

Background: Protein kinase D1 (PKD1) regulates actin reorganization processes at the leading edge.
Results: PKD1 phosphorylates VASP at two serine residues, Ser-157 and Ser-322.
Conclusion: VASP is a substrate for PKD1.
Significance: We provide an additional mechanism of how VASP functions can be regulated.
Keywords: Phosphorylation, Protein Kinase C (PKC), Protein Kinase D (PKD), Signal Transduction, Signaling, VASP, Kinase
Abstract
Enabled/Vasodilator-stimulated phosphoprotein (Ena/VASP) protein family members link actin dynamics and cellular signaling pathways. VASP localizes to regions of dynamic actin reorganization such as the focal adhesion contacts, the leading edge or filopodia, where it contributes to F-actin filament elongation. Here we identify VASP as a novel substrate for protein kinase D1 (PKD1). We show that PKD1 directly phosphorylates VASP at two serine residues, Ser-157 and Ser-322. These phosphorylations occur in response to RhoA activation and mediate VASP re-localization from focal contacts to the leading edge region. The net result of this PKD1-mediated phosphorylation switch in VASP is increased filopodia formation and length at the leading edge. However, such signaling when persistent induced membrane ruffling and decreased cell motility.
Introduction
The enabled/vasodilator-stimulated phosphoprotein (Ena/VASP)2 family of proteins is comprised of vasodilator-stimulated phosphoprotein (VASP), mammalian Enabled (Mena), and Ena-VASP-like (EVL) (1, 2). VASP proteins consist of a N-terminal Ena/VASP homology-1 (EVH-1) domain that binds to WASP, zyxin, and vinculin (3), a poly-proline region (PPR) that interacts with profilin and SH3 domain containing proteins such as Src and Abl (4), and a C-terminal EVH-2 domain that mediates binding to G-actin, F-actin, and oligomerization (5, 6). Ena/VASP family members are involved in many RhoGTPase-regulated and actin-related processes including epithelial cell adhesion, cell motility, and pathogen F-actin tail formation (summarized in Refs. 7, 8). VASP localizes to regions of dynamic actin re-organization such as the focal adhesion contacts, the leading edge or filopodia, where it promotes F-actin filament formation (9–11). Stress fiber assembly is promoted by VASP through its anti-capping (12), bundling (5), and anti-branching activities (13).
VASP functions are regulated by phosphorylations, although the exact roles of phosphorylations on functional outcomes are unclear. Known phosphorylation sites in VASP are amino acid residues Ser-157, Ser-239, and Thr-278. Phosphorylation of Ser-157 and Ser-239 is mediated by cAMP-dependent protein kinase (PKA) and cGMP-dependent protein kinase (PKG) (14, 15). Phosphorylation of Thr-278 is mediated by AMP-activated protein kinase (AMPK) (16). VASP phosphorylation can reduce its association with actin, has negative effects on actin polymerization, and modulates interaction with other proteins (17). For example Ser-157 phosphorylation abrogates interaction of VASP with the SH3 domains of Abl, Src, and αΙΙ spectrin and controls cellular distribution of VASP (4, 18). On the other hand, phosphorylations at Ser-239 and Thr-278 impair F-actin accumulation (19). In contrast, binding of focal adhesion proteins to the EVH-1 domain and of profilin to the PPR domain are independent of phosphorylation of these known phosphorylation sites in VASP (17, 20).
Actin remodeling at the leading edge of motile cells is a dynamic process that is regulated by many signaling cascades, several of which are not well defined, yet. Protein kinase D1 (PKD1), a member of the PKD family of serine/threonine kinases, can be located at the leading edge of migrating cells where it binds to F-actin structures (21, 22). Similar to Ena/VASP family members, available data suggest both positive and negative regulatory functions of PKD proteins in respect to cell motility and invasiveness of cells (discussed in (23, 24)). PKD1 can be activated by members of the RhoGTPase family, including RhoA as the major activator (25–28). PKD1 activation through RhoA affects actin reorganization processes at the leading edge with a net effect of decreasing directed cell migration (21, 22, 29). For example, active PKD1 decreases cofilin activity and formation of free actin barbed ends at the leading edge (26, 29). This is mediated by direct phosphorylation and activation of group II p21-activated kinases (PAK) such as PAK4, which is an upstream kinase of LIMK (29), but also by an inhibiting phosphorylation of the phosphatase slingshot-1L (SSH-1L) (26, 30). Other substrates for PKD1 at the leading edge that control actin reorganization processes include cortactin, Rin-1, and also the VASP family member EVL-1 (26, 31, 32).
Here we show that PKD1 phosphorylates VASP at two serine residues, Ser-157 and Ser-322 in the EVH-2 domain. These phosphorylations occur in response to RhoA activation and mediate VASP translocation from focal contacts to the leading edge region. PKD1-mediated phosphorylation of VASP also increases stress fiber formation resulting in increased lamellipodium formation and filopodia length. When persistent such signaling resulted in membrane ruffling and decreased cell migration.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, and Reagents
HeLa and Hek293T cells (obtained from ATCC, Manassas, VA) were maintained in DMEM with 10% FBS, and NMuMG cells (obtained from ATCC) were maintained in DMEM with 10 μg/ml insulin and 10% FBS. Anti-GST and anti-PKD1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), anti-HA, anti-FLAG (M2), and anti-actin from Sigma-Aldrich, anti-pMOTIF (PKD substrate antibody), anti-pS157-VASP and anti-pS239-VASP from Cell Signaling Technology (Danvers, MA). Anti-VASP was from BD Biosciences (San Jose, CA). A rabbit polyclonal antibody specific for VASP phosphorylated at Ser-322 (anti-pS322-VASP antibody) was raised by 21 Century Biochemicals (Marlboro, MA) using C-Ahx-TLPRMK[pS]SSSVT-amide and Acetyl-TLPRMK[pS]SSSVT-Ahx-C-amide as immunogen. The antibody was affinity purified using protein A columns. Secondary HRP-linked antibodies were from Millipore (Billerica, MA). Secondary antibodies (Alexa Fluor 488 F(ab′)2 fragment of goat-anti-mouse IgG or Alexa Fluor 594 F(ab′)2 fragment of goat-anti-rabbit) were from Invitrogen. GenJet Reagent II (SignaGen Laboratories, Ijamsville, MD) was used for transient transfection of NMuMG, and TransIT HeLaMonster (Mirus Bio, Madison, WI) for HeLa. Lysozyme and deoxycholic acid were from Fisher Scientific.
DNA Constructs
The expression plasmids for HA-tagged and GFP-tagged constitutively active PKD1 (PKD1.CA, PKD1.S738E.S742E mutation) were described before (29, 33). The EGFP-actin expression plasmid was from Clontech (Mountain View, CA). FLAG-tagged human VASP was amplified from a HeLa cDNA library using 5′-CGCGGATCCATGGACTATAAGGACGATGATGACAAAAGCGAGACGGTCATCTGTTCCAGC-3′ and 5′-CGCCTCGAGTCAGGGAGAACCCCGCTTCCTCAG-3′ as primers and cloned into pcDNA3.1 via BamHI and XhoI. GST-VASP was generated by cloning full-length human VASP into pGEX4-T1 via BamHI and XhoI. Cherry-VASP and GFP-VASP were generated by cloning full-length human VASP into pmCherry-N1 or pEGFP-N1 via BamHI and XhoI. Site-directed mutagenesis was carried out using the QuikChange kit (Stratagene, La Jolla, CA). 5′-CACATAGAGCGCCGGGTCGCCAATGCAGGAGGCCCACCT-3′ and 5′-AGGTGGGCCTCCTGCATTGGCGACCCGGCGCTCTATGTG-3′ were used as primers to generate a VASP.S157A mutation, 5′-CACATAGAGCGCCGGGTCGAAAATGCAGGAGGCCCACCT-3′ and 5′-AGGTGGGCCTCCTGCATTTTCGACCCGGCGCTCTATGTG-3′ to generate a VASP.S157E mutation, 5′-CCTTGCCAAGGATGAAGGCGTCTTCTTCGGTGACCACTTCC-3′ and 5′-GGAAGTGGTCACCGAAGAAGACGCCTTCATCCTTGGCAAGG-3′ to generate a VASP.S322A mutation, and 5′-CCTTGCCAAGGATGAAGGAGTCTTCTTCGGTGACCACTTCC-3′ and 5′-GGAAGTGGTCACCGAAGAAGACTCCTTCATCCTTGGCAAGG-3′ to generate a VASP.S322E mutation. The 4xSRE-TK-luc reporter plasmid was described before (34). pEBG vector or pEBG-RhoA.CA and use and sequences for lentiviral PKD1/2-shRNA were described before (26).
