

Background: We study NF-κB-activated CTCF in EGF-induced wound healing.
Results: Corneal epithelial wound healing was significantly impaired because of the lack of CTCF activity in NFκB-p50−/− mice. EGF-induced NFκB activation regulated CTCF by interacting with the CTCF promoter to increases motility, migration, and wound healing.
Conclusion: CTCF is an NFκB-p50-interactive target in EGF-induced corneal epithelial wound healing.
Significance: NF-κB-controlled CTCF activation plays important roles in EGF-regulated wound healing.
Keywords: Cornea, Epidermal Growth Factor (EGF), Epithelial Cell, Gene Regulation, NF-κB, Wound Healing
Abstract
Epidermal growth factor (EGF) plays an important role in corneal epithelial migration and proliferation to improve the wound healing process. This study aimed to understand the role of NFκB in EGF-induced corneal epithelial wound healing through regulation of CTCF activity, which plays important roles in cell motility and migration to promote wound healing. The effect of NFκB p50 on corneal epithelial wound healing was investigated by comparing the eyes of wild-type and p50 knockout mice. We found that there was a significant retardation in corneal epithelial wound healing in the corneas of p50 knockout mice. Wound closure rates were measured in human corneal epithelial cells transfected with an NFκB activation-sensitive CTCF expression construct to demonstrate the effect of human CTCF expression under the control of EGF-induced NFκB activation on wound healing. EGF stimulation activated NFκB, which directly triggered the expression of the exogenous human CTCF in transfected cells and, subsequently, promoted human corneal epithelial cell motility, migration, and wound healing. Overexpression of CTCF in corneal epithelial cells and mouse corneas significantly enhanced the wound healing process. Furthermore, the effect of overexpressing NFκB p50 in corneal epithelial cells on the promotion of wound healing was abolished by knockdown of CTCF with CTCF-specific shRNA. Thus, a direct regulatory relationship between EGF-induced NFκB p50 and CTCF activation affecting corneal epithelial wound healing has been established, indicating that CTCF is, indeed, a NFκB p50-targeted and effective gene product in the core transcriptional network downstream from the growth factor-induced NFκB signaling pathway.
Introduction
The corneal epithelial layer protects the eye structures behind it from environmental insults and infections to maintain the intact function of the vision system (1). Corneal epithelial cells undergo a self-renewal process to replace the surface layer cells and repair corneal surface wounds dependent on the stimulation of growth factors. EGF is one of the growth factors that play important roles in corneal epithelial self-renewal and wound healing (2–9). EGF stimulates intracellular signaling pathways, including the NF-κB, PI3K/AKT, and MAPK/Erk cascades, to regulate cell cycle progression and to activate transcription factors that control the genetic responses (10–14). Earlier studies demonstrate that the mitogenic effect of EGF on the proliferation of corneal epithelial cells requires suppression of the eye-specific Pax6 expression (15). The effect of EGF on suppressing Pax6 expression is through activation of CTCF,2 an epigenetic CCCTC binding factor and zinc finger protein (16). In corneal epithelial cells, we found that EGF induces NF-κB subtype-specific signaling cascades to regulate CTCF activity and to promote cell proliferation (14). Our recent study demonstrates that CTCF is required for the EGF-induced alteration of focal adhesion and increases in cell motility and migration (17). However, the mechanisms of how EGF-induced activation of NF-κB and its subtypes regulate transcription activity of CTCF to effect corneal epithelial wound healing in the eye are still under investigation.
EGF activates transcription factors, such as NF-κB, CTCF, and other immediate early genes, upon exposure of mammalian cells to the growth factor (12, 18–23). NF-κB is an important gene regulator in the Rel transcription factor family involving inflammatory responses, developmental processes, cellular growth, and apoptosis (24, 25). CTCF is another gene regulator that plays important roles in the epigenetic regulation of genes. It functions as an insulator sensitive to DNA methylation to epigenetically control DNA imprinting and X chromosome inactivation during development (26–28). Many studies demonstrate that CTCF also play a role as a transcription activator and repressor. Recent studies indicate that CTCF is involved in the regulation of cell migration in cancer cell proliferation, tumor suppression, and apoptosis (29–31). In corneal epithelial cells, EGF-induced activation of the NF-κB pathway regulates cell fate in a subtype-specific fashion through interactions with CTCF that function as a downstream component in the core transcriptional network (14, 32). We found that, in corneal epithelial cells, CTCF is a targeted gene of the growth factor-induced pathways, including the Erk, AKT, and NF-κB signaling cascades. Activation of these signaling pathways by stimulation of EGF, insulin, and other stresses subsequently regulates the expression levels of CTCF to determine the corneal epithelial fate in the process of wound healing (12–14, 32).
As previously described, EGF is an important growth factor in corneal epithelial wound healing. It facilitates corneal epithelial wound repair by promoting migration and proliferation in both in vivo and in vitro model systems (1, 15, 33–35). The question that remains to be answered is whether CTCF is one of the key factors that directly switch EGF-induced activation of NF-κB signaling to genetic responses that subsequently change corneal epithelial cell stages, resulting in the acceleration of wound healing. On the corneal surface, corneal epithelial wound healing requires proper activities of cell migration that are essential for successful re-epithelialization in the process of corneal epithelial self-renewal (1). We demonstrate that EGF-induced CTCF activation accelerates corneal epithelial cell migration, which is favorable for wound healing and tissue repair in the cornea (15, 16). However, the results obtained for EGF-induced NFκB subtype activation are sometimes contradictory, and the role of CTCF in corneal epithelial wound healing remains unclear. This study aimed to advance our understanding of how the EGF-induced NFκB subtype p50 directly activates CTCF to increase cell motility and migration in human corneal epithelial cells to promote corneal epithelial wound healing. We further revealed an EGF-induced activation of the NFκB p50 subtype that interacts with CTCF in the promoter region, resulting in the activation of CTCF and facilitating corneal epithelial wound healing.
