reporter constructs in F344 rESCs. Constructs containing the promoter regions of the rat miR-21 and miR-29a genes showed higher reporter activity upon differentiation of F344 rESCs, and reporter activity was reduced upon deletion of the NF-κB motifs (Fig. 8E). These results indicate a positive regulation of miR-21 and miR-29a expression by NF-κB during rESC differentiation.
Because PKCi treatment as well as depletion of PKCζ is associated with repression of miR-21 and miR-29a in rESCs, we next tested the importance of miR-21 and miR-29a in inducing rESCs differentiation. To that end, we infected PKCζkd F344 rESCs with lentiviral particles expressing pre-miR-21 and pre-miR-29a and found that induction of miR-21 and miR-29a expression in PKCζkd F344 rESCs leads to a loss of undifferentiated ESC colony morphology (Fig. 8F), repression of pluripotency genes, and induction of differentiation markers (G). Collectively, our results strongly indicate that induction of miR-21 and miR-29a contributes to rESC differentiation, whereas repression of miR-21 and miR-29a expression because of an impaired PKCζ-NF-κB regulatory axis contributes to the maintenance of rESC self-renewal.
DISCUSSION
In this study, we examined the role of PKC signaling in pluripotency in the rat. We made three important new findings. First, we found that PKCi culture conditions maintain rESCs in the undifferentiated state. In the PKCi condition, we successfully cultured rESCs for up to 22 consecutive passages without observing differentiation (data not shown). Also, we found that the PKCi culture condition does not compromise the developmental potency and germ line competence of rESCs. Second, we found that the PKCi culture conditions can be used for de novo derivation of rESCs from rat blastocysts. Third, we noticed that the efficiency of PKCi-mediated derivation of new rESCs lines varies depending upon rat strains. For example, the establishment of new rESC lines from DA rats in the PKCi culture condition was relatively inefficient, and addition of the MEK inhibitor PD0325901 increased the efficiency. However, the molecular reasons behind the strain-specific outcome of rESC derivation in the PKCi culture condition are unknown to us at this moment.
We found that the PKCi culture condition could also facilitate reprogramming of REFs to iPSCs. Interestingly, unlike de novo derivation of rESCs from DA blastocysts, PKCi alone (without the presence of the MEK inhibitor) was sufficient to reprogram REFs from DA rats. These results indicate that, despite the strong expression of pluripotency genes in both rESCs and riPSCs in the PKCi culture condition, the efficacy of de novo derivation of rESCs from blastocysts versus derivation of riPSCs from differentiated cells could vary upon PKC inhibition.
Our molecular analyses revealed that, similar to mESCs (10), rESC differentiation is also associated with NF-κB activation. Furthermore, similar to mESCs, PKCi treatment inhibits PKCζ phosphorylation and NF-κB activation in rESCs and represses a PKCζ-NF-κB-miR-21/miR-29a differentiation axis in rESCs. The conserved role of the PKCζ-NF-κB signaling axis in balancing self-renewal versus differentiation in both mouse and rat ESCs indicate that this signaling pathway could be targeted to facilitate the derivation of pluripotent stem cells from other mammalian species.
Another significant finding of this study is maintenance of an ESC-specific epigenetic signature upon PKC inhibition. The presence of characteristic epigenetic marks at the active pluripotency genes along with the presence of repressive H3K27me3 modification at the developmental regulator genes indicate that PKCi-maintained rESCs do not bypass the requirement of a proper ESC-specific epigenetic state. However, it is unclear whether maintenance of the ESC-specific epigenetic signature is a cause for PKCi-mediated maintenance of pluripotency or simply a reflection of the pluripotent state. Thus, the efficacy of PKC inhibition in maintaining pluripotency in ESCs, which lacks specific epigenetic components, merits further study.
In this study, we validated maintenance of pluripotency in 2i/LIF-derived and PKCi-maintained rESCs via chimera generation and germ line transmission. Also, PKCi-derived and -maintained rESCs and riPSCs from both F344 and DA rats generated chimeras or developed teratoma, inferring their pluripotent state. We also found that PKCi culture is a better condition than 2i/LIF culture to repress expression of TSC-specific factors like CDX2 in rESCs (Fig. 6, A and B). Because expression of CDX2 induces trophoblast fate in ESCs (34), it is possible that high levels of TSC-specific genes in rESCs might negatively affect their pluripotent state. However, comprehensive future analyses through germ line transmission efficiency, tetraploid aggregation, and transplantation studies are necessary for definitive conclusions.