In Vitro Kinase Assays
Kinase assays with GST fusion proteins were carried out by adding 250 ng of active, purified PKD1 (Millipore, Billerica, MA) to 2 μg of purified GST or GST-fusion protein in a volume of 40 μl kinase buffer (50 mm Tris, pH 7.4, 10 mm MgCl2, and 2 mm DTT) supplemented with 100 μm ATP. The kinase reaction (30 min, room temperature) was stopped by adding 2× Laemmli buffer.
Immunoblotting, Immunoprecipitation, and PAGE
Cells were washed twice with ice-cold PBS (140 mm NaCl, 2.7 mm KCl, 8 mm Na2HPO4, 1.5 mm KH2PO4, pH 7.2) and lysed with lysis buffer A (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 5 mm EDTA, pH 7.4) containing Protease Inhibitor Cocktail (PIC, Sigma-Aldrich). Lysates were briefly vortexed and incubated on ice for 30 min. Following centrifugation at 13,000 rpm (15 min, 4 °C) protein concentration of lysates was determined. Proteins of interest were immunoprecipitated by 1 h of incubation with a specific antibody (2 μg) followed by 30 min of incubation with protein G-Sepharose (GE Healthcare, Piscataway, NJ). Immune complexes were washed three times with TBS (50 mm Tris-HCl, pH 7.4, 150 mm NaCl) and resolved in 20 μl of TBS and 2× Laemmlibuffer. Samples were subjected to SDS-PAGE, proteins were transferred to nitrocellulose membranes and visualized by immunostaining.
Immunofluorescence
Cells were transfected as indicated in 8-well ibiTreat μ-Slides (Ibidi, Martinsried, Germany). The next day cells were washed twice with phosphate-buffered saline (PBS). Following fixation with 4% paraformaldehyde (15 min, 37 °C), cells were washed three times in PBS and then permeabilized with 0.1% Triton X-100 in PBS for 2 min at room temperature. Samples were blocked with 3% bovine serum albumin and 0.05% Tween 20 in PBS (blocking solution) for 30 min at room temperature, and then incubated overnight at 4 °C with primary antibodies (anti-FLAG 1:4000 for VASP or anti-VASP 1:1000) diluted in blocking solution. Cells were washed five times with PBS and incubated with secondary antibodies (Alexa Fluor 488 F(ab′)2 fragment of goat-anti-mouse IgG or Alexa Fluor 594 F(ab′)2 fragment of goat-anti-rabbit; both Invitrogen), diluted (1:800) in blocking solution for 2 h at room temperature. F-actin was stained together with secondary antibodies by incubating with phalloidin (Alexa Fluor 633-Phalloidin, shown as magenta pseudo color staining) in blocking solution. After extensive washes in PBS, cells were mounted in Ibidi mounting medium (Ibidi). Samples were examined using an IX81 DSU Spinning Disc Confocal from Olympus with a 40× objective.
Fluorescence Recovery After Photobleaching (FRAP)
For FRAP analysis cells were transfected and seeded in ibiTreat μ-Slides VI (Ibidi), and imaged in CEB buffer (2 mm MgCl2, 2 mm CaCl2, 150 mm NaCl, 2.5 mm KCl, 10 mm glucose, and 10 mm sodium HEPES, pH 7.5) (35). Cells were imaged using a Zeiss LSM 510 META laser-scanning confocal microscope (Carl Zeiss Microscopy) equipped with a Plan-APOCHROMAT 100x oil immersion objective (1.4 NA) and Zen (2009) software. Prior to performing FRAP on a given cell, an initial image was acquired with both 488 nm and 594 nm excitation to confirm co-transfection. For FRAP, cells were imaged with an argon/2 laser at 488 nm, and emission was collected using a band-pass 505–570IR filter. A series of 250 images was acquired every 0.495 s at 256 × 256 pixel resolution 0.35 μm x 0.35 μm pixel size; 3.21 μsec pixel dwell time). After the first image, a defined region encompassing part of the cell lamellipodia was bleached using the 405–30 nm diode laser (30 milliwatt) and 488 nm argon laser (30 milliwatt) lines simultaneously at 100% transmission for 200 iterations (12.84 μs pixel dwell time during the bleach phase). Fluorescence in the bleached region and in a similarly sized and positioned non-bleached (reference) region was quantified at every time point using the Zen software. At least 10 cells were analyzed per transfection condition. To correct for differences in expression level between cells, fluorescence for the bleached and reference regions were normalized to the level before bleaching. In addition, to correct for fluorescence loss due to repeated imaging, data at all time points were normalized to fluorescence in the reference region. Mathematically, FRAP curves were created using the equation Ft = (R0 · Bt)/(Rt · B0), where Ft is the normalized fluorescence at time point t, R0 and Rt are the fluorescence in the reference region at time points 0 and t, respectively and B0 and Bt are the fluorescence in the bleached region at time points 0 and t. This way of presenting FRAP data provides an accurate estimate of the immobile fraction (36). In each experiment, the normalized Ft were averaged and plotted. Using the resulting FRAP curve, the t1/2 of maximal recovery was determined, which is defined as the time point after bleaching at which the normalized fluorescence has increased to half the amount of the maximal recovery.
Purification of GST Fusion Proteins
GST fusion proteins were expressed in Escherichia coli by IPTG induction (4 h, 37 °C). Bacteria pellets were resuspended in suspension buffer (50 mm Tris, pH 8.0, 1 mm EDTA, 100 mm NaCl, 1 mm DTT, 100 μm PMSF, 0.02 mg/ml DNase I), lysed with 2 mg/ml lysozyme (20 min on ice) and incubated with 2 mg/ml deoxycholic acid (30 min at room temperature). Following sonication of the suspension, the supernatant was purified using glutathione-Sepharose beads (GE Healthcare). Beads were eluted with elution buffer (0.15 m NaCl, 2.5 mm MgCl2, 5% glycerol, 50 mm NaP, pH 7.6, 0.2% Tween-20, 1 mm DTT, 1 mm PMSF, 10 mm reduced glutathione), and dialyzed into dialysis buffer (0.15 m NaCl, 2.5 mm KCl, 1 m MgCl2·6 H2O, 10 mm NaP, pH 7.6, 10% glycerol, 1% Tween-20, 1 mm DTT) using 0.5 ml of dialysis cassettes from Thermo Scientific (Rockford, IL). For normalization, GST-fusion proteins were analyzed via SDS-PAGE and visualized with GelCode staining reagent (Thermo Scientific, Rockford, IL).
SRE Activity Assays
Cells were transiently transfected with 1 μg of 4xSRE-TK-luc Reporter construct, 0.1 μg of Renilla luciferase reporter, and 1 μg of the cDNA of interest in serum-free media for 24 h. Cell lysates were prepared by washing cells with PBS, scraping in 250 μl Passive Lysis Buffer (Promega, Madison, WI) and centrifugation (13,000 rpm, 10 min, 4 °C). Assays for luciferase activity were performed on cell lysates using the Dual-Luciferase Reporter Assay System (Promega) and a Veritas luminometer (Symantec, Cupertino, CA). Protein expression was controlled by immunoblot analysis.