EXPERIMENTAL PROCEDURES
Experimental Animals and Cell Cultures
Transgenic Mice
NF-κB p50 knockout transgenic mice (NF-κB 1−/−) and wild-type mice were obtained from The Jackson Laboratory (Bar Harbor, ME), and genotypes of these mice were confirmed by PCR analysis from prepared tail DNA. All animal experiments were conducted in accordance with the institutional guidelines of the Animal Care and Use Committee according to National Institutes of Health guidelines.
Cultures of Human Corneal Epithelial Cells
Human telomerase-immortalized corneal epithelial (HTCE) cells were cultured in a keratinocyte serum-free medium containing 120 μm calcium and supplemented with 0.4% bovine pituitary extract and 0.2 ng/ml EGF (Invitrogen). Human SV-40 large T-transformed corneal epithelial (HCE) cells were grown in Dulbecco's modified Eagle's medium/F-12 (1:1) containing 10% fetal bovine serum and 5 μg/ml insulin. Cells were cultured in an incubator supplied with 95% air and 5% CO2 at 37 °C. Culture media were replaced every 2 days, and cells were subcultured by treatment with 0.05% trypsin-EDTA. For EGF-induced experiments, cells were synchronized in growth factor-deprived medium for 24–48 h before EGF stimulation.
Overexpression and Knockdown of NFκB p50
Full-length cDNA encoding human p50 was cloned into the pcDNA4-to-A vector (Invitrogen), named pcDNA4-p50. Both the pcDNA4-p50 construct and the pcDNA4-to-A vector (control) were transfected into HTCE cells by FuGENE HD transfection reagent (Roche) for wound healing assays and Western blot analysis. For the experiments knocking down cellular NF-κB p50, p50-specific siRNA (GGGGCUAUAAUCCUGG-ACU (sense) and AGUCCAGGAUUAU-AGCCCC (antisense)) and control siRNA (Santa Cruz Biotechnology) were transfected into HCE cells that were subject to wound healing assays and Western blot analysis.
Infections of NF-κB and CTCF cDNAs to HCE Cells and Mouse Corneas
Lentiviral particles containing shRNA of CTCF tagged with a variant of green fluorescent protein (Turbo-GFP, Sigma-Aldrich, St. Louis, MO) were packaged in HEK-293T cells (17). The viral concentrations in the culture medium were titrated by PCR after cotransfection of HTCE cells with pCMV-VSV-G, psPAX2, and pGIPZshRNA-CTCF fused to the GFP for 72 h (Open Biosystems, Huntsville, AL). The culture medium containing the lentiviral particles secreted from HEK-293T cells was added to HTCE cells, and infected clones stably expressing shRNAs were selected in selective culture with puromycin (2 μg/ml). HTCE cells infected with a pGIPZ-shRNA-control vector packed in the lentivirus served as controls. In addition, expression of GFP from the pGIPZ-TurboGFP vector allowed measuring of the efficiency of the viral infection and distinguishing green from non-green cells. The green cells integrated with shRNA were visualized by fluorescence microscope (Nikon). For corneal wound healing studies, the cDNA encoding the full-length CTCF gene were introduced into a linearized Adeno-x-vector using the In-Fusion HD cloning system (Clontech). The recombinant adenovirus was packaged in HEK293 cells and amplified by transfecting PacI-digested vectors. The viral titer of 109 plaque-forming units was obtained from crude viral lysates. The recombinant adenoviruses Adeno-x-vector (for controls) or Adeno-x-CTCF were added to the culture medium at 2.5 × 108 plaque-forming units/ml. After 5 days of incubation, the eyeballs were transferred to a new dish containing normal defined keratinocyte serum-free (KSF) medium without virus and incubated for additional 10–15 days. The medium was changed every other day and photographed with a Nikon fluorescent microscope. The wound area was calculated with Nikon Tis NIS-Elements software.
Constructions of NF-κB TRE-Control and TRE-CTCF
NF-κB transcription response element (TREs) sties were inserted in five repeats upstream of the mini-CMV promoter followed by GFP or GFP plus full-length human CTCF cDNAs. The constructs were termed TRE-Control and TRE-CTCF, respectively. GFP served as a reporter of NF-κB activity. Lentiviral particles that contained NF-κB TRE-Control or TRE-CTCF were cotransfected with pCMV-VSV-G and psPAX2 into HEK-293T cells for packaging. The culture medium containing a high titer of lentivirus secreted from HEK-293T cells was collected and added to HTCE cells. All clones that were integrated with TRE-Control or TRE-CTCF were selected for 4 weeks by adding puromycin (2 μg/ml) to the culture medium to establish stable expression cell lines.
Wound Healing Assays
Two wound healing assays were performed, including corneal wound healing in cultured whole-eye organ and a scratch-induced directional wound-healing assay.
Corneal Wound Healing Assays
The corneas in cultured whole-eye organs were used for experiments of corneal epithelial wound healing. Under a dissecting microscope, the surface layer of the mouse corneas was debrided without damaging the basement membrane of the corneal epithelia using a corneal rust ring remover with a 0.5-mm burr (Algerbrush, The Alger Company, Inc., Lago Vista, TX). The whole eyeball was dissected and placed in culture wells (the corneas facing up) with the medium containing 10% fetal bovine serum and 1% antibiotic/antimycotic solution at 37 °C and 5% CO2 in a humidified incubator. The rate of epithelial healing in whole-eye organ culture was measured immediately after wounding. Eyeballs were taken from wild-type or NF-κB p50 knockout mice and allowed to heal under culture conditions. Lesions of the corneas were stained topically with fluorescein (fluorescein sodium 1.0% w/v) and photographed with an inverted microscope (Nikon). The corneal epithelial layer was removed in an area of 1.5-mm diameter (or 2-mm diameter for Ad-x-CTCF transduction assays) near the central cornea. Wounded corneas were incubated for 1–2 days under normal and EGF-induced conditions and up to 15 days in the absence of FBS and EGF. All animals used in our experiments were treated in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research using protocols approved and monitored by the LABioMed Animal Care Committee at the University of California Los Angeles School of Medicine.