Acknowledgment
We thank Dr. Michael J. Soares for providing mRNAs isolated from rat trophoblast stem cells.
This work was supported, in whole or in part, by National institutes of Health grants HL094892, HL106311, L104322, and HD062546 (to S. P.). This work was also supported by a gift from the Ronald. D. Deffenbaugh Foundation.

This article contains supplemental Tables 1–3.
- rESC
- rat embryonic stem cell
- LIF
- leukemia inhibitory factor
- mESC
- mouse embryonic stem cell
- TSC
- trophoblast stem cell
- iPSC
- induced pluripotent stem cell
- PKCi
- 3-[1-[3-(dimethylamino)propyl]-5-methoxy-1H-indol-3-yl]-4-(1H-indol-3-yl)-1H-pyrrole-2,5-dione
- riPSC
- rat induced pluripotent stem cell
- EB
- embryoid body
- SD
- Sprague-Dawley
- Dox
- doxycycline
- miRNA
- microRNA
- qRT-PCR
- quantitative RT-PCR
- 2i
- CHIR99021 and PD032590
- REF
- rat embryonic fibroblast
- DA
- Dark Agouti
- OSKM
- OCT4/SOX2/KLF4/c-MYC.
REFERENCES
- 1. Jacob H. J., Kwitek A. E. (2002) Rat genetics. Attaching physiology and pharmacology to the genome. Nat. Rev. Genet. 3, 33–42 [DOI] [PubMed] [Google Scholar]
- 2. Li P., Tong C., Mehrian-Shai R., Jia L., Wu N., Yan Y., Maxson R. E., Schulze E. N., Song H., Hsieh C. L., Pera M. F., Ying Q. L. (2008) Germline competent embryonic stem cells derived from rat blastocysts. Cell 135, 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buehr M., Meek S., Blair K., Yang J., Ure J., Silva J., McLay R., Hall J., Ying Q. L., Smith A. (2008) Capture of authentic embryonic stem cells from rat blastocysts. Cell 135, 1287–1298 [DOI] [PubMed] [Google Scholar]
- 4. Hong J., He H., Weiss M. L. (2012) Derivation and characterization of embryonic stem cells lines derived from transgenic Fischer 344 and Dark Agouti rats. Stem Cells Dev. 21, 1571–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tong C., Li P., Wu N. L., Yan Y., Ying Q. L. (2010) Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 467, 211–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawamata M., Ochiya T. (2010) Generation of genetically modified rats from embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan H. X., Wu H. P., Ashton C., Tong C., Ying Q. L. (2012) Rats deficient for p53 are susceptible to spontaneous and carcinogen-induced tumorigenesis. Carcinogenesis 33, 2001–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ying Q. L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. (2008) The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong J., He H., Bui P., Ryba-White B., Rumi M. A., Soares M. J., Dutta D., Paul S., Kawamata M., Ochiya T., Ying Q. L., Rajanahalli P., Weiss M. (2013) A focused microarray for screening rat embryonic stem cell lines. Stem Cells Dev. 22, 431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dutta D., Ray S., Home P., Larson M., Wolfe M. W., Paul S. (2011) Self-renewal versus lineage commitment of embryonic stem cells. Protein kinase C signaling shifts the balance. Stem Cells 29, 618–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carey B. W., Markoulaki S., Hanna J., Saha K., Gao Q., Mitalipova M., Jaenisch R. (2009) Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc. Natl. Acad. Sci. U.S.A. 106, 157–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Home P., Saha B., Ray S., Dutta D., Gunewardena S., Yoo B., Pal A., Vivian J. L., Larson M., Petroff M., Gallagher P. G., Schulz V. P., White K. L., Golos T. G., Behr B., Paul S. (2012) Altered subcellular localization of transcription factor TEAD4 regulates first mammalian cell lineage commitment. Proc. Natl. Acad. Sci. U.S.A. 109, 7362–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ray S., Dutta D., Rumi M. A., Kent L. N., Soares M. J., Paul S. (2009) Context-dependent function of regulatory elements and a switch in chromatin occupancy between GATA3 and GATA2 regulate Gata2 transcription during trophoblast differentiation. J. Biol. Chem. 284, 4978–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dutta D., Ray S., Vivian J. L., Paul S. (2008) Activation of the VEGFR1 chromatin domain. An angiogenic signal-ETS1/HIF-2α regulatory axis. J. Biol. Chem. 283, 25404–25413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Home P., Ray S., Dutta D., Bronshteyn I., Larson M., Paul S. (2009) GATA3 is selectively