Live Cell Imaging and Kymography
Fluorescence time laps analysis was performed using a spinning disc confocal microscope (Olympus) with a 60x NA 1.4 oil immersion lens (Olympus), which was driven by SlideBook5 (Olympus) and equipped with a digital CCD camera (ORCA-R2; HAMAMATSU). HeLa cells were transfected with GFP-Actin and Cherry-VASP as indicated, the next day trypsinized, re-seeded into μ-Slides (Ibidi) in phenol red-free media and after 12 h images were acquired, every 10 s for 20 min. Image analysis was performed using ImageJ (ImageJ, U.S. National Institutes of Health, Bethesda, MD). Kymographic analysis was performed using plug-ins provided by J. Rietdorf and A Seitz, EMBL Heidelberg. Images were prepared for publication using ImageJ and Adobe Photoshop.
Impedance-based Real-time Cell Migration Analysis
For the electric cell-substrate impedance sensing (ECIS) migration assay, cells were transfected as indicated and after 24 h seeded on 8W1E electrode arrays (Applied BioPhysics, Troy, NY). An electric fence with high current, which prevents cells from growing on the electrode surface, was applied prior to cell attachment. After 20 h, the fence was deactivated, cells washed with media for two times, and real-time migration was monitored by measuring changes in impedance over time. For xCELLigence directed cell migration assays, cells were transfected as indicated and after 24 h seeded on Transwell CIM-plate 16 plates (Roche, Indianapolis, IN). After 1 h of attachment, cell migration toward NIH-3T3 conditioned media was continuously monitored in real-time using the xCELLigence RTCA DP instrument (Roche).
RESULTS
PKD1 Phosphorylates VASP at Ser-157 and Ser-322 in Vitro
To determine if VASP is a target for PKD1, we co-expressed VASP and active PKD1 (PKD1.CA, PKD1.S738E.S742E) and utilized the anti-pMOTIF antibody which recognizes the phosphorylated PKD consensus motif and detects PKD1 substrates (26, 29, 37, 38). We found that in presence of active PKD1, VASP shows increased phosphorylation as well as an electrophoretic mobility shift from 46 to 50 kDa (Fig. 1A), which previously was described to be due to phosphorylation at Ser-157, but not Ser-239 and Thr-278 (2, 14). Of all these known VASP phosphorylation sites, only the amino acid sequence surrounding Ser-157 represents an ideal PKD motif (Fig. 1B). However, analysis of the VASP amino acid sequence also identified two other potential PKD1 phosphorylation sites, Ser-322 and Ser-379, both in the EVH-2 domain (Fig. 1B). First we tested if PKD1 can phosphorylate VASP at Ser-157. Therefore, we performed in vitro kinase (IVK) assays with purified proteins. By comparing phosphorylation of GST (control) with GST-VASP in kinase assays with baculovirus-expressed and purified PKD1 and analysis with anti-pS157-VASP and anti-pS239-VASP (used as negative control) antibodies, we found that active PKD1 mediates VASP phosphorylation at Ser-157, but not at Ser-239 (Fig. 1C). However, similar in vitro kinase assays using GST, GST-VASP, and a GST-VASP.S157A mutant as substrates, when analyzed with the anti-pMOTIF antibody showed that PKD1 phosphorylates VASP at least at one additional site (Fig. 1D). We focused on Ser-322 and Ser-379 as potential other phosphorylation sites. Comparison of homologies of different VASP species showed that the amino acid motif surrounding Ser-322 is more conserved between species than the motif surrounding Ser-379 (Fig. 1B). Therefore, we next analyzed if PKD1 can phosphorylate VASP at Ser-322. To test this we again performed in vitro kinase assays with purified bacterially expressed GST-VASP proteins. As previously shown, GST-VASP was an effective PKD1 target, and GST-VASP.S157A and GST-VASP.S322A single mutants, both showed reduced phosphorylation. A GST-VASP.S157A.S322A double mutant lacked phosphorylation by PKD1 (Fig. 1E, short and long exposures). We next generated a phosphospecific antibody directed against VASP phosphorylated at Ser-322. Use of this anti-pS322-VASP antibody showed that PKD1 directly mediates phosphorylation of VASP at Ser-322 as demonstrated in an in vitro kinase assay with purified components (Fig. 1F). The antibody specifically recognized VASP when phosphorylated at Ser-322 as shown with a VASP.S322A mutant in the same assay. Taken together, these data establish PKD1 as a kinase that directly phosphorylates VASP at Ser-157 and Ser-322 in vitro.
FIGURE 1.

PKD1 phosphorylates VASP at Ser-157 and Ser-322 in vitro. A, HeLa cells (0.65 × 106 cells, 6-cm dish) were transfected with vector control, HA-tagged constitutively active PKD1 (PKD1.CA) and FLAG-tagged VASP as indicated. VASP was immunoprecipitated (anti-FLAG), and precipitates were analyzed with the pMOTIF antibody that recognizes PKD-mediated phosphorylation. Samples were re-stained for VASP (anti-FLAG) and lysates were control stained for expressed PKD1.CA (anti-PKD1). B, depicted is the PKD consensus phosphorylation motif with arginine or lysine at −3 and leucine, valine, or isoleucine at −5 relative to the serine or threonine phosphorylation site. Also shown are known phosphorylation sites in human VASP (Ser-157, Ser-239, Thr-278) and potential PKD1 phosphorylation motifs at Ser-322 and Ser-379 in VASP of different species. C, PKD1 phosphorylates VASP at Ser-157 in an in vitro assay. Bacterially expressed and purified GST (negative control) or GST-VASP was incubated in a kinase reaction with purified active PKD1. Substrate phosphorylation was detected using the phosphosite-specific antibodies anti-pS157-VASP or anti-pS239-VASP as indicated. Control blots were performed for protein input (anti-PKD1, anti-GST). D, Ser-157 is not the only PKD1 phosphorylation site in VASP. Bacterially expressed and purified GST (negative control), GST-VASP, or GST-VASP.S157A was incubated in a kinase reaction with purified active PKD1. Substrate phosphorylation was detected using the pMOTIF antibody, which recognizes the phosphorylated PKD motif in PKD substrates. Control blots were performed for protein input (anti-PKD1, anti-GST). E, bacterially expressed and purified GST (negative control), GST-VASP, GST-VASP.S157A, GST-VASP.S322A, GST-VASP.S157A.S322A were incubated in a kinase reaction with purified active PKD1. Substrate phosphorylation was detected using the pMOTIF antibody, which recognizes the phosphorylated PKD motif in PKD substrates. Shown are short and long exposures. Control blots were performed for protein input (anti-PKD1, anti-VASP). F, PKD1 phosphorylates VASP at Ser-322 in an in vitro assay. Bacterially-expressed and purified GST-VASP or GST-VASP.S322A was incubated in a kinase reaction with purified active PKD1. Substrate phosphorylation was detected using the novel anti-pS322-VASP antibody specifically generated for this site. Control blots were performed for protein input (anti-PKD1, anti-GST).
Mimicking Ser-157 phosphorylation (VASP.S157E mutation) did not lead to Ser-322 phosphorylation (Fig. 2A), and mimicking Ser-322 phosphorylation (VASP.S322E mutation) did not lead to Ser-157 phosphorylation (Fig. 2B).
FIGURE 2.
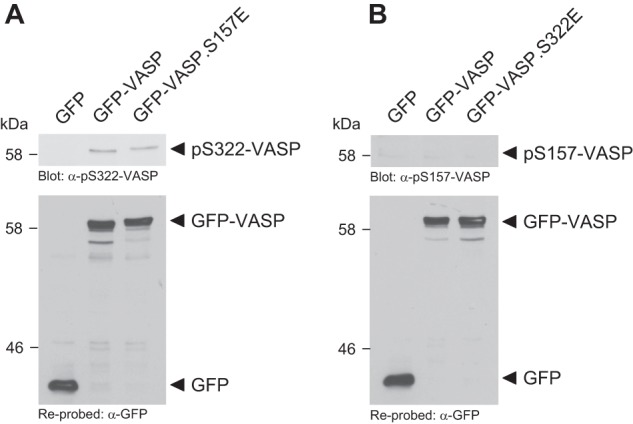
PKD1-mediated phosphorylations of VASP are independent of each other. A, mimicking Ser-157 phosphorylation does not prime for Ser-322 phosphorylation. HeLa cells were transfected with GFP-VASP, GFP-VASP.S157E, or vector control as indicated. Cells were lysed and analyzed by Western blot for phosphorylation at Ser-322 using a phosphospecific antibody. VASP expression was controlled by re-probing with anti-GFP. B, mimicking Ser-322 phosphorylation does not prime for Ser-157 phosphorylation. HeLa cells were transfected with GFP-VASP, GFP-VASP.S322E, or vector control as indicated. Cells were lysed and analyzed by Western blot for phosphorylation at Ser-157 using a phosphospecific antibody. VASP expression was controlled by re-probing with anti-GFP.