Scratch-induced Directional Wound Healing Assay
Corneal epithelial cells were seeded at 3 × 105 cells/well in 12-well plates and grown to 100% confluence. A cross-stripe scratch wound was made on the cell surface with a yellow micropipette tip. The wound area was measured by calculating the average values at multiple points (at least 10 points/wound along the edges) using commercial software (NIS-Element, Nikon, Tokyo, Japan) and photographed with an inverted microscope (Eclipse Ti, Nikon) during the healing period. The microscope was able to record exactly the same area at each time point by memorizing the x-y directions through a computer-controlled and motorized head stage. The width of the wounded area was measured, and the rate of wound closure was calculated using the units of micrometers/hour.
Cell Migration Assays
The cell migration assay was performed following the instructions of the manufacturer (Transwell, Corning Inc., Corning, NY). The migration chamber culture insert contained a polyethylene terephthalate membrane 6.5 mm in diameter with an 8-μm pore size. HTCE cells expressing NF-κB TRE-CTCF or TRE-Control (5 × 104) were seeded in the culture insert (upper chamber) with plain medium and incubated for 24 h. EGF (20 ng/ml) or the sham was added to the culture insert, and the cells were incubated for 48 h. Migrated cells that grew on the culture well (bottom chamber) were counted and photographed with an inverted fluorescence microscope (Nikon). The cells were fixed in 4% paraformaldehyde, stained with 0.3% crystal violet, and photographed. The dye in the cells was then dissolved in 10% acetic acid, and the absorbance of the dissolved dye was measured at a wavelength of 600 nm.
Live Cell Imaging and Cell Motility Analysis
The Motility of HTCE cells expressing NF-κB TRE-CTCF and TRE-Control was measured using an inverted microscope (Eclipse Ti, Nikon) with the following functions: time-lapse videos of the phase contrast/fluorescent live images, built-in total internal reflection fluorescence and FRET, perfect focus system, and a digital charge-coupled device (CCD) camera at a time interval of 2 min for each photo. The system was equipped with a heated chamber at 37 °C and flushed with mixed 5% CO2 that kept the cells under normal culture conditions. Live cells were recorded for a period of 0.5–3 h. Cell motility was examined by tracking cell movements and distances (millimeters/hour) using an inverted microscope with a motorized head stage and software (Tis NIS-Elements, Nikon).
Immunocytochemistry and Western Blot Analyses
Immunocytochemistry experiments were performed following a protocol as described previously (36). Briefly, mouse eyeballs were fixed with 4% paraformaldehyde and sectioned into 8-μm sections. The tissue section was perforated with 0.3% Triton X-100 in PBS (PBS-T). After being blocked with 2% BSA and 5% normal serum in PBS-T, the sections were incubated with primary antibody against CTCF (Millipore) in 1% BSA-0.1% Triton X-100-PBS for 16 h at 4 °C. Cy3-conjugated secondary antibody was applied in 1% BSA- 0.1% Triton X-100-PBS for 1 h at room temperature. Stained tissues were mounted with shield mounting medium (Vector Laboratories Inc.) and photographed using the Nikon Eclipse Ti inverted microscope with a ×60 oil total internal reflection fluorescence lens. Western blot analyses were performed by lysing corneal epithelial cells (2 × 105) in SDS sample buffer that contained 62.5 mm Tris-HCl (pH 6.8), 2% (w/v) SDS, 10% glycerol, 50 mm DTT, and 0.01% (w/v) bromphenol blue. Proteins in the lysates were denatured by boiling for 5 min and being size-fractionated in 8–10% PAGE gels. Proteins in PAGE gels were electrotransferred to PVDF membranes (Millipore) by using a semidry gel transferring apparatus (Bio-Rad, CA). The PVDF membranes were exposed to the blocking buffer containing 5% nonfat milk in TBS and 0.1% Tween 20 (TBS-T) for 1 h at 22 °C and then incubated with the respective primary antibodies at 4 °C overnight. HRP-conjugated secondary antibody was applied in TBS-T buffer for 1 h at 22 °C. Western blot analyses were developed by an ECL Plus system (Santa Cruz Biotechnology) and visualized by exposure to x-ray films.
RESULTS
Retarded Corneal Epithelial Wound Healing in NF-κB p50−/− mice
Recent reports demonstrate that corneal epithelial wound healing is affected by a deficiency of IκB (in the NF-κB pathway) in the eyes of IκB knockout mice (37). We predicted that the NF-κB subtype p50 deficiency would result in a delay of corneal epithelial cell wound healing. In this study, we utilized eyes from NF-κB p50 knockout mice to test the effect of NF-κB p50 on corneal epithelial wound healing. The corneas from the same litter of WT, heterozygous (p50+/−), and p50 knockout (p50−/−) mice were characterized by specific PCR genotyping (Fig. 1A). The corneas from the eyes of these mice were subjected to the defined surface injuries by using an Algerbrush corneal rust ring. The eyeballs containing the corneas with wounds in the epithelial layer (shown in green in fluorescein staining) were incubated in normal KSF medium during tests of wound healing. We found that the rate of corneal epithelial wound healing was significantly slower in NF-κB p50−/− mice than in wild-type mice after 16 h of incubation (Fig. 1, B and C), whereas there were no obvious changes found in the eyes of their p50+/− heterozygous siblings (data not shown). The result reveals for the first time that NF-κB p50-induced signaling pathway activation plays a functional role in growth factor-stimulated corneal epithelial wound healing.
FIGURE 1.