expressed in the trophectoderm of peri-implantation embryo and directly regulates Cdx2 gene expression. J. Biol. Chem. 284, 28729–28737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang M. Y., Kim D., Kim C. H., Kang H. C., Yang E., Moon J. I., Ko S., Park J., Park K. S., Lee K. A., Hwang D. Y., Chung Y., Lanza R., Kim K. S. (2010) Direct reprogramming of rat neural precursor cells and fibroblasts into pluripotent stem cells. PLoS ONE 5, e9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He H., McHaney M., Hong J., Weiss M. L. (2009) Cloning and characterization of 3.1-kb promoter region of the Oct4 gene from the Fischer 344 rat. Open Stem Cell J. 1, 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liskovykh M., Chuykin I., Ranjan A., Safina D., Popova E., Tolkunova E., Mosienko V., Minina J. M., Zhdanova N. S., Mullins J. J., Bader M., Alenina N., Tomilin A. (2011) Derivation, characterization, and stable transfection of induced pluripotent stem cells from Fischer344 rats. PLoS ONE 6, e27345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bibikova M., Laurent L. C., Ren B., Loring J. F., Fan J. B. (2008) Unraveling epigenetic regulation in embryonic stem cells. Cell Stem Cell 2, 123–134 [DOI] [PubMed] [Google Scholar]
- 20. Boyer L. A., Plath K., Zeitlinger J., Brambrink T., Medeiros L. A., Lee T. I., Levine S. S., Wernig M., Tajonar A., Ray M. K., Bell G. W., Otte A. P., Vidal M., Gifford D. K., Young R. A., Jaenisch R. (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 [DOI] [PubMed] [Google Scholar]
- 21. Pasini D., Bracken A. P., Hansen J. B., Capillo M., Helin K. (2007) The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 27, 3769–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 23. Epsztejn-Litman S., Feldman N., Abu-Remaileh M., Shufaro Y., Gerson A., Ueda J., Deplus R., Fuks F., Shinkai Y., Cedar H., Bergman Y. (2008) De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15, 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J. Y., Pu M. T., Hirasawa R., Li B. Z., Huang Y. N., Zeng R., Jing N. H., Chen T., Li E., Sasaki H., Xu G. L. (2007) Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol. Cell. Biol. 27, 8748–8759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loh Y. H., Zhang W., Chen X., George J., Ng H. H. (2007) Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21, 2545–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang C. S., Li Z., Rana T. M. (2011) microRNAs modulate iPS cell generation. RNA 17, 1451–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi Y. J., Lin C. P., Ho J. J., He X., Okada N., Bu P., Zhong Y., Kim S. Y., Bennett M. J., Chen C., Ozturk A., Hicks G. G., Hannon G. J., He L. (2011) miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 13, 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh S. K., Kagalwala M. N., Parker-Thornburg J., Adams H., Majumder S. (2008) REST maintains self-renewal and pluripotency of embryonic stem cells. Nature 453, 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou R., Hu G., Gong A. Y., Chen X. M. (2010) Binding of NF-κB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res. 38, 3222–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niu J., Shi Y., Tan G., Yang C. H., Fan M., Pfeffer L. M., Wu Z. H. (2012) DNA damage induces NF-κB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J. Biol. Chem. 287, 21783–21795 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Li J., Wang K., Chen X., Meng H., Song M., Wang Y., Xu X., Bai Y. (2012) Transcriptional activation of microRNA-34a by NF-κB in human esophageal cancer cells. BMC Mol. Biol. 13, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cai X., Hagedorn C. H., Cullen B. R. (2004) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10, 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong D., Teixeira A., Oikonomopoulos S., Humburg P., Lone I. N., Saliba D., Siggers T., Bulyk M., Angelov D., Dimitrov S., Udalova I. A., Ragoussis J. (2011) Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 12, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niwa H., Toyooka Y., Shimosato D., Strumpf D., Takahashi K., Yagi R., Rossant J. (2005) Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 123, 917–929 [DOI] [PubMed] [Google Scholar]