PKD1-mediated Phosphorylations of VASP Occur in Vivo and Are Induced by RhoA
To determine if PKD1 can phosphorylate VASP at both sites in vivo, we co-expressed VASP, VASP.S157A, VASP.S322A, or the VASP.S157A.S322A double mutant with constitutively active PKD1 in NMuMG cells and determined their phosphorylation using the anti-pMOTIF antibody (Fig. 3A). The data obtained in cell culture confirmed data from in vitro kinase assays showing that a VASP.S157A.S322A double mutant cannot be phosphorylated by PKD1. We also compared wild type VASP or above mutants in absence or presence of active PKD1 in a second cell line (HeLa), this time probing for phosphorylations at Ser-157 or Ser-322 using phosphospecific anti-pS157-VASP or anti-pS322-VASP antibodies. In HeLa, active PKD1 also induced the phosphorylation of VASP at both sites, although in this cell line Ser-322 showed slight basal phosphorylation (Fig. 3B). While phosphorylation of Ser-157 led to a mobility shift, phosphorylation of Ser-322 did not affect VASP mobility.
FIGURE 3.
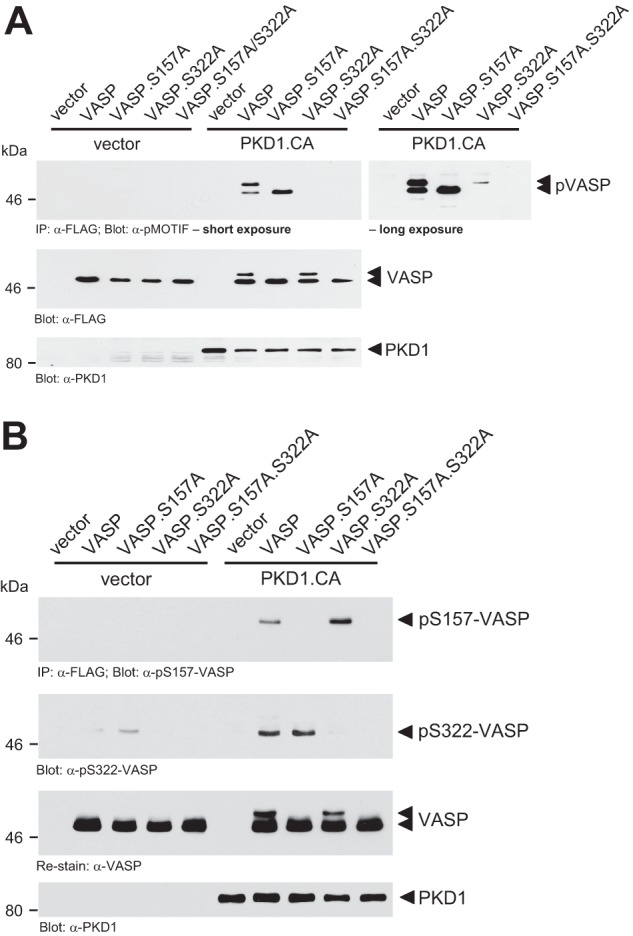
PKD1-mediated phosphorylations of VASP occur in vivo. A, NMuMG cells (0.5 × 106 cells, 6-cm dish) were transfected with vector control, HA-tagged constitutively active PKD1 (PKD1.CA) and FLAG-tagged VASP, VASP.S157A, VASP.S322A, or VASP.S157A.S322A as indicated. VASP was immunoprecipitated (anti-FLAG) and analyzed for PKD1-mediated phosphorylation using the pMOTIF antibody. Shown are short and long exposures. Samples were re-probed with anti-FLAG for equal expression of wild type or mutant VASP. Control Western blots were performed for PKD1.CA expression (anti-PKD1). B, HeLa cells (0.65 × 106 cells, 6-cm dish) were transfected as indicated. VASP was immunoprecipitated (anti-FLAG) and analyzed for phosphorylation at Ser-157 or Ser-322 using anti-pS157-VASP or anti-pS322-VASP antibodies. Samples were re-probed with anti-FLAG for equal loading of wild type or mutant VASP. Control Western blots were performed for PKD1.CA expression (anti-PKD1).
Next we determined the induction of such signaling by a PKD1 activating stimulus. In HeLa cells the RhoGTPase RhoA is a potent activator of PKD1 (25). We found that in response to active RhoA, VASP is phosphorylated at both, Ser-157 and Ser-322 and that this is dependent on PKD (Fig. 4A). Moreover, inhibition of PKD activity using the PKD inhibitor CID755673 blocked the RhoA-induced phosphorylation of endogenous VASP at Ser-322 and partially blocked phosphorylation at Ser-157 (Fig. 4B). Taken together this indicates that PKD-mediated phosphorylations of endogenous VASP can occur downstream of RhoA.
FIGURE 4.
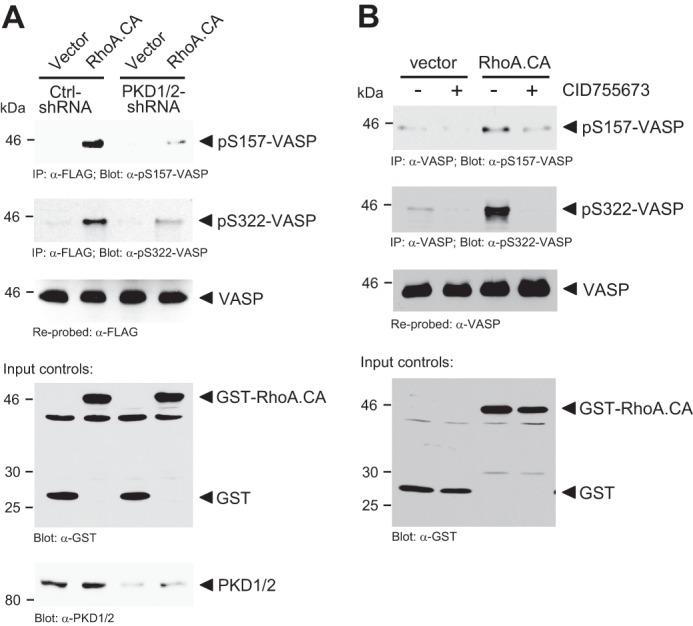
PKD1-mediated phosphorylations of VASP occur in response to active RhoA. A, HeLa cells (0.35 × 106 cells, 6-cm dish) were lentivirally infected with scrambled control or PKD-shRNA as indicated. The next day, cells were additionally transfected with combinations of VASP, vector control (pEBG), or active RhoA.CA (pEBG-RhoA.CA) as indicated. VASP was immunoprecipitated (anti-FLAG) and analyzed for phosphorylation at Ser-157 or Ser-322 using indicated phosphospecific antibodies. Samples were re-probed with anti-FLAG for VASP. Control Western blots were performed for PKD1 knockdown (anti-PKD1/2) and for RhoA.CA expression (anti-GST). B, HeLa cells (0.35 × 106 cells, 6-cm dish) were transfected with vector control (pEBG) or active RhoA.CA (pEBG-RhoA.CA) and treated with PKD inhibitor (CID755673, 20 μm) as indicated. Endogenous VASP was immunoprecipitated (anti-VASP) and analyzed for phosphorylation at Ser-157 or Ser-322 using indicated phosphospecific antibodies. Samples were re-probed with anti-VASP. Control Western blots were performed for RhoA.CA expression (anti-GST).