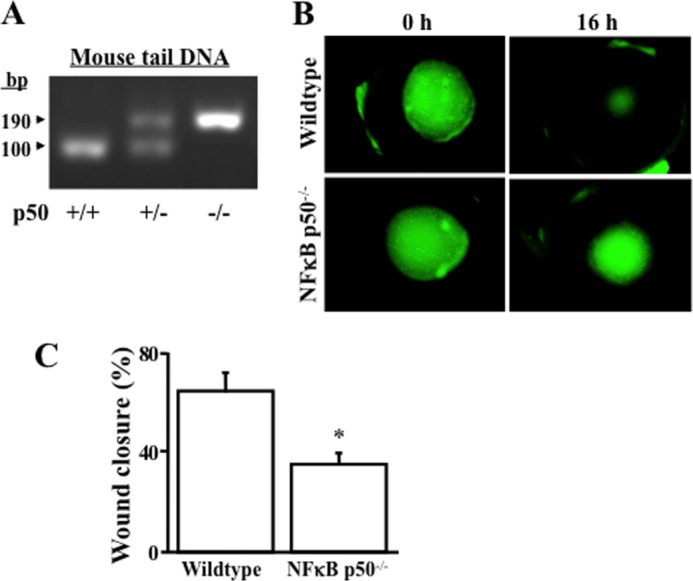
Effect of NFκB p50 deficiency on corneal epithelial wound healing. A, knockout of NFκB p50 verified by genotyping in NFκB p50−/− mice obtained from the Jackson Laboratory. B, retarded corneal epithelial wound healing in corneas of NFκB p50−/− knockout mice. C, statistical analysis of corneal epithelial wound healing rates in the eyes of wild-type and NFκB p50−/− knockout mice. The corneal epithelial layer was debrided without damaging the basement membrane by using a corneal rust ring remover with a 0.5-mm burr. Lesions of the corneal surface were stained topically with fluorescein (green) and photographed with an inverted microscope (Nikon). *, p < 0.05. Data were collected from 27 independent experiments.
Expression of the NF-κB Subtype p50 Promoted Wound Healing
To answer the question whether the NF-κB subtype p50 plays a role in wound healing in corneal epithelial cells, we applied two opposite approaches in HCE cells, including knocking down NF-κB p50 using specific siRNA and overexpressing NF-κB p50 cDNA. We found that knockdown of NF-κB p50 markedly delayed wound closure of human corneal epithelial cells (Fig. 2, A and B). Western blot analysis verified the decrease of NF-κB p50 expression in NF-κB p50 knocked-down HCE cells (Fig. 2C). Conversely, overexpression of NF-κB p50 by transfecting NF-κB p50 cDNA into HCE cells resulted in accelerated corneal epithelial wound closure when compared with vector-transfected control cells (Fig. 2, D and E). The increased protein level of NF-κB p50 in p50-overexpressing cells was also verified (Fig. 2F). These results indicate that increased and decreased cellular activities of the NF-κB subtype p50 promoted and suppressed wound healing of HCE cells, respectively.
FIGURE 2.

Effect of altering NFκB p50 activity on HCE cell wound healing. A, effect of knocking down NFκB p50 on EGF-induced wound closure. B, significant retardation of wound closure in NFκB p50 knockdown cells in the absence and presence of EGF induction. C, Western blot analysis of NFκB p50 expression in NFκB p50 knockdown cells. D, effect of overexpressing NFκB p50 on EGF-induced wound closure. E, significant acceleration of EGF-induced wound closure in NFκB p50-overexpressed (OE) cells. F, Western blot analysis of NFκB p50 expression in NFκB p50-overexpressed cells. Expression levels of NFκB p50 were altered to be suppressed and enhanced by knocking down HCE cells by knocking down NFκB p50 mRNA and overexpressing full-length cDNA encoding NFκB p50, respectively.
Suppression of NF-κB p50 Overexpression-induced Acceleration of Wound Healing by Knocking Down CTCF
We reported that expression of CTCF is regulated by the NF-κB subtype p50 in human corneal epithelial cells (14). In this study, NF-κB p50-specific cDNA and shRNA were used to overexpress and knock down the p50 in these cells, respectively. Overexpression of CTCF markedly increased the rate of HCE cell wound healing following a 24-h period (Fig. 3, A and B). In contrast, knockdown of CTCF mRNA with CTCF-specific shRNA significantly suppressed the HTCE cell wound healing rate within 30 h (Fig. 3, C and D). Knockdown of p50 effectively suppressed expression of CTCF in HCE cells (Fig. 4A, left panel). In contrast, overexpression of NF-κB p50 markedly increased CTCF expression (Fig. 4A, right panel). The correlation of NF-κB p50 protein level and CTCF expression was demonstrated in these cells. The effect of EGF-induced activation of NF-κB p50 on CTCF expression was examined next in the corneas of wild-type and NF-κB p50−/− mice. Immunostaining experiments revealed that EGF failed to stimulate CTCF expression in the basal layer in the NF-κB p50-deficient corneal epithelium of p50−/− mice compared with the corneas of wild-type mice (Fig. 4B). A control experiment was done by performing staining in PBS without antibodies. On the basis of the observation that EGF failed to stimulate CTCF expression in the p50−/− mouse corneas, we predicted that a blockage of CTCF should prevent NF-κB p50-accelerated wound healing. Full-length cDNA encoding the NF-κB p50 was introduced into HCE cells in which the CTCF was already suppressed by CTCF-specific shRNA. Western blot analysis revealed that knocking down CTCF mRNA markedly diminished the effect of NF-κB p50 overexpression on CTCF activation (Fig. 4C). We found that overexpression of NF-κB p50 in cells in which CTCF was knocked down yielded very different results in the process of wound healing. The wound closure process in these cells was accelerated effectively by overexpression of NF-κB p50. However, knockdown of CTCF mRNA by CTCF shRNA abolished or delayed effects of NF-κB p50 overexpression and EGF-induced wound closure rate in HTCE cells (Fig. 4D). Statistical analysis further verified that knockdown of CTCF significantly suppressed the NF-κB p50 overexpression and EGF-induced effects on acceleration of corneal epithelial wound closure (Figs. 4E). The results indicate that CTCF plays a role in mediating the NF-κB p50-induced acceleration of wound healing.