PKD1-mediated Phosphorylations Target VASP to the Leading Edge
Phosphorylation of VASP at S157 previously was shown to contribute to its localization to the leading edge, but also to have minor impact on VASP-induced F-actin assembly (19). In HeLa cells, endogenous VASP was localized to focal contacts as well as to the leading edge (Fig. 5A). Inhibition of PKD with the PKD-specific inhibitor CID755673 led to a loss of VASP localization to the leading edge, but retained VASP at focal contacts, indicating that PKD1-mediated phosphorylations contribute to redistribution of VASP to the leading edge (Fig. 5B). To further determine how Ser-157 and Ser-322 phosphorylations impact VASP localization we utilized a mutational approach. We compared the localization of phosphorylation-mimicking mutants such as VASP.S157E, VASP.S322E, and VASP.S157E.S322E (Fig. 6, A–J). The VASP.S157E mutant, consistent with previous publications, showed increased localization to the lamellipodium edge (Fig. 6B). When overexpressed, wild type and mutant VASP, all showed cytosolic expression. In addition, VASP.S322E mutant, similar to wild type VASP, was localized to focal contacts, but also at the leading edge (Fig. 6, A and C). The double mutant exclusively localized to the leading edge region, where it co-localized with F-actin (Fig. 6, J, M, P). To determine if Ser-322 phosphorylation is required for VASP location to the periphery we also investigated the location of VASP.S322A and VASP.S157E.S322A mutants. Both were localized to the focal contacts (Fig. 6, K, L, N, O, Q, R). Taken together, this indicates that phosphorylation of VASP at both serines is needed to drive it from focal contacts to the leading edge.
FIGURE 5.
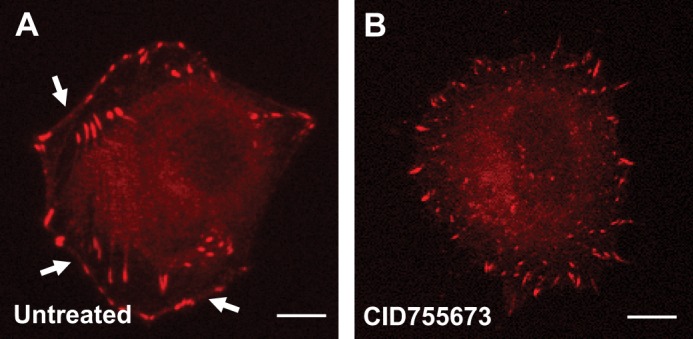
PKD activity is necessary for localization of VASP to the cell periphery. A and B, HeLa cells were treated with the PKD inhibitor CID755673 or left untreated. After 16 h, samples were fixed and analyzed for localization of endogenous VASP using immunofluorescence. Shown is a representative cell under each condition. In untreated cells, arrows indicate localization of VASP at the leading edge/cell periphery (bar is 10 μm).
FIGURE 6.
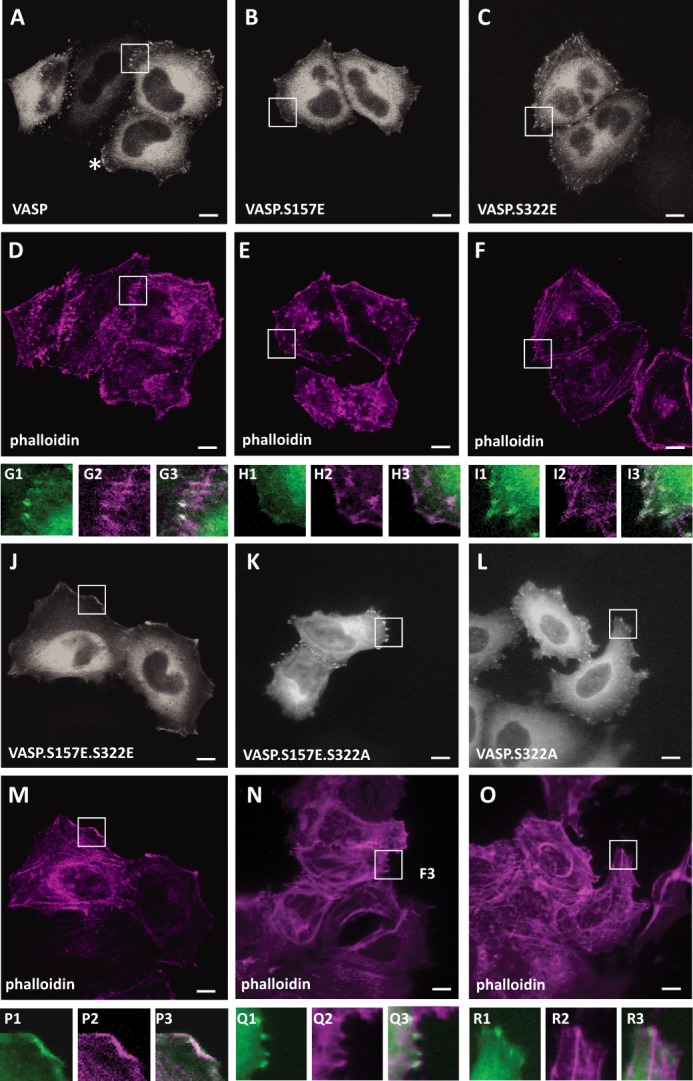
Ser-157 and Ser-322 phosphorylation is necessary for localization of VASP to the cell periphery. A–R, HeLa cells were transfected with GFP-tagged VASP, VASP.S157E, VASP.S322E, VASP.S157E.S322E, VASP.S157E.S322A, or VASP.S322A as indicated. After 24 h cells were fixed, and F-actin was stained with phalloidin. Localization of GFP or GFP-tagged proteins was determined using immunofluorescence analysis (bar is 10 μm). Insets are 2.5-fold enhanced and shown in G–I and P–R. VASP is stained in gray (green in insets), phalloidin in deep red. G3, H3 I3, P3, Q3, R3 show an overlay. In A–O, changes in cellular localization were observed in all cells analyzed (typically n = 100). All data were confirmed in at least three independent experiments.
PKD1 Increases VASP-mediated F-actin Polymerization
VASP at the leading edge can contribute to F-actin filament extension. Therefore, we tested if VASP phosphorylation by PKD1 has an impact on F-actin polymerization in cells. In in vitro pyrene actin polymerization assays, using only purified VASP and PKD1, alone or in combination, we did not observe significant changes in actin polymerization, when compared with actin alone (not shown). This indicates that VASP or PKD1-phosphorylated VASP does not serve as a nucleation-promoting factor (NPF) per se. To address if the phosphorylation of VASP by PKD1 affects F-actin accumulation in cells, we performed a serum response factor (SRF) gene reporter assay using a SRE (serum response element)-dependent luciferase reporter assay (Fig. 7, A and B). Such assays have been shown to quantify the ratio of G-actin to F-actin by stimulation of SRF and can be used as tool to quantify the effects of actin binding proteins downstream of RhoA and to monitor filament assembly in living cells (39, 40). In similar assays, VASP overexpression has been shown to induce F-actin assembly and SRE-driven luciferase activity (41). Expression of active PKD1 in cells alone increased SRE activity, most likely acting through endogenous VASP. When comparing VASP and VASP.S157A, VASP.S322A and VASP.S157A.S322A mutants for their effects on actin assembly, we found that in presence of active PKD1 only wild type VASP but none of the mutants showed increased actin assembly (Fig. 7A). Moreover, when compared with wild type VASP, the phosphorylation mimicking mutants VASP.S157E and VASP.S322E showed increased F-actin assembly. Mimicking both phosphorylations (VASP.S157E.S322E) increased F-actin accumulation even further, indicating that both phosphorylations may act additive (Fig. 7B).
FIGURE 7.