FIGURE 3.

Effects of altered CTCF activity on HCE and HTCE cell wound healing. A, effect of overexpression (OE) of CTCF on corneal epithelial cell wound closure. B, significant acceleration of wound closure in CTCF-overexpressing cells. C, effect of knocking down CTCF on corneal epithelial cell wound closure. D, significant retardation of wound closure in CTCF knockdown cells. HCE and HTCE cells were infected with lentiviral vector containing either cDNA encoding full-length CTCF or CTCF-specific shRNA to overexpress or knock down CTCF, respectively. *, p < 0.05. Data were collected from four sets of independent experiments.
FIGURE 4.

Effect of suppressing CTCF on overexpression of NFκB p50-enhanced wound healing. A, effects of knocking down NFκB p50 mRNA and overexpression (OE) of p50 cDNA on CTCF activity. B, lack of EGF-induced CTCF expression in the corneas of NFκB p50−/− knockout mice detected by immunostaining. The corneas obtained from wild-type and NFκB p50−/− knockout mice with/without stimulation of EGF were compared in immunostaining sections. C, effect of knocking down CTCF on EGF-induced CTCF expression detected by Western blot analysis. D, effects of overexpressing NFκB p50 on wound closure in CTCF knockdown cells following a time course. E, a significant delay of NFκB p50 overexpression and EGF-induced wound closure by knockdown of CTCF. *, p < 0.05. Data were collected from four sets of independent experiments. Non-silencing (NS) shRNA was used as a control.
EGF-induced Overexpression of NF-κB-controlled CTCF
To further study whether the effect of NF-κB on EGF-induced wound healing is mediated by CTCF, we generated a NF-κB-regulated exogenous CTCF overexpression construct. As shown in Fig. 5A, upper panel, five repeats of NF-κB TREs were inserted into a specific location in an expression vector upstream from the mini-CMV promoter. GFP (TRE-control) and GFP-linked CTCF (TRE-CTCF) cDNAs were inserted individually into the vector immediately downstream of the mini-CMV promoter to allow cellular NF-κB to directly regulate the expression of CTCF in stably transfected cells. The TRE-control and TRE-CTCF constructs were introduced individually into HTCE cells by lentivirus-mediated gene transfer. We found that EGF and TNFα stimulated the expression of GFP in both TRE-control- and TRE-CTCF-expressing HTCE cells (Fig. 5B, lower panel). Activation of the mini-CMV promoter was upon NF-κB binding to the NF-κB TRE regions, resulting in transcription of GFP and CTCF simultaneously. As expected, TRE-CTCF-expressing cells showed a green color under a fluorescent microscope and produced exogenous CTCF proteins. Thus, the expression of GFP and CTCF controlled by NF-κB activation in response to EGF or TNFα stimulation could be visualized in these cells. Western blot analysis showed that the expression of CTCF was increased significantly upon stimulation of EGF in TRE-CTCF-expressing HTCE cells compared with TRE-control-expressing HTCE cells (Fig. 5B). The results demonstrate that EGF- and TNF-α-induced activation of NF-κB promoted expression of exogenous CTCF through an NF-κB-controlled mechanism and verified the interaction of NF-κB with its downstream target, CTCF, in HTCE cells.
FIGURE 5.

Monitoring effect of EGF-induced NFκB activation on control of CTCF expression. A, effects of EGF- and TNF-α-induced changes of intracellular NFκB activity on exogenous CTCF expression. The upper panel illustrates the NFκB activity-controlled CTCF in vivo expression system containing the TRE-control and TRE-CTCF constructs. The lower panel shows EGF- and TNF-α-induced expression of exogenous GFP and GFP + CTCF in lentivirus-infected HTCE cells. mCMV, mini-CMV. B, EGF-induced significant increase in exogenous CTCF expression. Expression of exogenous CTCF in cells that were transfected with an NFκB activity-controlled CTCF construct was detected by Western blot analysis in the absence and presence of EGF or TNF-α. *, p < 0.05. Data were collected from four independent experiments. Non-silencing (NS) shRNA was used as a control.
Effect of NF-κB-controlled CTCF Overexpression on EGF-induced Wound Healing
Next, we examined the effect of EGF-induced exogenous CTCF expression controlled by NF-κB on motility, migration, and wound closure of HTCE cells. We found that cell migration was increased significantly in TRE-CTCF-expressing HTCE cells in the absence and presence of EGF stimulation compared with the TRE-control-expressing counterpart (Figs. 5B and 6A). In addition, we tracked the cells to measure cell motility changes under a computer-controlled fluorescent microscope with a motorized head stage in an attached culture chamber for 50 min (Fig. 6C). Statistical analysis revealed that EGF stimulated an increase in cell motility up to 30% in TRE-CTCF-expressing HTCE cells compared with TRE-control-expressing cells (p < 0.05, n = 86, Fig. 6D). The rate of wound closure was also accelerated significantly in TRE-CTCF-expressing HTCE cells compared with cells that were expressing TRE-control (Figs. 6, D and F). The results provide further evidence, in three experiments, that EGF-activated NF-κB up-regulated expression of CTCF to promote increases in cell motility and migration, resulting in acceleration of corneal epithelial cell wound healing.
FIGURE 6.