VASP phosphorylation is necessary for actin polymerization. A, Hek293T cells (0.5 × 106 cells/well, 6-well plate) were transfected with vector control, HA-tagged constitutively active PKD1 (PKD1.CA) and VASP or VASP mutants as indicated, as well as SRE promoter luciferase and Renilla luciferase reporters. Induced luciferase activity was measured. Error bars shown represent standard deviations. * indicates statistical significance (p < 0.05) as compared with vector control. p values were acquired with the Student's t test using Graph Pad software. Protein expression was controlled by Western blotting with anti-FLAG (VASP), anti-PKD1 (PKD1), or anti-tubulin (loading control) antibodies. B, Hek293T cells (0.5 × 106 cells/well, 6-well plate) were transfected with vector control, GFP-tagged VASP or VASP.S157E.S322E mutant as indicated, as well as SRE promoter luciferase and Renilla luciferase reporters. Induced luciferase activity was measured. Error bars shown represent standard deviations. * indicates statistical significance (p < 0.05) as compared with vector control; ** indicates statistical significance (p < 0.05) as compared with wild type VASP. p values were acquired with the Student's t test using Graph Pad software. Protein expression and loading was controlled by Western blotting with anti-GFP (VASP) or anti-β-actin (loading control) antibodies.
We then measured β-actin recovery after photo bleach in cells expressing VASP or the VASP.S157E.S322E mutant (Fig. 8A). Cells expressing wild type VASP showed a halftime of maximal recovery (t1/2) of 32.14 ± 3.4 s at sites of VASP localization, whereas cells expressing the mutant VASP showed a halftime of recovery of 15.12 ± 1.9 s. Moreover, in cells expressing VASP the GFP-β-actin immobile fraction was at ∼19.6% ± 4.1%, whereas the immobile fraction in cells expressing VASP.S157E.S322E was significantly lower at ∼12.1% ± 4.0% (Fig. 8B). This indicates that structures formed at location of VASP show less actin dynamics than structures formed at location of the VASP.S157E.S322E mutant.
FIGURE 8.
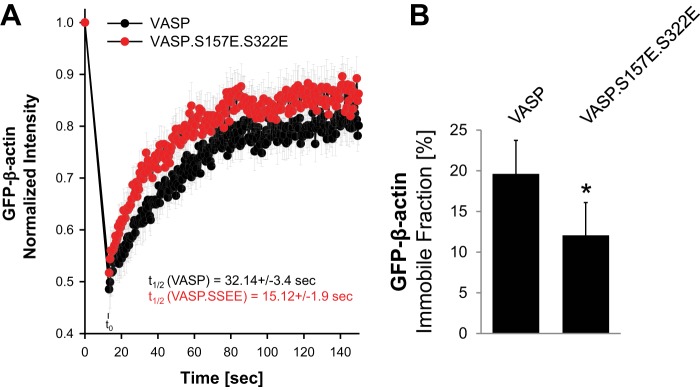
β-Actin recovery and mobility are different in cells expressing VASP or the VASP. S157E.S322E mutant. A and B, HeLa cells were co-transfected with GFP-β-actin and mCherry-VASP or mCherry-VASP.S157E.S322E. FRAP analysis was performed to measure actin recovery and mobility. At least 10 cells were analyzed per transfection condition. * indicates statistical significance (p < 0.05).
PKD1-mediated Phosphorylation of VASP Induces Leading Edge Formation and Elongated Filopodia
Next we compared membrane protrusions at the sites of VASP or VASP.S157E.S322E localization. Therefore, we analyzed cells expressing GFP-actin and Cherry-tagged wild type VASP or VASP.S157E.S322E mutant by live cell imaging. Cell movement was captured for 20 min as indicated under “Experimental Procedures.” Cherry-VASP was mainly located at the focal contacts but also in the cytosol (Fig. 9A). We then performed kymographic analyses, in which we determined membrane protrusion at the site of VASP localization (Fig. 9B). Wild type VASP mainly located at the focal contacts. In cells expression wild type VASP continuous membrane protrusion was only observed at the site where a potential lamellipodium was formed (Fig. 9B, kymographs of area B3 and B4; red asterisk).
FIGURE 9.

PKD1-mediated phosphorylation of VASP induces leading edge formation and elongated filopodia. A–F, HeLa cells were co-transfected with GFP-actin and Cherry-VASP or the Cherry-VASP.S157E.S322E mutant as indicated. The next day cells were reseeded into ibidi μ-Slides in phenol-red free media, and after 12 h live cell imaging was performed as described under “Experimental Procedures.” A1, A2, C1, and C2 show single channel pictures for Cherry-VASP, Cherry-VASP.S157E.S322E or GFP-actin. The bar represents 10 μm. The insets in A and C show typical localization of VASP (at the focal contacts) or VASP.S157E.S322E (red, VASP, green, actin, yellow, overlay). The bar shows 10 μm. B and D show kymographic analysis (t = 1200 s, length of line in B = 17.3 μm, in C = 18.2 μm) of several areas at sites of VASP or VASP mutant location. E shows single pictures of an area from C showing rapid formation, protrusion, and retraction of filopodia at sites of Cherry-VASP.S157E.S322E localization. Pictures shown are at a difference of 20 s. Cherry-VASP.S157E.S322E is in red pseudocolor (at t = 0), GFP-actin is in gray in all pictures. F and G show quantifications of distances from nuclei to the leading edge (F) or length of filopodia formed (G). Analysis was performed using Image J. p values were acquired with the student's t test using Graph Pad software. In F, n represents the number of cells (50) analyzed for nucleus to leading edge distance, combined from three independent experiments. In G, n represents the numbers of filopodia analyzed for their length from 10 different cells, combined from three independent experiments.
In contrast to wild type VASP, the expression of a VASP.S157E.S322E mutant led to the formation of large lamellipodium-like areas (Fig. 9, C and D). In all of the sites of VASP.S157E.S322E location at the leading edge within the 20 min time range we observed constant membrane protrusion and retraction, along with filopodia formation, but no persistent membrane protrusion that would be needed for directed cell migration (Fig. 9D, kymographs of line scans D2-D7, Fig. 9E). Quantitative analysis of cells (n = 50) showed that both the distance between nucleus and edge of cells (Fig. 9F) and the relative filopodia length (Fig. 9G) are increased in cells expressing VASP with phosphorylation mimicking mutations. This indicates that phosphorylation of VASP by PKD1 re-localizes VASP from focal contacts to the leading edge where it induces filopodia formation.
Persistent PKD1-mediated VASP Phosphorylations Induce Membrane Ruffling and Negatively Affect Cell Migration
Results so far were surprising, since PKD1 previously was described to inhibit cofilin-driven directed cell migration, but by phosphorylating VASP it seems to contribute to lamellipodium and filopodia formation. One explanation of how this could lead to decrease cell motility is that (if persistent) PKD1/VASP signaling leads to loss of directionality due to formation of multiple lamellipodium-like structures. Another explanation is that filopodia-formed show increased filament elongation and instability of focal contacts, when PKD1 signaling is persistently ON. An expected result of such signaling would be increased membrane ruffling and decreased cell migration. Therefore, we next determined the functional consequences of persistent PKD1/VASP signaling on cell migration. As predicted, the expression of active PKD1 led to the localization of wild type VASP to the leading edge region, where it induced membrane ruffling (Fig. 10, A–D). A VASP.S157A.S322A mutant in presence of active PKD1 did not induce membrane ruffling (Fig. 10, E–H) and did not co-localize with active PKD1.
FIGURE 10.

PKD1-mediated VASP phosphorylations induce membrane ruffling and negatively affect cell migration. A–H, HuMEC cells were co-transfected with GFP-tagged active PKD1 (GFP-PKD1.CA) and VASP, or the VASP.S157A.S322A mutant as indicated. 16 h after transfection, cells were fixed, and F-actin was stained with phalloidin. Localization of proteins was determined using immunofluorescence analysis (bar is 10 μm). Insets are 2.5-fold enhanced. Changes in cellular localization were observed in all cells (typically n = 50) analyzed. All data were confirmed in at least three independent experiments. I and J, HeLa cells were transfected with vector control, VASP or VASP phosphorylation mimicking mutant as indicated (I), or with vector or PKD1.CA in combination with VASP or the VASP.S157A.S322A mutant (J). Real-time cell migration was monitored with an impedance-based assay system (ECIS) over a time period of 5 h. K, schematic of the here proposed PKD1-mediated signaling events downstream active RhoA that lead to VASP localization to the leading edge of cells, to filopodia formation and cause F-actin filament extension. If persistent such signaling results in membrane ruffling and eventually to decreased cell migration.