Effect of NFκB-controlled CTCF activity on EGF-induced wound healing. A, effect of NFκB-controlled CTCF activation on EGF-induced cell migration. B, significant increase in migration by EGF-induced and NFκB-controlled CTCF activation. HTCE cell migration was measured by transwell migration assays. * and **, p < 0.05 between the TRE-control and TRE-CTCF groups in the absence and presence of EGF, respectively (n = 4). C, EGF-activated cell motility enhanced by NFκB-controlled CTCF activation. D, statistical significance of motility increase by NFκB-controlled CTCF activation in EGF-induced cells. HCE cell motility was measured by using the cell tracking function of a Nikon fluorescent microscope. *, p < 0.05 between the TRE-control and TRE-CTCF groups in response to EGF stimulation (n = 86). E, EGF-activated wound closure enhanced by NFκB-controlled CTCF activation. F, statistical significance of accelerated wound closure by NFκB-controlled CTCF activation in EGF-induced cells. HCE cell wound closure was measured by a scratch-induced directional wound healing assay. *, p < 0.05 between the TRE-control and TRE-CTCF groups in response to EGF stimulation (n = 4).
Effect of Ad-x-CTCF Transduction on Corneal Epithelial Wound Healing
To test whether CTCF overexpression would accelerate corneal epithelial wound healing in vivo in the absence and presence of NFκB p50, eyeballs obtained from wild-type and p50 knockout mice (p50−/−) were infected with adenoviral Ad-x-CTCF and Ad-x-vector (for controls). Both adenoviral Ad-x-CTCF and Ad-x-vector containing a GFP expression marker were tested in HCE cells for efficiency of the gene transfer before they were used for eyeball infections. We found that the transferring efficiencies for both constructs reached more than 95% within 2 days, as determined by Western blot analyses (data not shown). Corneas transduced with Ad-x-CTCF and Ad-x-vector were wounded, and the healing process was monitored every day up to 15 days. There were significantly increased wound healing rates in 10 days in wild-type mouse corneas overexpressing CTCF compared with Ad-x-vector-infected controls (Fig. 7, A and B). There were marked delays in wound healing rate in p50−/− corneas compared with wild-type corneas with/without overexpression of Ad-x-CTCF (Fig. 7, C and D). Acceleration of corneal epithelial wound healing transduced by Ad-x-CTCF was observed in corneas of wild-type mice, and transduction of CTCF had a lesser effect on the wound healing in corneas from p50 knockout mice, indicating that the effect of CTCF on corneal epithelial wound healing is associated with the NFκB-p50 subtype pathway.
FIGURE 7.

Effect of adenovirus-mediated CTCF transduction on corneal epithelial wound healing. A, effect of overexpressing Ad-x-CTCF on mouse corneal epithelial wound healing. B, significant increase in wound healing rate in Ad-x-CTCF-transduced corneas. C, effects of overexpressing Ad-x-CTCF on corneal epithelial wound healing of p50−/− knockout mice. D, comparison of wound healing in corneas of wild-type and p50−/− knockout mice with/without overexpression of CTCF. A green color indicates cells expressing GFP after adenoviral transduction of the corneal surface. Arrows indicate nude regions of the corneal surface without epithelial coverage. The left eye of each mouse was subject to experiments, and the right eye served as the control. *, p < 0.05 (n = 4).
DISCUSSION
One of the major stimuli to stimulate corneal epithelial wound healing is EGF, which promotes cell migration and proliferation (1). As demonstrated in human corneal epithelial cells, EGF elicits complex responses at early times by inducing specific cellular signaling pathways that transfer the signals to the nucleus and activate the transcription factor at later times (3, 14, 15, 33, 38). In previous studies, we found that EGF-induced formations of the NFκB p65/p50 heterodimer and p50/50 homodimer activate CTCF transcription by binding to a κB site located in the promoter region of the CTCF gene and that an increase in CTCF activity in corneal epithelial cells promotes cell motility and migration (14, 17). These results indicate that NFκB subtypes interact directly with the CTCF gene in the promoter region. However, it is unknown whether this interaction has functional significance in corneal epithelial wound healing. In this study, we provide direct evidence, for the first time, of EGF-induced CTCF activation through the NFκB subtype p50 and show that it subsequently regulates corneal epithelial wound healing in HTCE cells and in the corneas of mice. The effect of NFκB p50 on EGF-induced wound healing was examined in HTCE cells when the cellular level of p50 was knocked down by NFκB p50 siRNA or enhanced by overexpression of p50. Suppressed and enhanced expression of NFκB p50 resulted in deceleration and acceleration of cell wound healing, respectively. This observation is consistent with a report indicating that NFκB p50 activation promotes migration in breast cancer cells (39). There is also a parallel relationship between knockdown/overexpression of NFκB p50 and decrease/increase in CTCF activities in HTCE cells. In addition, overexpression of NFκB p50 enhanced CTCF expression, but it failed to activate CTCF in the presence of CTCF shRNA transfection (Fig. 4). Further evidence that the effect of overexpression of NFκB p50 on promoting HTCE cell wound healing was retarded by knocking down CTCF mRNA indicates that CTCF is indeed a downstream target that plays a key role in EGF-induced NFκB signaling cascades to regulate corneal epithelial wound healing.
An in vivo model system containing the TRE-control and TRE-CTCF expression constructs was engineered with multiple κB binding sites to sense EGF-induced changes of intracellular NFκB activity that control expression levels of GFP and CTCF. Upon stimulation of EGF and TNF-α, exogenous GFP and CTCF proteins were expressed in transfected cells under the control of cellular NFκB p50 activities (Fig. 5). Thus, transfection of TRE-CTCF into HTCE cells enabled us to monitor the effects of EGF-induced NFκB activation and NFκB-controlled CTCF activity on corneal epithelial wound healing. It appeared that there were more green cells in TRE-control-transfected cells compared with TRE-CTCF-expressing cells. The different level of GFP expression may be a result of the size difference because of linking the CTCF cDNA to GFP in the TRE-CTCF construct. In addition, TNF-α-induced cells displayed a stronger expression of GFP than EGF-induced cells, indicating that there were probably more NF-κB activations induced by TNF-α in these cells.