We next tested if PKD1-mediated phosphorylation of VASP can have a negative impact on cell migration. Therefore, we performed real-time wound healing assays using the ECIS system. First we compared the migratory potential of cells expressing vector control, wild type VASP or the phosphorylation-mimicking mutant VASP.S157E.S322E over a time period of 5 h. We found that cells expressing the phosphorylation-mimicking VASP mutant showed decreased migratory potential as compared with wild type VASP (Fig. 10I). Similar effects were observed in impedance-based transwell assays measuring real-time directed cell migration toward a chemoattractant using the xCELLigence system (data not shown). We also compared the motility of cells when VASP or the VASP.S157A.S322A double mutant was co-expressed with active PKD1 or vector control. The presence of active PKD1 and subsequent phosphorylation of VASP significantly decreased HeLa cell motility, whereas a double blunting mutant rescued cell motility in the presence of active PKD1 (Fig. 10J).
Taken together, our data suggest that in response to RhoA signaling PKD1-mediated phosphorylation of VASP localizes VASP to the leading edge. When persistent, this signaling results in increased filopodia formation and length, leading to membrane ruffling and decreased cell migration (Fig. 10K).
DISCUSSION
Protein kinase D1 interacts with F-actin and regulates actin reorganization at several levels (22). For example, in response to RhoA activation it modulates the activity of cofilin through phosphorylation of PAK4 in its activation loop (29), but also through inactivation of the cofilin phosphatase SSH1L (26, 30). The net effect of such signaling is a decrease in the pool of active cofilin, resulting in less formation of F-actin free barbed ends (22, 26). Other PKD1 targets, whose phosphorylation mediate its effects on the motile phenotype include Rhotekin (42), RIN1 (32), E-cadherin (43), β-catenin (44), EVL-1 (31), Hsp27 (37), and cortactin (21).
We here investigated if the Ena/VASP family protein VASP is a target for PKD1 and found that PKD1 in response to active RhoA can directly phosphorylate VASP at two serine residues, Ser-157 and Ser-322 (Figs. 1, 2, 3, and 4). While Ser-322 so far only was identified as a PKD1 site, phosphorylation of VASP at Ser-157 has been shown to be mediated by several other kinases. It is likely that RhoA also mediates phosphorylation at Ser-157 via these kinases. For example, S157 was previously shown to be phosphorylated by PKA. However, PKD1-mediated phosphorylation at this residue is independent of PKA since forskolin, a vasodilator that raises the levels of cAMP to activate PKA, or treatment of cells with the PKA inhibitor H89 had no effects on PKD1-mediated VASP phosphorylations (data not shown). Further, forskolin did neither activate PKD1, nor did it lead to phosphorylation of VASP at Ser-322 (data not shown).
We also show that PKD1 phosphorylates VASP at a second serine residue, Ser-322. In contrast to phosphorylation at Ser-157, phosphorylation of VASP at Ser-322 did not induce a significant mobility shift in VASP in SDS-PAGE (Figs. 1, 3). Moreover, none of the phosphorylations primed for the other (Fig. 2). The two other previously described phosphorylation sites in VASP, Ser-239 and Thr-278, which both have been shown to impair actin-driven filament formation (19), are not located within a PKD1 phosphorylation motif (Fig. 1B). Moreover, PKD1-induced VASP phosphorylation did not induce binding of VASP to 14-3-3, or to focal contact proteins such as vinculin and zyxin (not shown).
Our data indicate that phosphorylation by PKD1 drives VASP from the focal contacts to the leading edge region (Figs. 5, 6, 9, 10). This is consistent with previous data showing that phosphorylation of Ser-157 can localize VASP to the leading edge, but has only minor impact on VASP-induced F-actin assembly (19). The Ser-322 phosphorylation site in VASP is located in the EVH-2 domain. The EVH-2 domain is required for efficient targeting of VASP to lamellipodia and filopodia (10), in which VASP is stably-associated with growing filaments (45). This domain can bind to G-actin and F-actin and also mediates the tetramerization of VASP, which is important for bundling of F-actin filaments and stabilization of actin structures (5). We did not observe any effects of PKD-mediated phosphorylations on multimerization of VASP (not shown).
The morphology and dynamics of membrane protrusions (filopodia or lamellipodium) can be altered by regulation of length of actin filaments and their stability and organization. In fibroblasts, genetic deletion or sequestration of Ena/VASP proteins led to faster cell motility with more persistent lamellipodial protrusions. The actin filaments within these lamellipodia were shorter and more branched, suggesting that filament capping activity was increased. Conversely, membrane targeting of Ena/VASP protein led to longer filaments with fewer branches (46). Moreover, VASP proteins localize to spreading, but not retracting lamellipodia (47). The increased actin polymerization activity that we observed (Figs. 7 and 8) when a phosphorylation mimicking mutant was compared with wild type VASP, directly correlated with increased length of protrusions such as filopodia and membrane ruffling (Figs. 9 and 10). One possible explanation for our data is that at the leading edge in response to PKD1-mediated phosphorylations VASP's anti-capping activity may lead to longer and branched filaments that contribute to membrane ruffling.
In summary, our data implicate that PKD1-mediated phosphorylations at Ser-157 and Ser-322 can drive VASP to the leading edge, where it induces filopodia formation and elongation. When persistent such signaling can result in membrane ruffling and decreased motility (Fig. 10).
Acknowledgments
We thank our colleagues in the Storz laboratory for helpful discussions.
This work was supported by National Institutes of Health Grants NS069753 (to P. Z. A.), GM086435, and CA140182 (to P. S.). This work was also supported by Bankhead-Coley Grant (10BG11) from the Florida Department of Health (to P. S.).
- Ena/VASP
- enabled/vasodilator-stimulated phosphoprotein
- AMPK
- AMP-activated protein kinase
- ECIS
- electric cell-substrate impedance sensing
- EVL
- Ena-VASP-like
- LIMK
- LIM domain kinase
- Mena
- mammalian-enabled
- PAK4
- p21-activated kinase 4
- PKA
- cAMP-dependent protein kinase
- PKD
- protein kinase D
- PKG
- cGMP-dependent protein kinase
- SSH1L
- slingshot 1L
- FRAP
- fluorescence recovery after photobleaching.