Corneal epithelial wound healing is largely dependent on the motility and migration capability of the basal layer cells. As the first step, the effect of EGF-induced NFκB activation to regulate exogenous CTCF on corneal epithelial cell migration and motility were studied by measuring a transwell-migrated cell population and continuous cell tracking. An increase in expression of exogenous CTCF in response to EGF-induced NFκB activation significantly increased cell migration and motility. The basal migration rate of TRE-CTCF-expressing cells was also faster than TRE-control expressing HTCE cells, even in the absence of EGF stimulation. As expected, we found that EGF induced a significant increase in the wound healing rate in HTCE cells expressing TRE-CTCF compared with TRE-control-expressing cells. The result of the wound healing study is consistent with the finding that TRE-CTCF transfection and expression significantly increased migration and motility of TRE-CTCF-expressing HTCE cells (Fig. 6). EGF stimulation activated NFκB p50 activity in the cornea of wild-type mice, but it was not seen in NFκB p50 knockout mouse corneas. In the absence of EGF, corneal epithelial wound healing was much slower in the eyeballs of both wild-type and p50−/− knockout mice (Fig. 7). Overexpression of CTCF by adenovirus-mediated CTCF transduction enhanced corneal epithelial wound healing in both wild-type and p50−/− knockout mice, which is consistent with the results observed in wound closure assays in HCE cells. The observation provides useful evidence to support that CTCF is a downstream component in the NFκB pathway. It also suggests that the effect of CTCF on wound healing is not exclusively regulated by NFκB p50. In fact, CTCF can be activated by other proliferative signaling pathways as well, such as the Erk pathway (15). In addition, at a posttranslational level, it has been shown that CTCF is also a target protein subject to modifications of SUMOylation and phosphorylation (40).
Finally, this study demonstrates for the first time that corneal epithelial wound healing is delayed significantly in the corneas of NFκB p50 knockout mice. There is more than 30% delay in corneal epithelial wound healing in the corneas of NFκB p50 knockout mice, suggesting that the NFκB signaling pathway plays important roles in growth factor-promoted corneal epithelial self-renewal and corneal protective functions.
This work was supported, in whole or in part, by National Institutes of Grants R01-EY015281 and EY022364 (to L. L.).
- CTCF
- CCCTC binding factor
- HTCE
- human telomerase-immortalized corneal epithelial
- HCE
- human SV-40 large T-transformed corneal epithelial
- TRE
- transcription response element.
REFERENCES
- 1. Lu L., Reinach P. S., Kao W. W. (2001) Corneal epithelial wound healing. Exp. Biol. Med. 226, 653–664 [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y., Akhtar R. A. (1996) Effect of epidermal growth factor on phosphatidylinositol 3-kinase activity in rabbit corneal epithelial cells. Exp. Eye Res. 63, 265–275 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y., Akhtar R. A. (1997) Epidermal growth factor stimulation of phosphatidylinositol 3-kinase during wound closure in rabbit corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 38, 1139–1148 [PubMed] [Google Scholar]
- 4. Zhang Y., Akhtar R. A. (1998) Epidermal growth factor stimulates phospholipase D independent of phospholipase C, protein kinase C or phosphatidylinositol-3 kinase activation in immortalized rabbit corneal epithelial cells. Curr. Eye Res. 17, 294–300 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y., Liou G. I., Gulati A. K., Akhtar R. A. (1999) Expression of phosphatidylinositol 3-kinase during EGF-stimulated wound repair in rabbit corneal epithelium. Invest. Ophthalmol. Vis. Sci. 40, 2819–2826 [PubMed] [Google Scholar]
- 6. Islam M., Akhtar R. A. (2000) Epidermal growth factor stimulates phospholipase Cγ1 in cultured rabbit corneal epithelial cells. Exp. Eye Res. 70, 261–269 [DOI] [PubMed] [Google Scholar]
- 7. Islam M., Akhtar R. A. (2001) Upregulation of phospholipase Cγ1 activity during EGF-induced proliferation of corneal epithelial cells. Effect of phosphoinositide-3 kinase. Invest. Ophthalmol. Vis. Sci. 42, 1472–1478 [PubMed] [Google Scholar]
- 8. Kang S. S., Li T., Xu D., Reinach P. S., Lu L. (2000) Inhibitory effect of PGE2 on EGF-induced MAP kinase activity and rabbit corneal epithelial proliferation. Invest. Ophthalmol. Vis. Sci. 41, 2164–2169 [PubMed] [Google Scholar]
- 9. Kang S. S., Wang L., Kao W. W., Reinach P. S., Lu L. (2001) Control of SV-40 transformed RCE cell proliferation by growth-factor-induced cell cycle progression. Curr. Eye Res. 23, 397–405 [DOI] [PubMed] [Google Scholar]
- 10. Madhani H. D., Fink G. R. (1998) The riddle of MAP kinase signaling specificity. Trends Genet. 14, 151–155 [DOI] [PubMed] [Google Scholar]
- 11. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 12. Li T., Lu L. (2007) Functional role of CCCTC binding factor (CTCF) in stress-induced apoptosis. Exp. Cell Res. 313, 3057–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou H., Gao J., Lu Z. Y., Lu L., Dai W., Xu M. (2007) Role of c-Fos/JunD in protecting stress-induced cell death. Cell Prolif. 40, 431–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu L., Wang L., Li T., Wang J. (2010) NF-κB subtypes regulate CCCTC binding factor affecting corneal epithelial cell fate. J. Biol. Chem. 285, 9373–9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li T., Lu L. (2005) Epidermal growth factor-induced proliferation requires down-regulation of Pax6 in corneal epithelial cells. J. Biol. Chem. 280, 12988–91295 [DOI] [PubMed] [Google Scholar]
- 16. Li T., Lu Z., Lu L. (2004) Regulation of eye development by transcription control of CCCTC binding factor (CTCF). J. Biol. Chem. 279, 27575–27583 [DOI] [PubMed] [Google Scholar]
- 17. Wang L., Deng S. X., Lu L. (2012) Role of CTCF in EGF-induced migration of immortalized human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 53, 946–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herrlich P., Ponta H., Rahmsdorf H. J. (1992) DNA damage-induced gene expression. Signal transduction and relation to growth factor signaling. Rev. Physiol. Biochem. Pharmacol. 119, 187–223 [DOI] [PubMed] [Google Scholar]
- 19. Holbrook N. J., Liu Y., Fornace A. J., Jr. (1996) Signaling events controlling the molecular response to genotoxic stress. EXS 77, 273–288 [DOI] [PubMed] [Google Scholar]
- 20. Belandia B., Latasa M. J., Villa A., Pascual A. (1998) Thyroid hormone negatively regulates the transcriptional activity of the β-amyloid precursor protein gene. J. Biol. Chem. 273, 30366–30371 [DOI] [PubMed] [Google Scholar]
- 21. Büscher M., Rahmsdorf H. J., Litfin M., Karin M., Herrlich P. (1988) Activation of the c-fos gene by UV and phorbol ester. Different signal transduction pathways converge to the same enhancer element. Oncogene 3, 301–311 [PubMed] [Google Scholar]
- 22. Devary Y., Rosette C., DiDonato J. A., Karin M. (1993) NF-κB activation by ultraviolet light not dependent on a nuclear signal. Science 261, 1442–1445 [DOI] [PubMed] [Google Scholar]
- 23. Li T., Dai W., Lu L. (2002) Ultraviolet-induced junD activation and apoptosis in myeloblastic leukemia ML-1 cells. J. Biol. Chem. 277, 32668–32676 [DOI] [PubMed] [Google Scholar]
- 24. Courtois G., Gilmore T. D. (2006) Mutations in the NF-κB signaling pathway. Implications for human disease. Oncogene 25, 6831–6843 [DOI] [PubMed] [Google Scholar]
- 25. Dutta J., Fan Y., Gupta N., Fan G., Gélinas C. (2006) Current insights into the regulation of programmed cell death by NF-κB. Oncogene 25, 6800–6816 [DOI] [PubMed] [Google Scholar]
- 26. Baniahmad A., Steiner C., Köhne A. C., Renkawitz R. (1990) Modular structure of a chicken lysozyme silencer. Involvement of an unusual thyroid hormone receptor binding site. Cell 61, 505–514 [DOI] [PubMed] [Google Scholar]
- 27. Bell K. D., Campbell R. J., Bourne W. M. (2000) Pathology of late endothelial failure. Late endothelial failure of penetrating keratoplasty. Study with light and electron microscopy. Cornea 19, 40–46 [DOI] [PubMed] [Google Scholar]
- 28. Hark A. T., Schoenherr C. J., Katz D. J., Ingram R. S., Levorse J. M., Tilghman S. M. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489 [DOI] [PubMed] [Google Scholar]
- 29. Qi C. F., Martensson A., Mattioli M., Dalla-Favera R., Lobanenkov V. V., Morse H. C., 3rd. (2003) CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc. Natl. Acad. Sci. U.S.A. 100, 633–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rasko J. E., Klenova E. M., Leon J., Filippova G. N., Loukinov D. I., Vatolin S., Robinson A. F., Hu Y. J., Ulmer J., Ward M. D., Pugacheva E. M., Neiman P. E., Morse H. C., 3rd, Collins S. J., Lobanenkov V. V. (2001) Cell growth inhibition by the multifunctional multivalent zinc-finger factor CTCF. Cancer Res. 61, 6002–6007 [PubMed] [Google Scholar]
- 31. Docquier F., Farrar D., D'Arcy V., Chernukhin I., Robinson A. F., Loukinov D., Vatolin S., Pack S., Mackay A., Harris R. A., Dorricott H., O'Hare M. J., Lobanenkov V., Klenova E. (2005) Heightened expression of CTCF in breast cancer cells is associated with resistance to apoptosis. Cancer Res. 65, 5112–5122 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Lu L. (2011) Activation of oxidative stress-regulated Bcl-3 suppresses CTCF in corneal epithelial cells. PLoS ONE 6, e23984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang J., Lin A., Lu L. (2010) Effect of EGF-induced HDAC6 activation on corneal epithelial wound healing. Invest. Ophthalmol. Vis. Sci. 51, 2943–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu K. P., Ding Y., Ling J., Dong Z., Yu F. S. (2004) Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 45, 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yin J., Yu F. S. (2009) ERK1/2 mediate wounding- and G-protein-coupled receptor ligands-induced EGFR activation via regulating ADAM17 and HB-EGF shedding. Invest. Ophthalmol. Vis. Sci. 50, 132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang L., Payton R., Dai W., Lu L. (2011) Hyperosmotic stress-induced ATF-2 activation through Polo-like kinase 3 in human corneal epithelial cells. J. Biol. Chem. 286, 1951–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen L., Meng Q., Kao W., Xia Y. (2011) IκB kinase β regulates epithelium migration during corneal wound healing. PLoS ONE 6, e16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilson S. E., Lloyd S. A., He Y. G. (1992) EGF, basic FGF, and TGFβ-1 messenger RNA production in rabbit corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 33, 1987–1995 [PubMed] [Google Scholar]
- 39. Helbig G., Christopherson K. W., 2nd, Bhat-Nakshatri P., Kumar S., Kishimoto H., Miller K. D., Broxmeyer H. E., Nakshatri H. (2003) NF-κB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J. Biol. Chem. 278, 21631–21638 [DOI] [PubMed] [Google Scholar]
- 40. Wang J., Wang Y., Lu L. (2012) De-SUMOylation of CCCTC binding factor (CTCF) in hypoxic stress-induced human corneal epithelial cells. J. Biol. Chem. 287, 12469–12479 [DOI] [PMC free article] [PubMed] [Google Scholar]