REFERENCES
- 1. Krause M., Dent E. W., Bear J. E., Loureiro J. J., Gertler F. B. (2003) Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19, 541–564 [DOI] [PubMed] [Google Scholar]
- 2. Kwiatkowski A. V., Gertler F. B., Loureiro J. J. (2003) Function and regulation of Ena/VASP proteins. Trends Cell Biol. 13, 386–392 [DOI] [PubMed] [Google Scholar]
- 3. Ball L. J., Kühne R., Hoffmann B., Häfner A., Schmieder P., Volkmer-Engert R., Hof M., Wahl M., Schneider-Mergener J., Walter U., Oschkinat H., Jarchau T. (2000) Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J. 19, 4903–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lambrechts A., Kwiatkowski A. V., Lanier L. M., Bear J. E., Vandekerckhove J., Ampe C., Gertler F. B. (2000) cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J. Biol. Chem. 275, 36143–36151 [DOI] [PubMed] [Google Scholar]
- 5. Bachmann C., Fischer L., Walter U., Reinhard M. (1999) The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J. Biol. Chem. 274, 23549–23557 [DOI] [PubMed] [Google Scholar]
- 6. Kühnel K., Jarchau T., Wolf E., Schlichting I., Walter U., Wittinghofer A., Strelkov S. V. (2004) The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. U.S.A. 101, 17027–17032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krause M., Bear J. E., Loureiro J. J., Gertler F. B. (2002) The Ena/VASP enigma. J. Cell Sci. 115, 4721–4726 [DOI] [PubMed] [Google Scholar]
- 8. Reinhard M., Jarchau T., Walter U. (2001) Actin-based motility: stop and go with Ena/VASP proteins. Trends Biochem. Sci. 26, 243–249 [DOI] [PubMed] [Google Scholar]
- 9. Reinhard M., Jouvenal K., Tripier D., Walter U. (1995) Identification, purification, and characterization of a zyxin-related protein that binds the focal adhesion and microfilament protein VASP (vasodilator-stimulated phosphoprotein). Proc. Natl. Acad. Sci. U.S.A. 92, 7956–7960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bear J. E., Gertler F. B. (2009) Ena/VASP: towards resolving a pointed controversy at the barbed end. J. Cell Sci. 122, 1947–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trichet L., Sykes C., Plastino J. (2008) Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J. Cell Biol. 181, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barzik M., Kotova T. I., Higgs H. N., Hazelwood L., Hanein D., Gertler F. B., Schafer D. A. (2005) Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J. Biol. Chem. 280, 28653–28662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skoble J., Auerbuch V., Goley E. D., Welch M. D., Portnoy D. A. (2001) Pivotal role of VASP in Arp2/3 complex-mediated actin nucleation, actin branch-formation, and Listeria monocytogenes motility. J. Cell Biol. 155, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Butt E., Abel K., Krieger M., Palm D., Hoppe V., Hoppe J., Walter U. (1994) cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 269, 14509–14517 [PubMed] [Google Scholar]
- 15. Zhuang S., Nguyen G. T., Chen Y., Gudi T., Eigenthaler M., Jarchau T., Walter U., Boss G. R., Pilz R. B. (2004) Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J. Biol. Chem. 279, 10397–10407 [DOI] [PubMed] [Google Scholar]
- 16. Blume C., Benz P. M., Walter U., Ha J., Kemp B. E., Renné T. (2007) AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J. Biol. Chem. 282, 4601–4612 [DOI] [PubMed] [Google Scholar]
- 17. Harbeck B., Hüttelmaier S., Schluter K., Jockusch B. M., Illenberger S. (2000) Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction with actin. J. Biol. Chem. 275, 30817–30825 [DOI] [PubMed] [Google Scholar]
- 18. Benz P. M., Blume C., Moebius J., Oschatz C., Schuh K., Sickmann A., Walter U., Feller S. M., Renné T. (2008) Cytoskeleton assembly at endothelial cell-cell contacts is regulated by αII-spectrin-VASP complexes. J. Cell Biol. 180, 205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benz P. M., Blume C., Seifert S., Wilhelm S., Waschke J., Schuh K., Gertler F., Münzel T., Renné T. (2009) Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J. Cell Sci. 122, 3954–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferron F., Rebowski G., Lee S. H., Dominguez R. (2007) Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 26, 4597–4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eiseler T., Hausser A., De Kimpe L., Van Lint J., Pfizenmaier K. (2010) Protein kinase D controls actin polymerization and cell motility through phosphorylation of cortactin. J. Biol. Chem. 285, 18672–18683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eiseler T., Schmid M. A., Topbas F., Pfizenmaier K., Hausser A. (2007) PKD is recruited to sites of actin remodelling at the leading edge and negatively regulates cell migration. FEBS Lett. 581, 4279–4287 [DOI] [PubMed] [Google Scholar]
- 23. Machesky L. M. (2000) Putting on the brakes: a negative regulatory function for Ena/VASP proteins in cell migration. Cell 101, 685–688 [DOI] [PubMed] [Google Scholar]
- 24. Wang Q. J. (2006) PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol. Sci. 27, 317–323 [DOI] [PubMed] [Google Scholar]
- 25. Cowell C. F., Yan I. K., Eiseler T., Leightner A. C., Döppler H., Storz P. (2009) Loss of cell-cell contacts induces NF-κB via RhoA-mediated activation of protein kinase D1. J. Cell Biochem. 106, 714–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eiseler T., Döppler H., Yan I. K., Kitatani K., Mizuno K., Storz P. (2009) Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 11, 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan J., Rey O., Rozengurt E. (2006) Activation of protein kinase D3 by signaling through Rac and the α subunits of the heterotrimeric G proteins G12 and G13. Cell Signal 18, 1051–1062 [DOI] [PubMed] [Google Scholar]
- 28. Yuan J., Slice L. W., Rozengurt E. (2001) Activation of protein kinase D by signaling through Rho and the α subunit of the heterotrimeric G protein G13. J. Biol. Chem. 276, 38619–38627 [DOI] [PubMed] [Google Scholar]
- 29. Spratley S. J., Bastea L. I., Döppler H., Mizuno K., Storz P. (2011) Protein kinase D regulates cofilin activity through p21-activated kinase 4. J. Biol. Chem. 286, 34254–34261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peterburs P., Heering J., Link G., Pfizenmaier K., Olayioye M. A., Hausser A. (2009) Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 69, 5634–5638 [DOI] [PubMed] [Google Scholar]
- 31. Janssens K., De Kimpe L., Balsamo M., Vandoninck S., Vandenheede J. R., Gertler F., Van Lint J. (2009) Characterization of EVL-I as a protein kinase D substrate. Cell Signal 21, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziegler S., Eiseler T., Scholz R. P., Beck A., Link G., Hausser A. (2011) A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol. Biol. Cell 22, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Storz P., Toker A. (2003) Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 22, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hausser A., Storz P., Hübner S., Braendlin I., Martinez-Moya M., Link G., Johannes F. J. (2001) Protein kinase C mu selectively activates the mitogen-activated protein kinase (MAPK) p42 pathway. FEBS Lett. 492, 39–44 [DOI] [PubMed] [Google Scholar]
- 35. Hardy L. R. (2012) Current Protocols Neuroscience Chapter 2, Unit 2, page 17 [DOI] [PubMed] [Google Scholar]
- 36. Phair R. D., Misteli T. (2000) High mobility of proteins in the mammalian cell nucleus. Nature 404, 604–609 [DOI] [PubMed] [Google Scholar]
- 37. Döppler H., Storz P., Li J., Comb M. J., Toker A. (2005) A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 280, 15013–15019 [DOI] [PubMed] [Google Scholar]
- 38. Hausser A., Storz P., Märtens S., Link G., Toker A., Pfizenmaier K. (2005) Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat. Cell Biol. 7, 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- 40. Sotiropoulos A., Gineitis D., Copeland J., Treisman R. (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98, 159–169 [DOI] [PubMed] [Google Scholar]
- 41. Grosse R., Copeland J. W., Newsome T. P., Way M., Treisman R. (2003) A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J. 22, 3050–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pusapati G. V., Eiseler T., Rykx A., Vandoninck S., Derua R., Waelkens E., Van Lint J., von Wichert G., Seufferlein T. (2012) Protein kinase D regulates RhoA activity via rhotekin phosphorylation. J. Biol. Chem. 287, 9473–9483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jaggi M., Rao P. S., Smith D. J., Wheelock M. J., Johnson K. R., Hemstreet G. P., Balaji K. C. (2005) E-cadherin phosphorylation by protein kinase D1/protein kinase C{mu} is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 65, 483–492 [PubMed] [Google Scholar]
- 44. Du C., Jaggi M., Zhang C., Balaji K. C. (2009) Protein kinase D1-mediated phosphorylation and subcellular localization of β-catenin. Cancer Res. 69, 1117–1124 [DOI] [PubMed] [Google Scholar]
- 45. Applewhite D. A., Barzik M., Kojima S., Svitkina T. M., Gertler F. B., Borisy G. G. (2007) Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol. Biol. Cell 18, 2579–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bear J. E., Svitkina T. M., Krause M., Schafer D. A., Loureiro J. J., Strasser G. A., Maly I. V., Chaga O. Y., Cooper J. A., Borisy G. G., Gertler F. B. (2002) Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell 109, 509–521 [DOI] [PubMed] [Google Scholar]
- 47. Rottner K., Behrendt B., Small J. V., Wehland J. (1999) VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1, 321–322 [DOI] [PubMed] [Google Scholar]